
Dietary Supplementation with 23-Hydroxy Ursolic Acid Reduces the Severity and Incidence of Acute Experimental Autoimmune Encephalomyelitis (EAE) in a Murine Model of Multiple Sclerosis

 , , ,
, , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Highlights
- Dietary 23-Hydroxy ursolic acid (23-OH UA) suppressed the severity of ataxia by 52% and EAE disease severity by 48%.
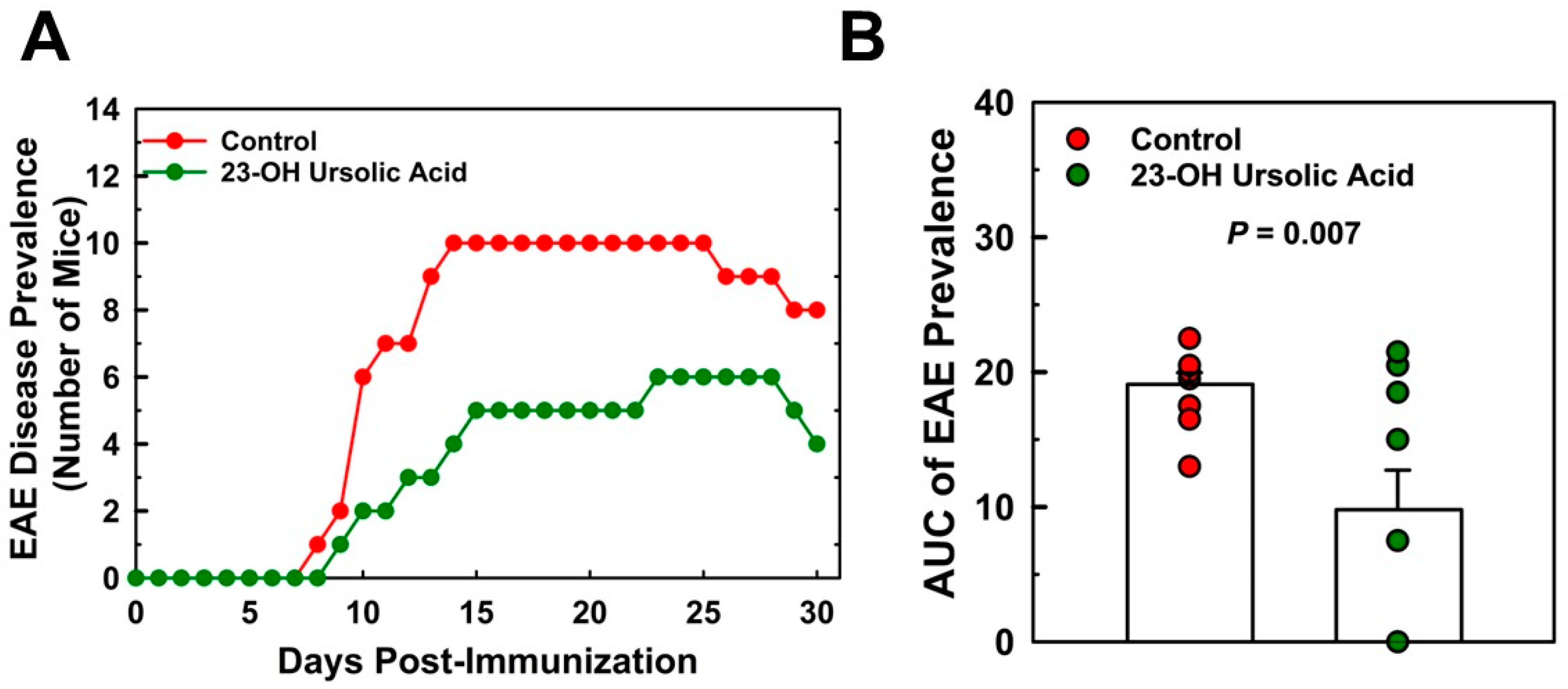
- Dietary 23-OH UA reduced the incidence of disease by over 49% in mice, but did not affect the time to disease onset.
- EAE disease-associated weight loss was strikingly ameliorated in 23-OH UA-fed mice.
