
Calorie Restriction Using High-Fat/Low-Carbohydrate Diet Suppresses Liver Fat Accumulation and Pancreatic Beta-Cell Dedifferentiation in Obese Diabetic Mice

 , , ,
, , , 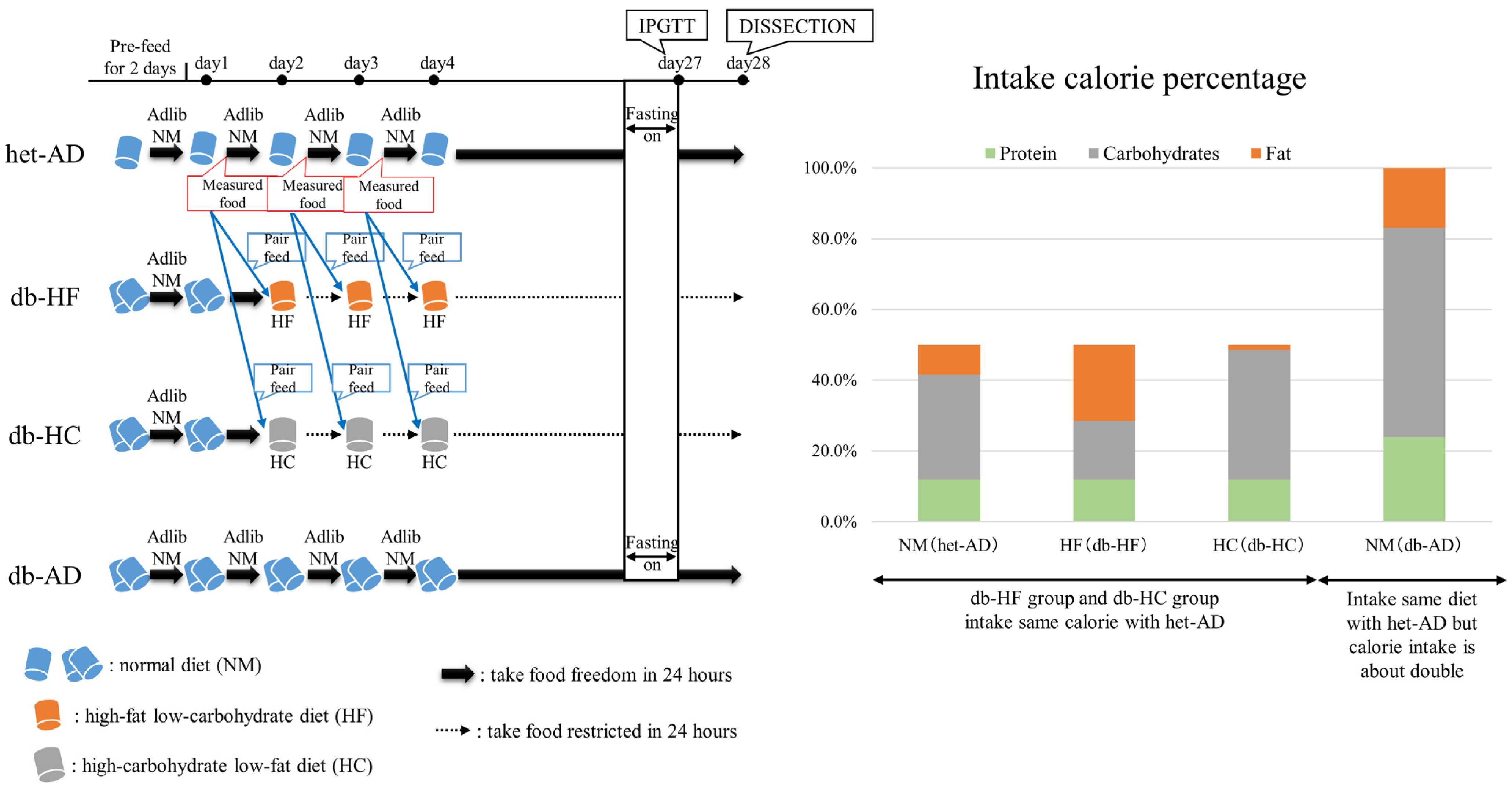{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Intraperitoneal Glucose Tolerance Test (IPGTT)
2.3. Pancreatic Islets Isolation
2.4. Glucose-Stimulated Insulin Secretion (GSIS)
2.5. Hematoxylin and Eosin (HE) Staining
2.6. Biochemical Examination of Blood and Liver
2.7. qPCR
2.8. RNA Sequencing (RNA-Seq)
2.9. Fluorescent Immunostaining
2.10. Statistical Analyses
3. Results
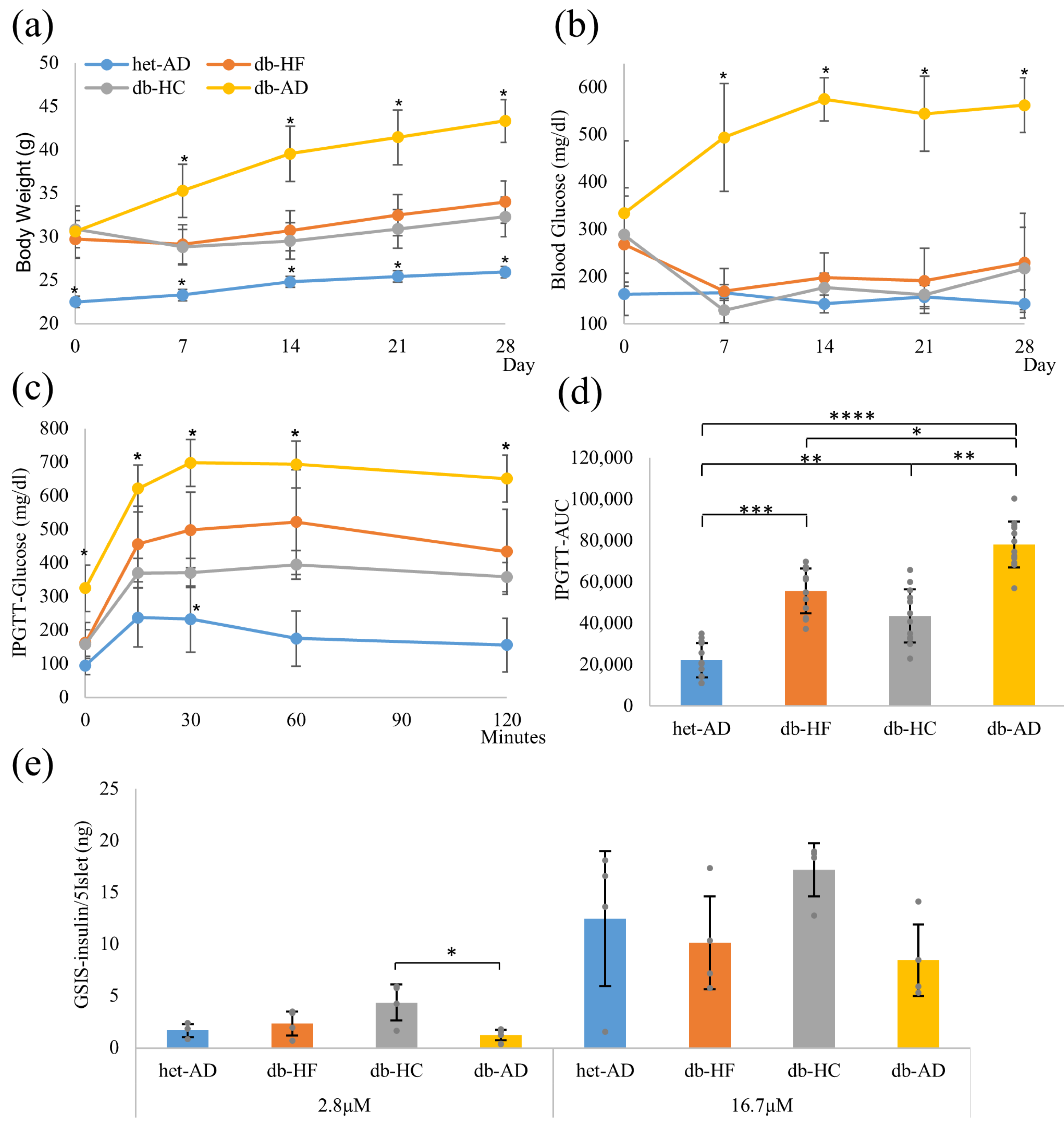
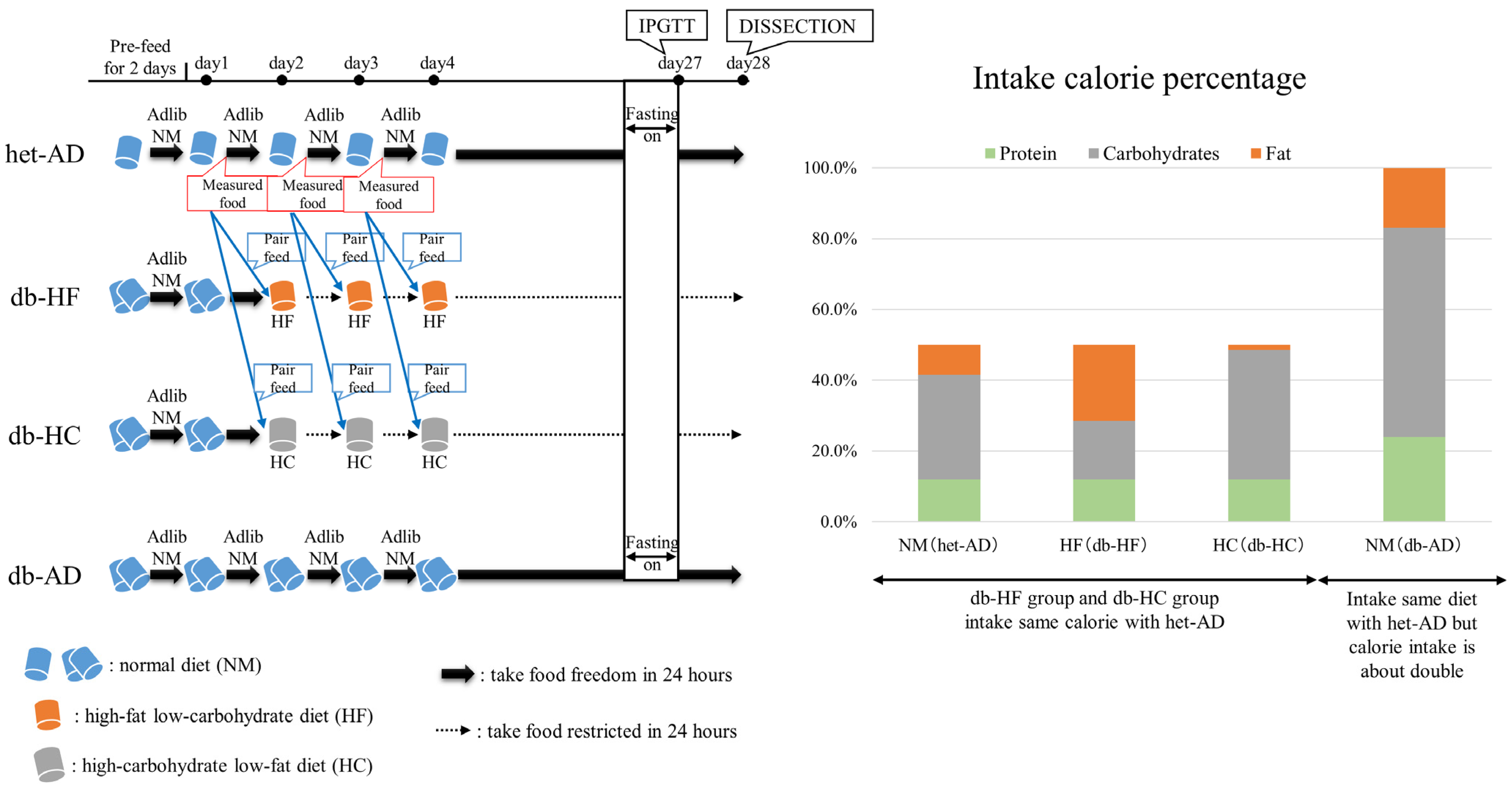
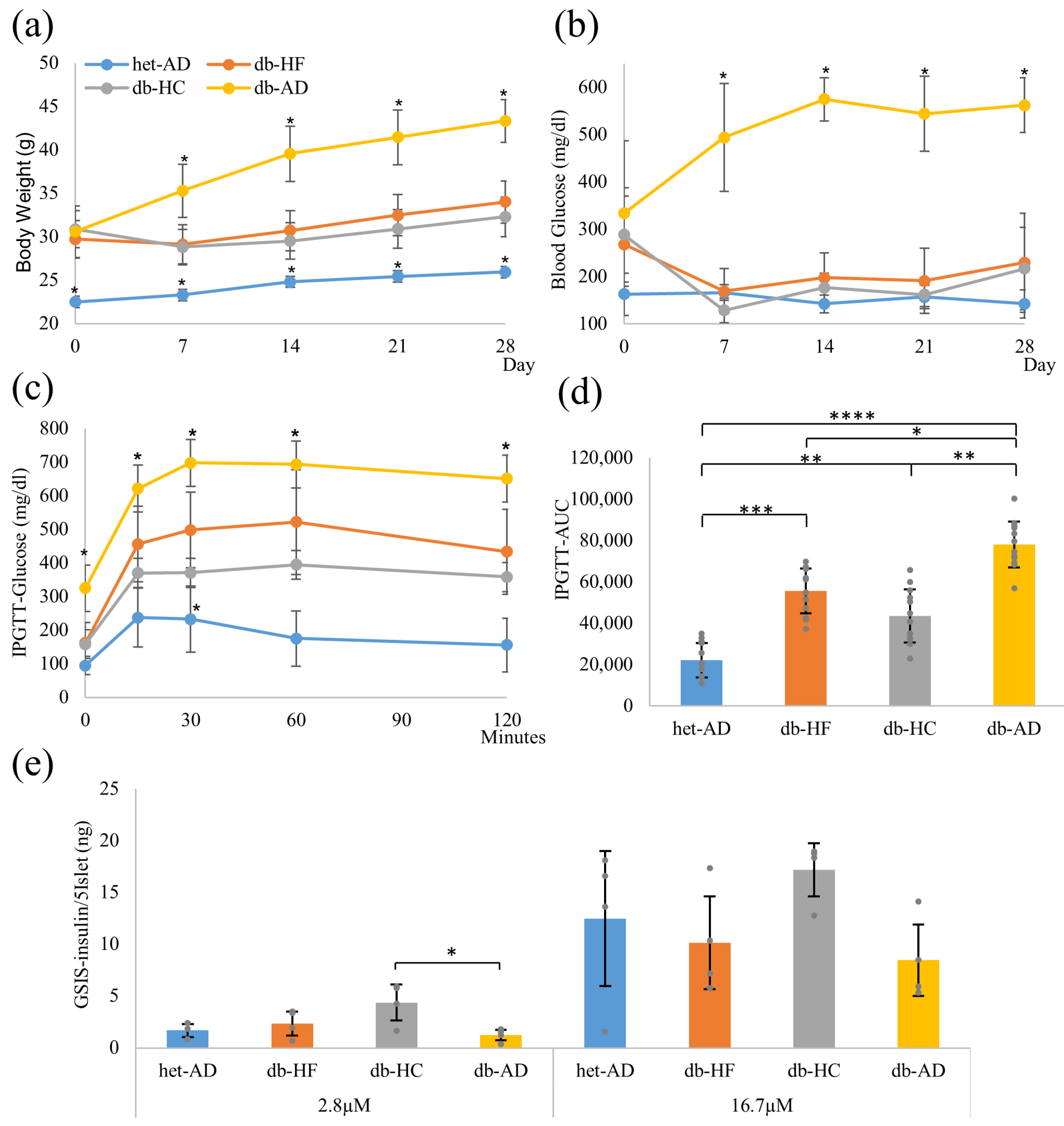
3.1. Calorie Restriction Lowered the Body Weight, Glucose Tolerance, and Insulin Secretion in Obese Mice, Regardless of the Carbohydrate/Fat Food Ratio
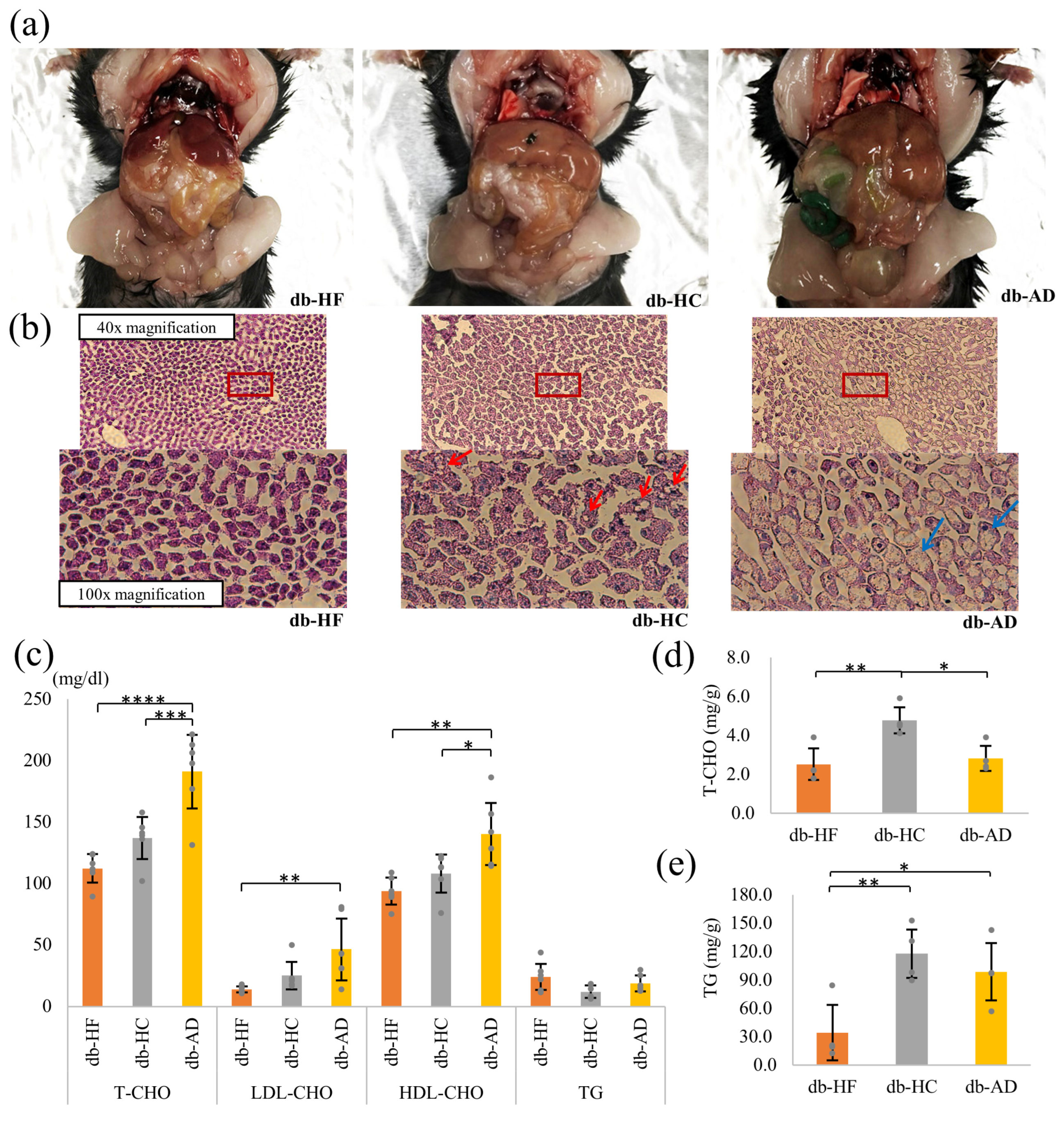
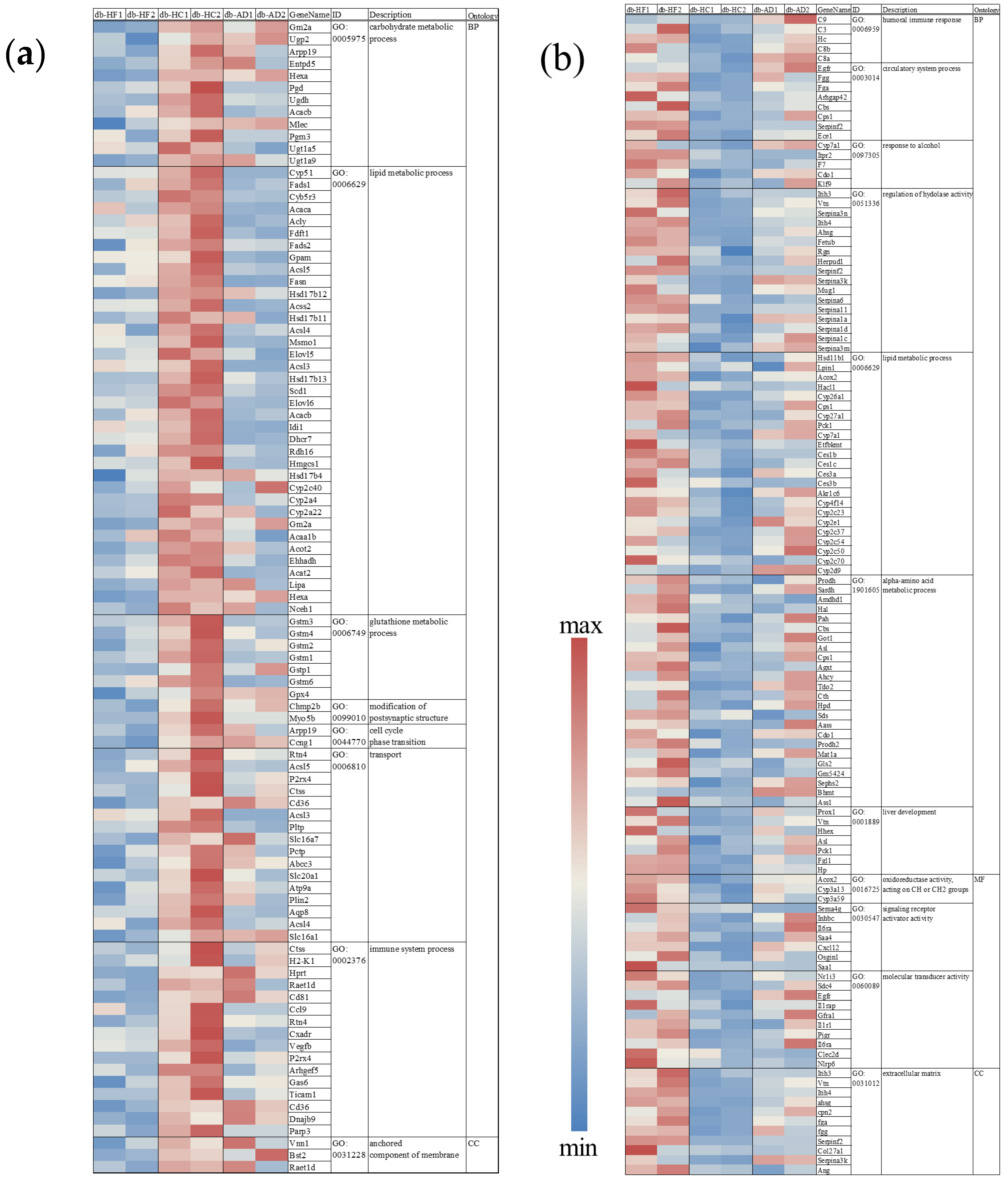
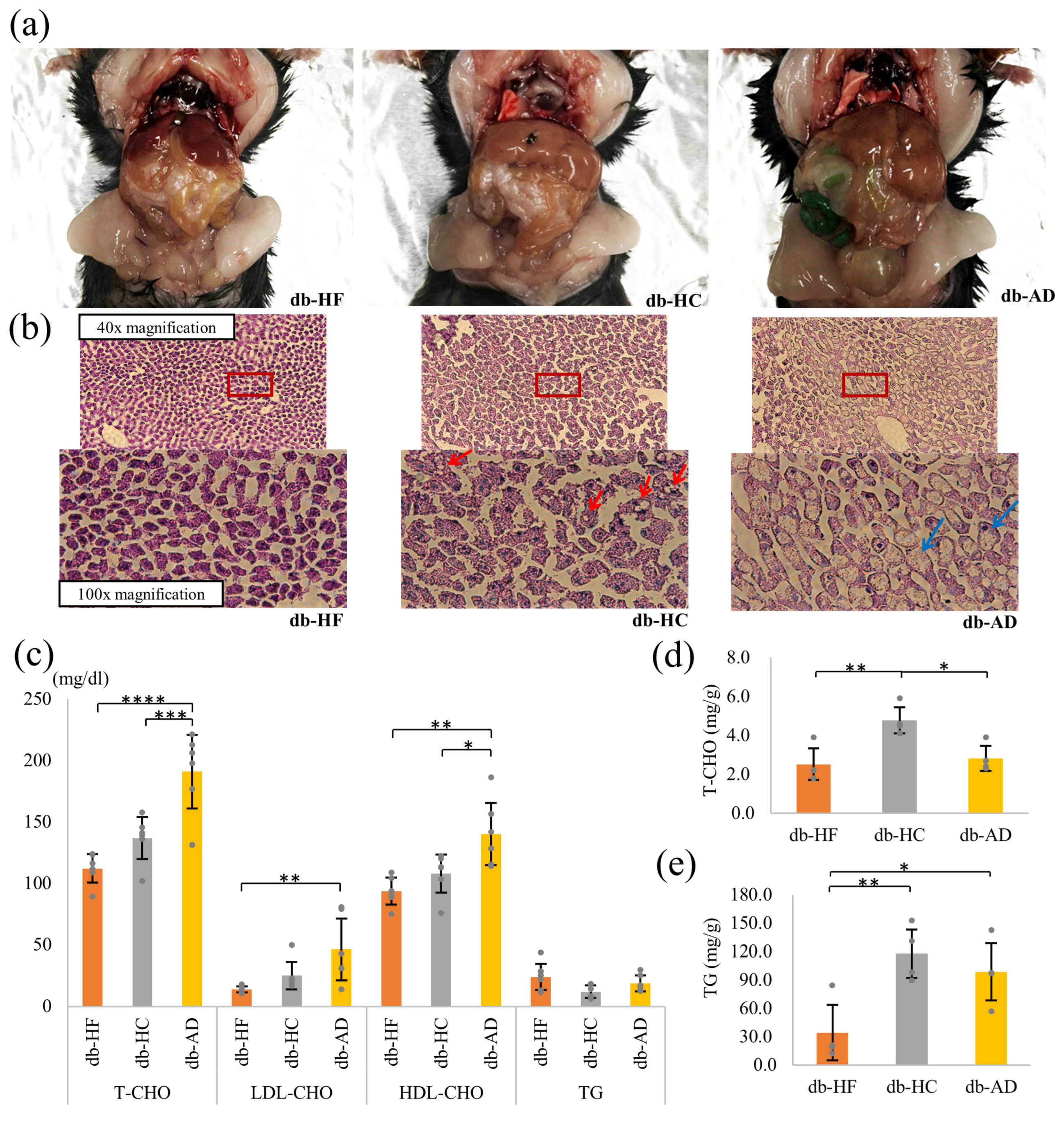
3.2. Calorie Restriction Using High-Carbohydrate Low-Fat Food Led to Hypercholesterolemia and Fat Accumulation in the Liver
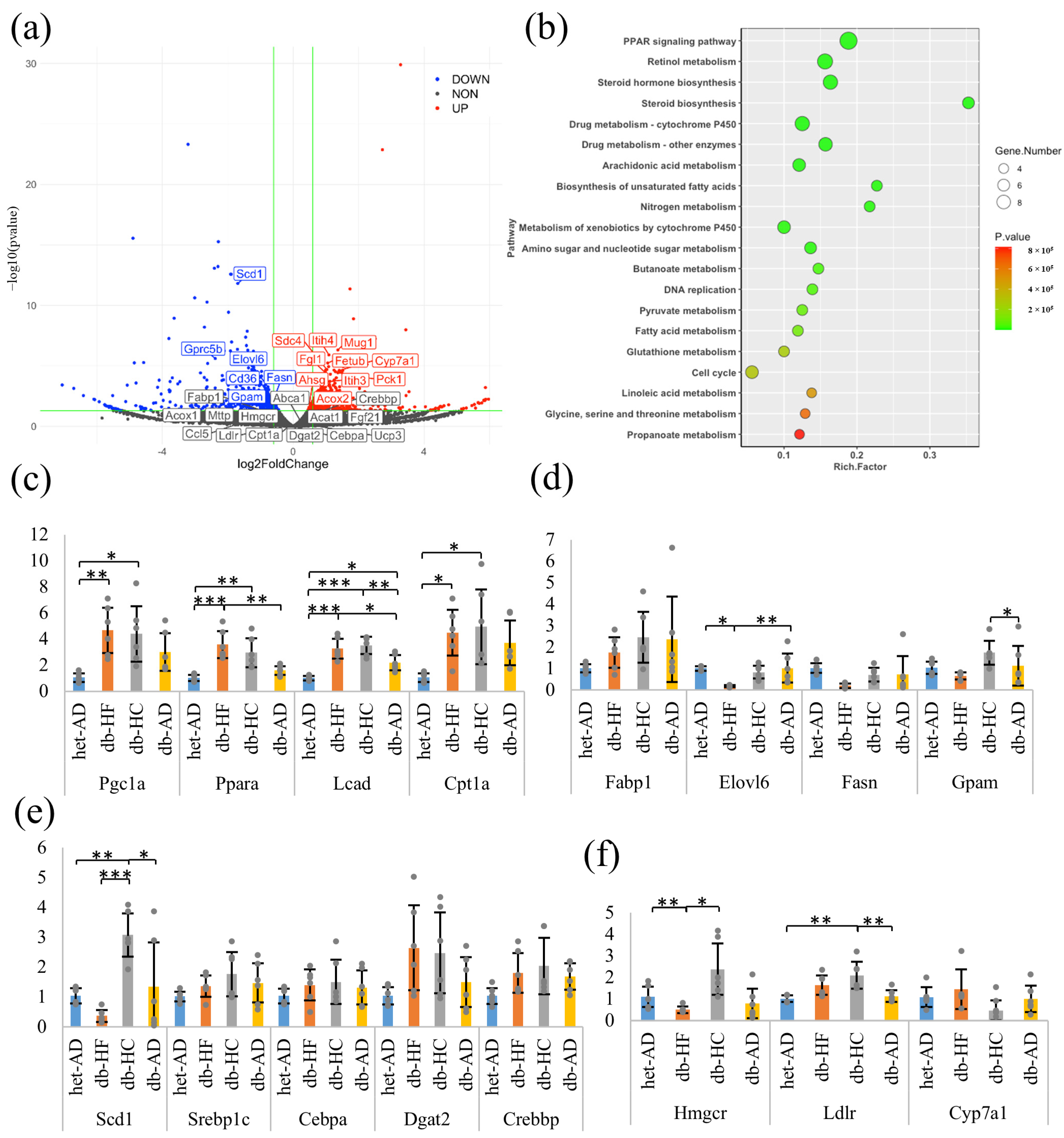
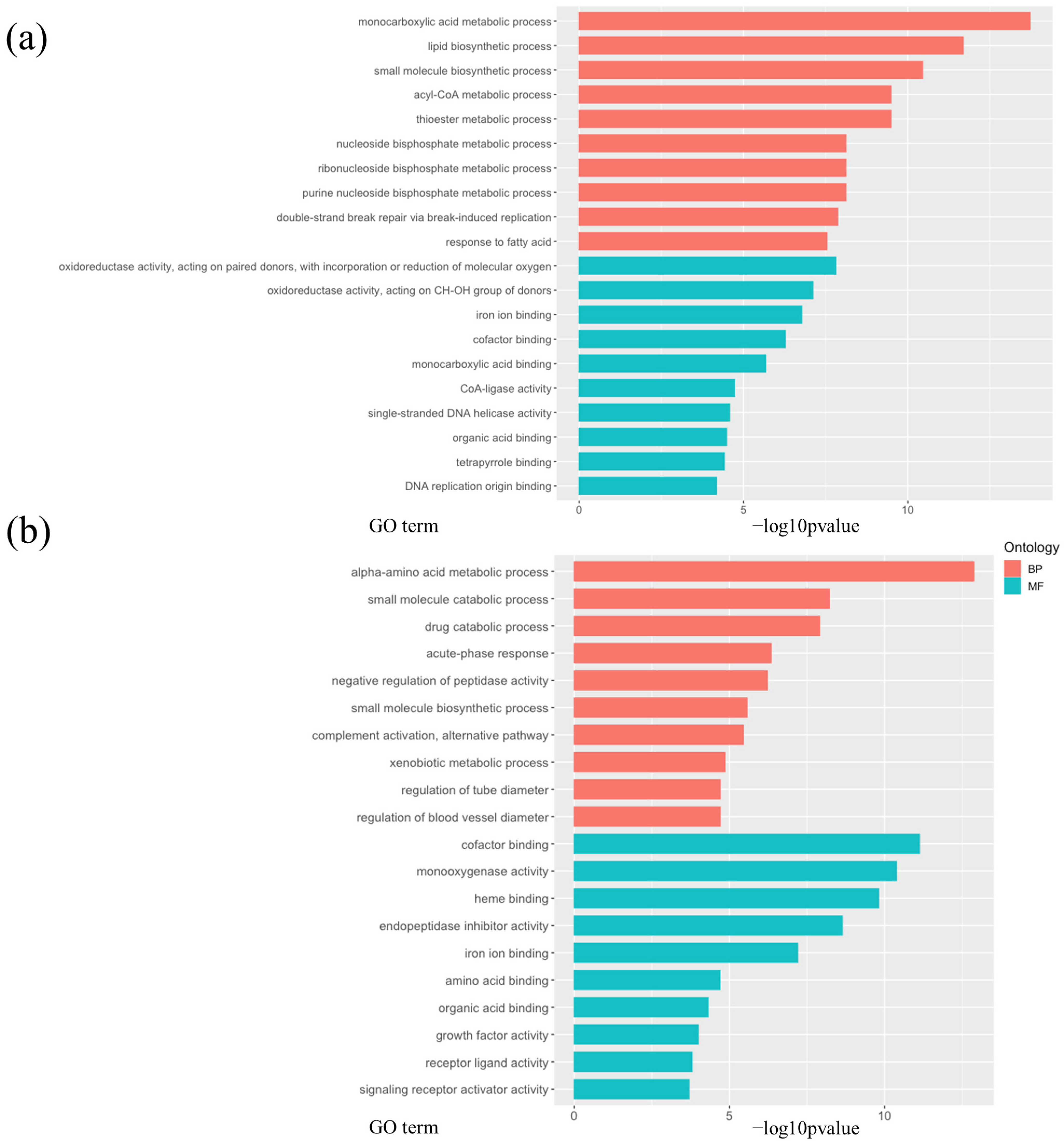
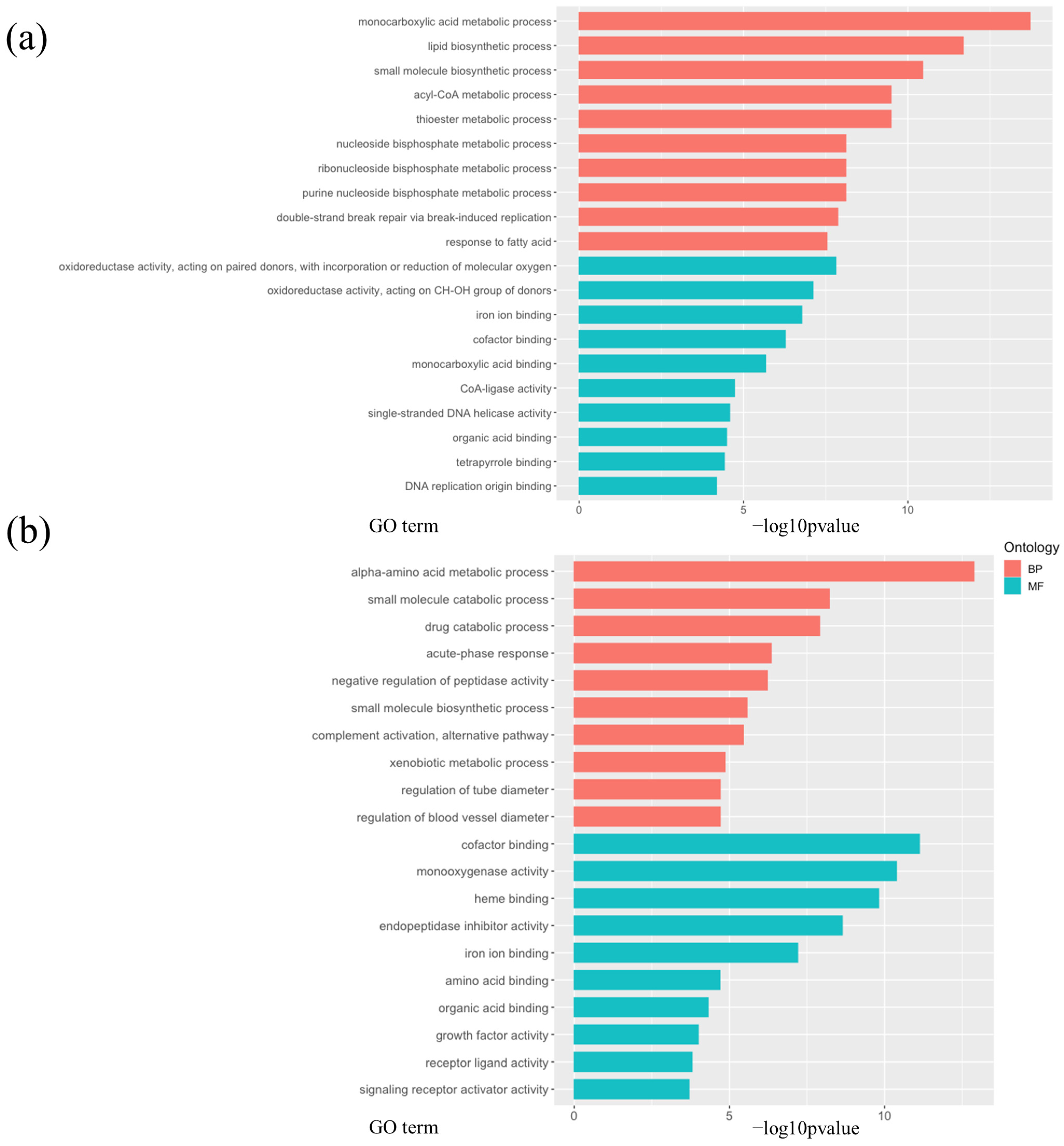
3.3. Calorie Restriction Using High-Carbohydrate Low-Fat Food Resulted in Increased Fatty Acid Elongation, Desaturation, and the Transportation of Long-Chain Fatty Acid in the Liver
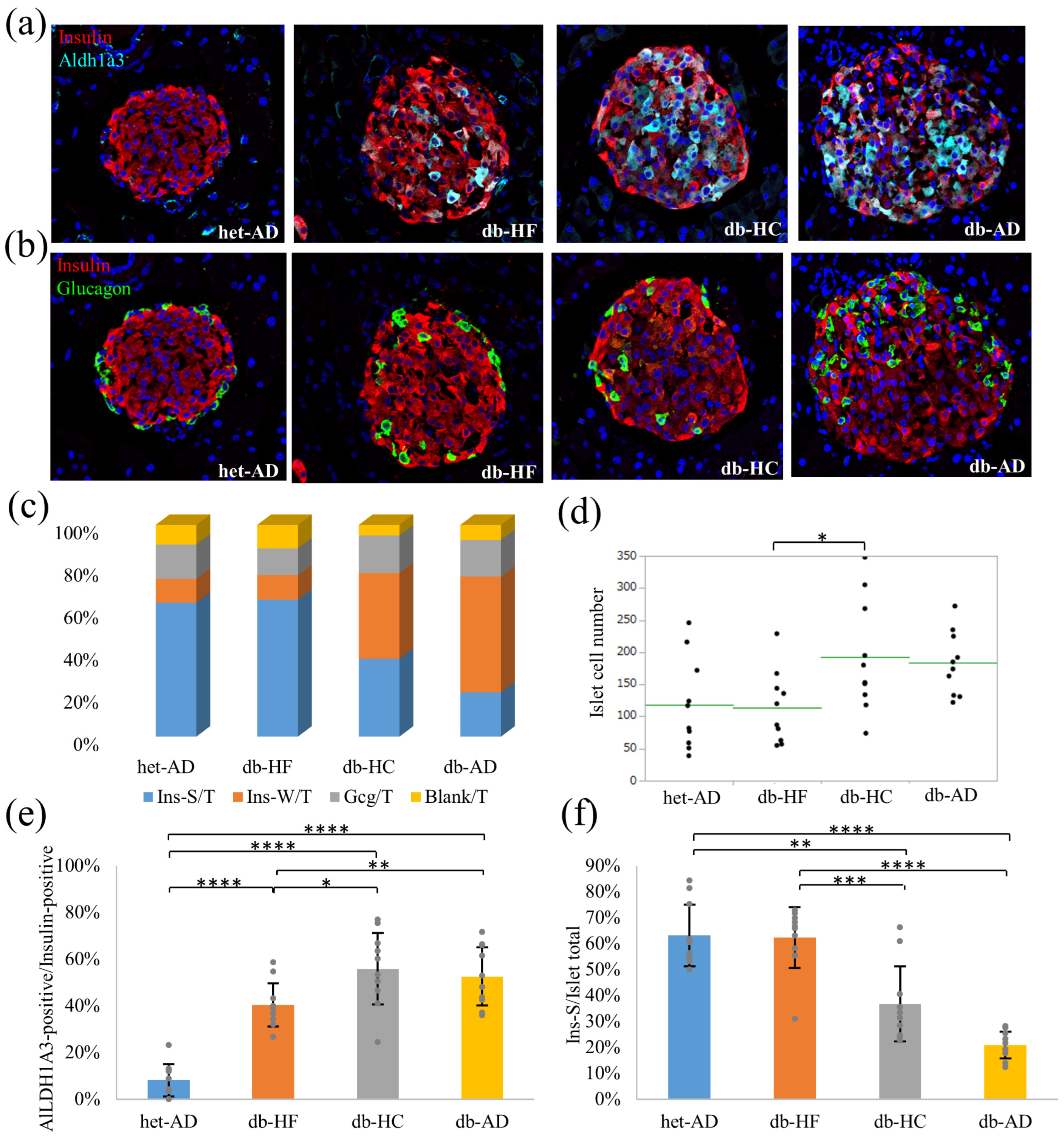
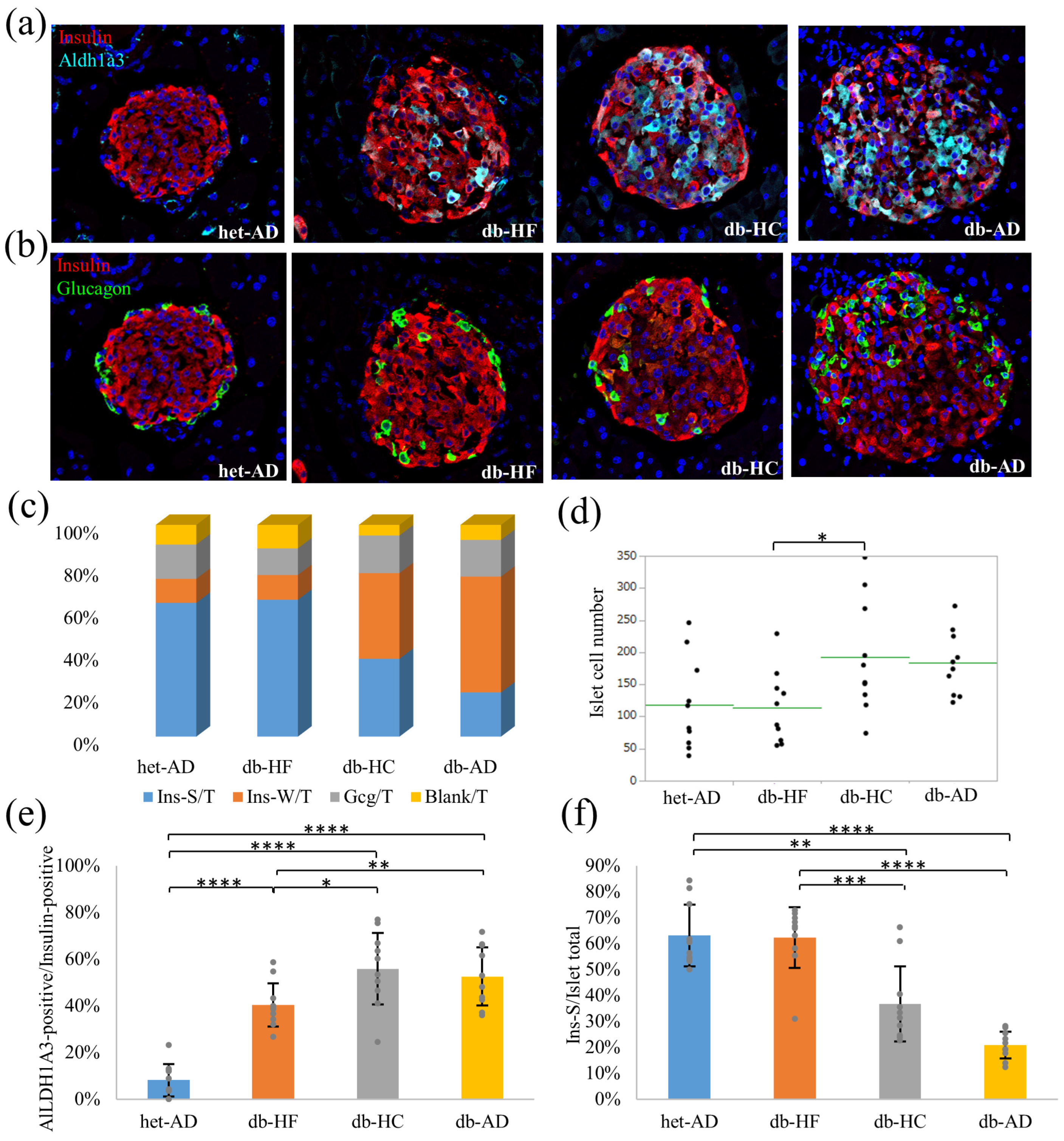
3.4. Calorie Restriction Using Low-Carbohydrate High-Fat Food Better Restored the β-Cell Dedifferentiation Than High-Carbohydrate Low-Fat Food
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- IDF Diabetes Atlas. Available online: https://diabetesatlas.org/resources/ (accessed on 27 December 2023).
- Gillies, C.L.; Abrams, K.R.; Lambert, P.C.; Cooper, N.J.; Sutton, A.J.; Hsu, R.T.; Khunti, K. Pharmacological and lifestyle interventions to prevent or delay type 2 diabetes in people with impaired glucose tolerance: Systematic review and meta-analysis. BMJ 2007, 334, 299. [Google Scholar] [CrossRef] [PubMed]
- Coppell, K.J.; Kataoka, M.; Williams, S.M.; Chisholm, A.W.; Vorgers, S.M.; Mann, J.I. Nutritional intervention in patients with type 2 diabetes who are hyperglycaemics despite optimised drug treatment-Lifestyle Over and Above Drugs in Diabetes (LOADD) study: Randomised controlled trial. BMJ 2010, 341, c3337. [Google Scholar] [CrossRef] [PubMed]
- Pak, H.H.; Haws, S.A.; Green, C.L.; Koller, M.; Lavarias, M.T.; Richardson, N.E.; Yang, S.E.; Dumas, S.N.; Sonsalla, M.; Bray, L.; et al. Fasting drives the metabolic, molecular and geroprotective effects of a calorie-restricted diet in mice. Nat. Metab. 2021, 3, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Prentki, M.; Nolan, C.J. Islet beta cell failure in type 2 diabetes. J. Clin. Investig. 2006, 116, 1802–1812. [Google Scholar] [CrossRef] [PubMed]
- Talchai, C.; Xuan, S.H.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic beta Cell Dedifferentiation as a Mechanism of Diabetic beta Cell Failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Cinti, F.; Bouchi, R.; Kim-Muller, J.Y.; Ohmura, Y.; Sandoval, P.R.; Masini, M.; Marselli, L.; Suleiman, M.; Ratner, L.E.; Marchetti, P.; et al. Evidence of beta-Cell Dedifferentiation in Human Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; York, N.W.; Nichols, C.G.; Remedi, M.S. Pancreatic beta Cell Dedifferentiation in Diabetes and Redifferentiation following Insulin Therapy. Cell Metab. 2014, 19, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Sheng, C.J.; Li, F.; Lin, Z.W.; Zhang, M.N.; Yang, P.; Bu, L.; Sheng, H.; Li, H.; Qu, S. Reversibility of beta-Cell-Specific Transcript Factors Expression by Long-Term Caloric Restriction in db/db Mouse. J. Diabetes Res. 2016, 2016, 6035046. [Google Scholar] [CrossRef] [PubMed]
- Ishida, E.; Kim-Muller, J.Y.; Accili, D. Pair Feeding, but Not Insulin, Phloridzin, or Rosiglitazone Treatment, Curtails Markers of beta-Cell Dedifferentiation in db/db Mice. Diabetes 2017, 66, 2092–2101. [Google Scholar] [CrossRef]
- Khandelwal, R.; Dassanayake, A.S.; Conjeevaram, H.S.; Singh, S.P. Non-alcoholic fatty liver disease in diabetes: When to refer to the hepatologist? World J. Diabetes 2021, 12, 1479–1493. [Google Scholar] [CrossRef]
- Adams, L.A.; Waters, O.R.; Knuiman, M.W.; Elliott, R.R.; Olynyk, J.K. NAFLD as a Risk Factor for the Development of Diabetes and the Metabolic Syndrome: An Eleven-Year Follow-up Study. Am. J. Gastroenterol. 2009, 104, 861–867. [Google Scholar] [CrossRef]
- Tanase, D.M.; Gosav, E.M.; Costea, C.F.; Ciocoiu, M.; Lacatusu, C.M.; Maranduca, M.A.; Ouatu, A.; Floria, M. The Intricate Relationship between Type 2 Diabetes Mellitus (T2DM), Insulin Resistance (IR), and Nonalcoholic Fatty Liver Disease (NAFLD). J. Diabetes Res. 2020, 2020, 3920196. [Google Scholar] [CrossRef]