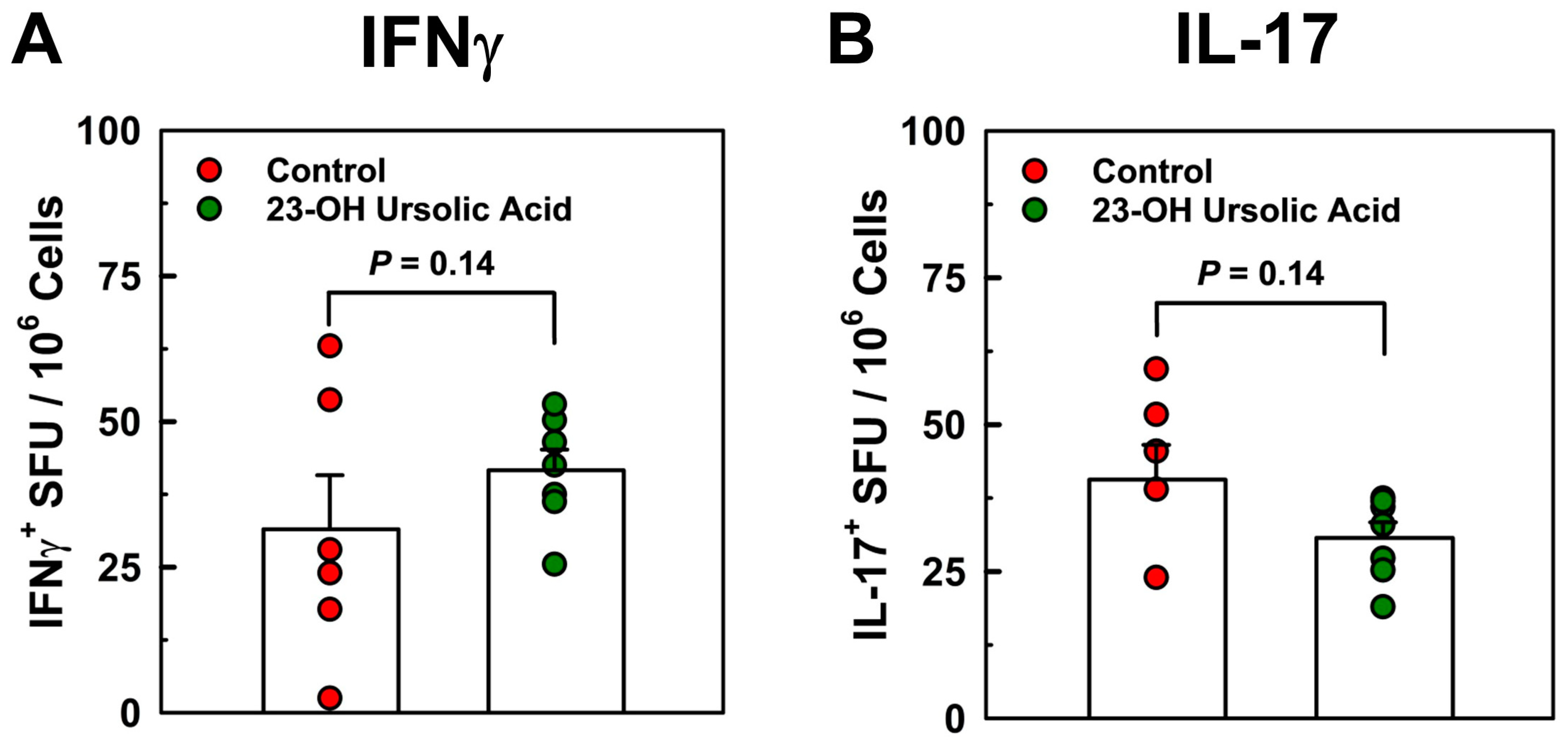
- Dietary 23-OH UA did not appear to regulate peripheral T cell responses.
- Dietary 23-OH UA may represent an effective oral adjunct therapy for the prevention and treatment of relapsing–remitting MS.