- Seppala-Lindroos, A.; Vehkavaara, S.; Hakkinen, A.M.; Goto, T.; Westerbacka, J.; Sovijarvi, A.; Halavaara, J.; Yki-Jarvinen, H. Fat accumulation in the liver is associated with defects in insulin suppression of glucose production and serum free fatty acids independent of obesity in normal men. J. Clin. Endocrinol. Metab. 2002, 87, 3023–3028. [Google Scholar] [CrossRef]
- Feinman, R.D.; Pogozelski, W.K.; Astrup, A.; Bernstein, R.K.; Fine, E.J.; Westman, E.C.; Accurso, A.; Frassetto, L.; Gower, B.A.; McFarlane, S.I.; et al. Dietary carbohydrate restriction as the first approach in diabetes management: Critical review and evidence base. Nutrition 2015, 31, 1–13. [Google Scholar] [CrossRef]
- Noakes, T.D.; Windt, J. Evidence that supports the prescription of low-carbohydrate high-fat diets: A narrative review. Br. J. Sports Med. 2017, 51, 133–139. [Google Scholar] [CrossRef]
- Samuel, V.T.; Liu, Z.X.; Qu, X.Q.; Elder, B.D.; Bilz, S.; Befroy, D.; Romanelli, A.J.; Shulman, G.I. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. 2004, 279, 32345–32353. [Google Scholar] [CrossRef]
- Taylor, R. Type 2 diabetes and remission: Practical management guided by pathophysiology. J. Intern. Med. 2021, 289, 754–770. [Google Scholar] [CrossRef]
- Watanabe, T.; Ozawa, A.; Masuda, S.; Yoshino, S.; Ishida, E.; Kondo, Y.; Matsumoto, S.; Katano-Toki, A.; Horiguchi, K.; Nakajima, Y.; et al. Transcriptional Regulation of the Angptl8 Gene by Hepatocyte Nuclear Factor-1 in the Murine Liver. Sci. Rep. 2020, 10, 9999. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Kim-Muller, J.Y.; Fan, J.; Kim, Y.J.R.; Lee, S.-A.; Ishida, E.; Blaner, W.S.; Accili, D. Aldehyde dehydrogenase 1a3 defines a subset of failing pancreatic β cells in diabetic mice. Nat. Commun. 2016, 7, 12631. [Google Scholar] [CrossRef]
- Schauer, P.R.; Kashyap, S.R.; Wolski, K.; Brethauer, S.A.; Kirwan, J.P.; Pothier, C.E.; Thomas, S.; Abood, B.; Nissen, S.E.; Bhatt, D.L. Bariatric Surgery versus Intensive Medical Therapy in Obese Patients with Diabetes. N. Engl. J. Med. 2012, 366, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.-H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Da Silva-Santi, L.; Antunes, M.; Caparroz-Assef, S.; Carbonera, F.; Masi, L.; Curi, R.; Visentainer, J.; Bazotte, R. Liver Fatty Acid Composition and Inflammation in Mice Fed with High-Carbohydrate Diet or High-Fat Diet. Nutrients 2016, 8, 682. [Google Scholar] [CrossRef] [PubMed]
- Göttlicher, M.; Widmark, E.; Li, Q.; Gustafsson, J.A. Fatty acids activate a chimera of the clofibric acid-activated receptor and the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 1992, 89, 4653–4657. [Google Scholar] [CrossRef]
- Kersten, S.; Stienstra, R. The role and regulation of the peroxisome proliferator activated receptor alpha in human liver. Biochimie 2017, 136, 75–84. [Google Scholar] [CrossRef]
- Kersten, S. Integrated physiology and systems biology of PPARalpha. Mol. Metab. 2014, 3, 354–371. [Google Scholar] [CrossRef]
- Forman, B.M.; Chen, J.; Evans, R.M. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors α and δ. Proc. Natl. Acad. Sci. USA 1997, 94, 4312–4317. [Google Scholar] [CrossRef]
- Oswal, D.P.; Balanarasimha, M.; Loyer, J.K.; Bedi, S.; Soman, F.L.; Rider, S.D.; Hostetler, H.A. Divergence between human and murine peroxisome proliferator-activated receptor alpha ligand specificities. J. Lipid Res. 2013, 54, 2354–2365. [Google Scholar] [CrossRef]
- Martin, P.G.P.; Guillou, H.; Lasserre, F.; Déjean, S.; Lan, A.; Pascussi, J.-M.; Sancristobal, M.; Legrand, P.; Besse, P.; Pineau, T. Novel aspects of PPARα-mediated regulation of lipid and xenobiotic metabolism revealed through a nutrigenomic study. Hepatology 2007, 45, 767–777. [Google Scholar] [CrossRef]
- Shimozawa, N. Induction of peroxisomal lipid metabolism in mice fed a high-fat diet. Mol. Med. Rep. 2011, 4, 1157–1162. [Google Scholar] [CrossRef]
- Kersten, S.; Seydoux, J.; Peters, J.M.; Gonzalez, F.J.; Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J. Clin. Investig. 1999, 103, 1489–1498. [Google Scholar] [CrossRef]
- Olefsky, J.M. Treatment of insulin resistance with peroxisome proliferator–activated receptor γ agonists. J. Clin. Investig. 2000, 106, 467–472. [Google Scholar] [CrossRef]
- Fujiwara, T.; Yoshioka, S.; Yoshioka, T.; Ushiyama, I.; Horikoshi, H. Characterization of New Oral Antidiabetic Agent CS-045: Studies in KK and ob/ob Mice and Zucker Fatty Rats. Diabetes 1988, 37, 1549–1558. [Google Scholar] [CrossRef] [PubMed]
- Miles, P.D.G.; Barak, Y.; He, W.; Evans, R.M.; Olefsky, J.M. Improved insulin-sensitivity in mice heterozygous for PPAR-γ deficiency. J. Clin. Investig. 2000, 105, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Patsouris, D.; Reddy, J.K.; Müller, M.; Kersten, S. Peroxisome proliferator-activated receptor alpha mediates the effects of high-fat diet on hepatic gene expression. Endocrinology 2006, 147, 1508–1516. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Hu, E.; Devine, J.; Beale, E.G.; Spiegelman, B.M. PPARγ2 Regulates Adipose Expression of the Phosphoenolpyruvate Carboxykinase Gene. Mol. Cell. Biol. 1995, 15, 351–357. [Google Scholar] [CrossRef]