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. EAE Induction
2.3. EAE Disease and Ataxia Scoring
2.4. Cytokine ELISPOT Assay
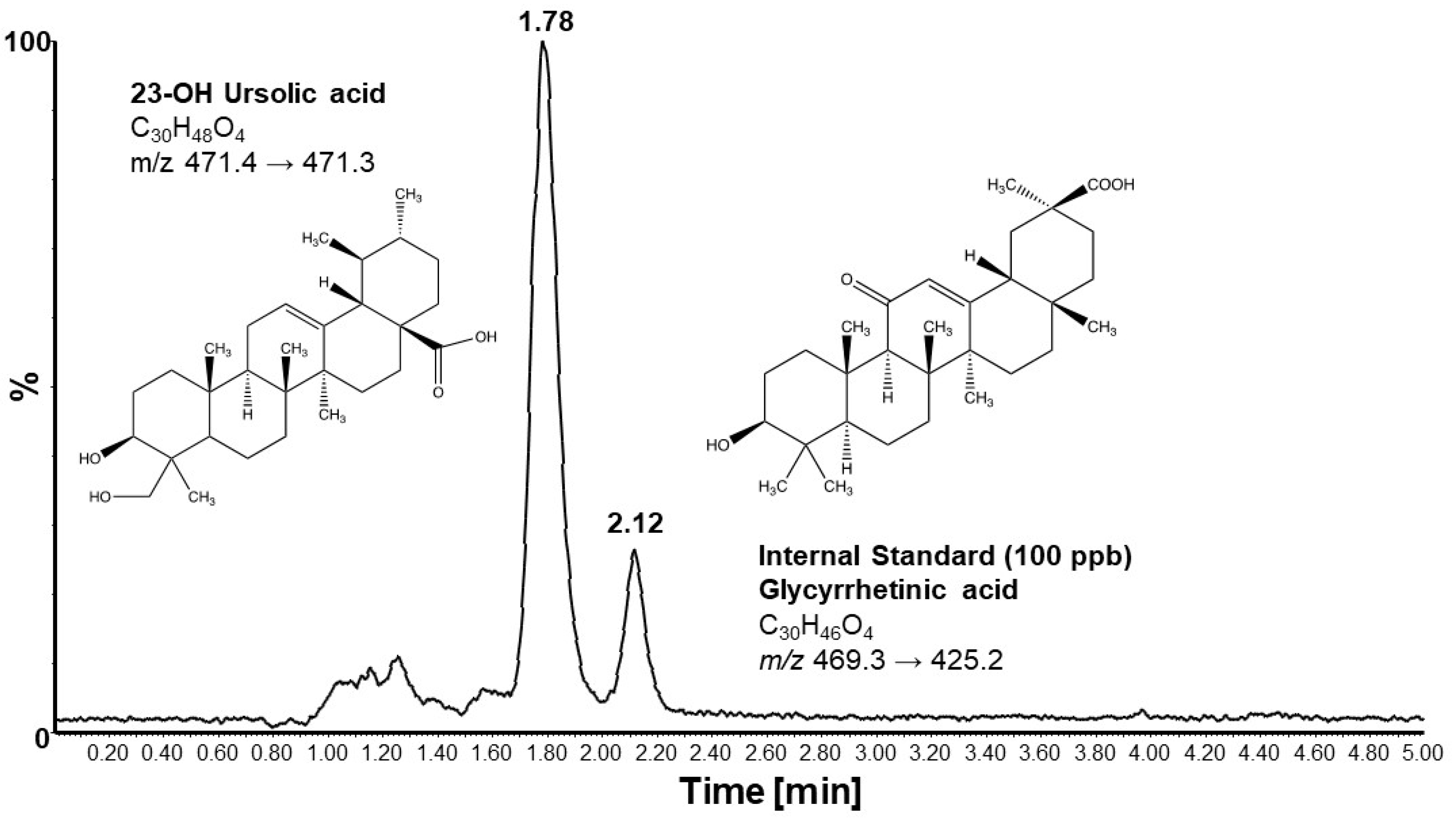
2.5. Liquid Chromatography–Mass Spectrometry
2.6. Statistical Analyses
3. Results
3.1. Dietary 23-OH UA Prevents Weight Loss Associated with EAE
3.2. Dietary 23-OH UA Reduces EAE Disease and Ataxia Severity
3.3. Dietary 23-OH UA Does Not Affect Peripheral Neuroantigen-Specific T Cell Responses
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Correale, J.; Gaitan, M.I.; Ysrraelit, M.C.; Fiol, M.P. Progressive multiple sclerosis: From pathogenic mechanisms to treatment. Brain 2017, 140, 527–546. [Google Scholar] [CrossRef]
- Walton, C.; King, R.; Rechtman, L.; Kaye, W.; Leray, E.; Marrie, R.A.; Robertson, N.; La Rocca, N.; Uitdehaag, B.; van der Mei, I.; et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult. Scler. 2020, 26, 1816–1821. [Google Scholar] [CrossRef]
- Barcellos, L.F.; Sawcer, S.; Ramsay, P.P.; Baranzini, S.E.; Thomson, G.; Briggs, F.; Cree, B.C.; Begovich, A.B.; Villoslada, P.; Montalban, X.; et al. Heterogeneity at the HLA-DRB1 locus and risk for multiple sclerosis. Hum. Mol. Genet. 2006, 15, 2813–2824. [Google Scholar] [CrossRef]
- Patsopoulos, N.A. Genetics of Multiple Sclerosis: An Overview and New Directions. Cold Spring Harb. Perspect. Med. 2018, 8, a028951. [Google Scholar] [CrossRef]
- Canto, E.; Oksenberg, J.R. Multiple sclerosis genetics. Mult. Scler. 2018, 24, 75–79. [Google Scholar] [CrossRef]
- Brownlee, W.J.; Hardy, T.A.; Fazekas, F.; Miller, D.H. Diagnosis of multiple sclerosis: Progress and challenges. Lancet 2017, 389, 1336–1346. [Google Scholar] [CrossRef]