- Badman, M.K.; Pissios, P.; Kennedy, A.R.; Koukos, G.; Flier, J.S.; Maratos-Flier, E. Hepatic Fibroblast Growth Factor 21 Is Regulated by PPARα and Is a Key Mediator of Hepatic Lipid Metabolism in Ketotic States. Cell Metab. 2007, 5, 426–437. [Google Scholar] [CrossRef]
- Honda, Y.; Kessoku, T.; Ogawa, Y.; Tomeno, W.; Imajo, K.; Fujita, K.; Yoneda, M.; Takizawa, T.; Saito, S.; Nagashima, Y.; et al. Pemafibrate, a novel selective peroxisome proliferator-activated receptor alpha modulator, improves the pathogenesis in a rodent model of nonalcoholic steatohepatitis. Sci. Rep. 2017, 7, 42477. [Google Scholar] [CrossRef]
- Botta, M.; Audano, M.; Sahebkar, A.; Sirtori, C.; Mitro, N.; Ruscica, M. PPAR Agonists and Metabolic Syndrome: An Established Role? Int. J. Mol. Sci. 2018, 19, 1197. [Google Scholar] [CrossRef]
- Diehl, A.M.; Day, C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef]
- Raza-Iqbal, S.; Tanaka, T.; Anai, M.; Inagaki, T.; Matsumura, Y.; Ikeda, K.; Taguchi, A.; Gonzalez, F.J.; Sakai, J.; Kodama, T. Transcriptome Analysis of K-877 (a Novel Selective PPARα Modulator (SPPARMα))-Regulated Genes in Primary Human Hepatocytes and the Mouse Liver. J. Atheroscler. Thromb. 2015, 22, 754–772. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A. Pemafibrate: First Global Approval. Drugs 2017, 77, 1805–1810. [Google Scholar] [CrossRef]
- Fruchart, J.-C. Selective peroxisome proliferator-activated receptorα modulators (SPPARMα): The next generation of peroxisome proliferator-activated receptor α-agonists. Cardiovasc. Diabetol. 2013, 12, 82. [Google Scholar] [CrossRef]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef]
- Sahebkar, A.; Chew, G.T.; Watts, G.F. New peroxisome proliferator-activated receptor agonists: Potential treatments for atherogenic dyslipidemia and non-alcoholic fatty liver disease. Expert Opin. Pharmacother. 2014, 15, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Diao, P.; Zhang, X.; Nakajima, T.; Kimura, T.; Tanaka, N. Clinically Relevant Dose of Pemafibrate, a Novel Selective Peroxisome Proliferator-Activated Receptor α Modulator (SPPARMα), Lowers Serum Triglyceride Levels by Targeting Hepatic PPARα in Mice. Biomedicines 2022, 10, 1667. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, A.; Eguchi, Y.; Yoneda, M.; Imajo, K.; Tamaki, N.; Suganami, H.; Nojima, T.; Tanigawa, R.; Iizuka, M.; Iida, Y.; et al. Randomised clinical trial: Pemafibrate, a novel selective peroxisome proliferator-activated receptor α modulator (SPPARMα), versus placebo in patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2021, 54, 1263–1277. [Google Scholar] [CrossRef]
- Kanno, K.; Koseki, M.; Chang, J.; Saga, A.; Inui, H.; Okada, T.; Tanaka, K.; Asaji, M.; Zhu, Y.; Ide, S.; et al. Pemafibrate suppresses NLRP3 inflammasome activation in the liver and heart in a novel mouse model of steatohepatitis-related cardiomyopathy. Sci. Rep. 2022, 12, 2996. [Google Scholar] [CrossRef]
- Subudhi, S.; Drescher, H.K.; Dichtel, L.E.; Bartsch, L.M.; Chung, R.T.; Hutter, M.M.; Gee, D.W.; Meireles, O.R.; Witkowski, E.R.; Gelrud, L.; et al. Distinct Hepatic Gene-Expression Patterns of NAFLD in Patients with Obesity. Hepatol. Commun. 2022, 6, 77–89. [Google Scholar] [CrossRef]
- Basyte-Bacevice, V.; Skieceviciene, J.; Valantiene, I.; Sumskiene, J.; Petrenkiene, V.; Kondrackiene, J.; Petrauskas, D.; Lammert, F.; Kupcinskas, J. SERPINA1 and HSD17B13 Gene Variants in Patients with Liver Fibrosis and Cirrhosis. J. Gastrointest. Liver Dis. 2019, 28, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Mariano, G.; Matamala, N.; Martínez, S.; Justo, I.; Marcacuzco, A.; Jimenez, C.; Monzón, S.; Cuesta, I.; Garfia, C.; Martínez, M.T.; et al. Liver organoids reproduce alpha-1 antitrypsin deficiency-related liver disease. Hepatol. Int. 2020, 14, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Meghadri, S.H.; Martinez-Delgado, B.; Ostermann, L.; Gomez-Mariano, G.; Perez-Luz, S.; Tumpara, S.; Wrenger, S.; Deluca, D.S.; Maus, U.A.; Welte, T.; et al. Loss of Serpina1 in Mice Leads to Altered Gene Expression in Inflammatory and Metabolic Pathways. Int. J. Mol. Sci. 2022, 23, 10425. [Google Scholar] [CrossRef]
- Kuai, X.; Lv, J.; Zhang, J.; Xu, M.; Ji, J. Serpin Family A Member 1 Is Prognostic and Involved in Immunological Regulation in Human Cancers. Int. J. Mol. Sci. 2023, 24, 11566. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.J.; Tang, L.Z.; Li, W.H.; Xu, Z.S.; Zhang, L.L.; Cheng, F.G.; Chen, H.X.; Wang, Z.H.; Luo, Y.C.; Dai, A.N.; et al. Serum syndecan-4 is associated with nonalcoholic fatty liver disease. J. Dig. Dis. 2021, 22, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Sutton, A.; Friand, V.; Brulé-Donneger, S.; Chaigneau, T.; Ziol, M.; Sainte-Catherine, O.; Poiré, A.; Saffar, L.; Kraemer, M.; Vassy, J.; et al. Stromal Cell–Derived Factor-1/Chemokine (C-X-C Motif) Ligand 12 Stimulates Human Hepatoma Cell Growth, Migration, and Invasion. Mol. Cancer Res. 2007, 5, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Meex, R.C.; Hoy, A.J.; Morris, A.; Brown, R.D.; Lo, J.C.; Burke, M.; Goode, R.J.A.; Kingwell, B.A.; Kraakman, M.J.; Febbraio, M.A.; et al. Fetuin B Is a Secreted Hepatocyte Factor Linking Steatosis to Impaired Glucose Metabolism. Cell Metab. 2015, 22, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- Wickramasekara, R.N.; Morrill, S.; Farhat, Y.; Smith, S.J.; Yilmazer-Hanke, D. Glutathione and Inter-alpha-trypsin inhibitor heavy chain 3 (Itih3) mRNA levels in nicotine-treated Cd44 knockout mice. Toxicol. Rep. 2018, 5, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, N.; Hatano, E.; Iguchi, K.; Sato, M.; Kawaguchi, H.; Ohtsu, I.; Sakurai, T.; Aizawa, N.; Iijima, H.; Nishiguchi, S.; et al. Elevated levels of circulating ITIH4 are associated with hepatocellular carcinoma with nonalcoholic fatty liver disease: From pig model to human study. BMC Cancer 2019, 19, 621. [Google Scholar] [CrossRef]
- Demchev, V.; Malana, G.; Vangala, D.; Stoll, J.; Desai, A.; Kang, H.W.; Li, Y.; Nayeb-Hashemi, H.; Niepel, M.; Cohen, D.E.; et al. Targeted Deletion of Fibrinogen Like Protein 1 Reveals a Novel Role in Energy Substrate Utilization. PLoS ONE 2013, 8, e58084. [Google Scholar] [CrossRef]
- Kozumi, K.; Kodama, T.; Murai, H.; Sakane, S.; Govaere, O.; Cockell, S.; Motooka, D.; Kakita, N.; Yamada, Y.; Kondo, Y.; et al. Transcriptomics Identify Thrombospondin-2 as a Biomarker for NASH and Advanced Liver Fibrosis. Hepatology 2021, 74, 2452–2466. [Google Scholar] [CrossRef] [PubMed]
- Haas, J.T.; Vonghia, L.; Mogilenko, D.A.; Verrijken, A.; Molendi-Coste, O.; Fleury, S.; Deprince, A.; Nikitin, A.; Woitrain, E.; Ducrocq-Geoffroy, L.; et al. Transcriptional network analysis implicates altered hepatic immune function in NASH development and resolution. Nat. Metab. 2019, 1, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Do, O.H.; Gunton, J.E.; Gaisano, H.Y.; Thorn, P. Changes in beta cell function occur in prediabetes and early disease in the Lepr db mouse model of diabetes. Diabetologia 2016, 59, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sundaram, K.; Mu, J.; Dryden, G.W.; Sriwastva, M.K.; Lei, C.; Zhang, L.; Qiu, X.; Xu, F.; Yan, J.; et al. High-fat diet-induced upregulation of exosomal phosphatidylcholine contributes to insulin resistance. Nat. Commun. 2021, 12, 213. [Google Scholar] [CrossRef] [PubMed]
- Tricò, D.; Moriconi, D.; Berta, R.; Baldi, S.; Quinones-Galvan, A.; Guiducci, L.; Taddei, S.; Mari, A.; Nannipieri, M. Effects of Low-Carbohydrate versus Mediterranean Diets on Weight Loss, Glucose Metabolism, Insulin Kinetics and β-Cell Function in Morbidly Obese Individuals. Nutrients 2021, 13, 1345. [Google Scholar] [CrossRef] [PubMed]
- Gower, B.A.; Goree, L.L.; Chandler-Laney, P.C.; Ellis, A.C.; Casazza, K.; Granger, W.M. A higher-carbohydrate, lower-fat diet reduces fasting glucose concentration and improves β-cell function in individuals with impaired fasting glucose. Metabolism 2012, 61, 358–365. [Google Scholar] [CrossRef] [PubMed]
- El Ouaamari, A.; Dirice, E.; Gedeon, N.; Hu, J.; Zhou, J.-Y.; Shirakawa, J.; Hou, L.; Goodman, J.; Karampelias, C.; Qiang, G.; et al. SerpinB1 Promotes Pancreatic β Cell Proliferation. Cell Metab. 2016, 23, 194–205. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, F.; Gong, Q.; Cui, A.; Zhuo, S.; Hu, Z.; Han, Y.; Gao, J.; Sun, Y.; Liu, Z.; et al. Hepatic ATF6 Increases Fatty Acid Oxidation to Attenuate Hepatic Steatosis in Mice Through Peroxisome Proliferator-Activated Receptor alpha. Diabetes 2016, 65, 1904–1915. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, X.; Ishida, E.; Yoshino, S.; Matsumoto, S.; Horiguchi, K.; Yamada, E. Calorie Restriction Using High-Fat/Low-Carbohydrate Diet Suppresses Liver Fat Accumulation and Pancreatic Beta-Cell Dedifferentiation in Obese Diabetic Mice. Nutrients 2024, 16, 995. https://doi.org/10.3390/nu16070995
Lei X, Ishida E, Yoshino S, Matsumoto S, Horiguchi K, Yamada E. Calorie Restriction Using High-Fat/Low-Carbohydrate Diet Suppresses Liver Fat Accumulation and Pancreatic Beta-Cell Dedifferentiation in Obese Diabetic Mice. Nutrients. 2024; 16(7):995. https://doi.org/10.3390/nu16070995
Chicago/Turabian StyleLei, Xiao, Emi Ishida, Satoshi Yoshino, Shunichi Matsumoto, Kazuhiko Horiguchi, and Eijiro Yamada. 2024. "Calorie Restriction Using High-Fat/Low-Carbohydrate Diet Suppresses Liver Fat Accumulation and Pancreatic Beta-Cell Dedifferentiation in Obese Diabetic Mice" Nutrients 16, no. 7: 995. https://doi.org/10.3390/nu16070995
APA StyleLei, X., Ishida, E., Yoshino, S., Matsumoto, S., Horiguchi, K., & Yamada, E. (2024). Calorie Restriction Using High-Fat/Low-Carbohydrate Diet Suppresses Liver Fat Accumulation and Pancreatic Beta-Cell Dedifferentiation in Obese Diabetic Mice. Nutrients, 16(7), 995. https://doi.org/10.3390/nu16070995