- Reich, D.S.; Lucchinetti, C.F.; Calabresi, P.A. Multiple Sclerosis. N. Engl. J. Med. 2018, 378, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Kingwell, E.; van der Kop, M.; Zhao, Y.; Shirani, A.; Zhu, F.; Oger, J.; Tremlett, H. Relative mortality and survival in multiple sclerosis: Findings from British Columbia, Canada. J. Neurol. Neurosurg. Psychiatry 2012, 83, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Scalfari, A.; Knappertz, V.; Cutter, G.; Goodin, D.S.; Ashton, R.; Ebers, G.C. Mortality in patients with multiple sclerosis. Neurology 2013, 81, 184–192. [Google Scholar] [CrossRef]
- Sospedra, M.; Martin, R. Immunology of multiple sclerosis. Annu. Rev. Immunol. 2005, 23, 683–747. [Google Scholar] [CrossRef] [PubMed]
- Huseby, E.S.; Liggitt, D.; Brabb, T.; Schnabel, B.; Ohlen, C.; Goverman, J. A pathogenic role for myelin-specific CD8(+) T cells in a model for multiple sclerosis. J. Exp. Med. 2001, 194, 669–676. [Google Scholar] [CrossRef] [PubMed]
- von Budingen, H.C.; Kuo, T.C.; Sirota, M.; van Belle, C.J.; Apeltsin, L.; Glanville, J.; Cree, B.A.; Gourraud, P.A.; Schwartzburg, A.; Huerta, G.; et al. B cell exchange across the blood-brain barrier in multiple sclerosis. J. Clin. Investig. 2012, 122, 4533–4543. [Google Scholar] [CrossRef] [PubMed]
- Krumbholz, M.; Derfuss, T.; Hohlfeld, R.; Meinl, E. B cells and antibodies in multiple sclerosis pathogenesis and therapy. Nat. Rev. Neurol. 2012, 8, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.K.; Ray, A.; Basu, S.; Karp, C.L.; Dittel, B.N. Pathogenic and regulatory roles for B cells in experimental autoimmune encephalomyelitis. Autoimmunity 2012, 45, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Steinman, L.; Martin, R.; Bernard, C.; Conlon, P.; Oksenberg, J.R. Multiple Sclerosis: Deeper Understanding of Its Pathogenesis Reveals New Targets for Therapy. Annu. Rev. Neurosci. 2002, 25, 491–505. [Google Scholar] [CrossRef]
- McFarland, H.F.; Martin, R. Multiple sclerosis: A complicated picture of autoimmunity. Nat. Immunol. 2007, 8, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Charabati, M.; Wheeler, M.A.; Weiner, H.L.; Quintana, F.J. Multiple sclerosis: Neuroimmune crosstalk and therapeutic targeting. Cell 2023, 186, 1309–1327. [Google Scholar] [CrossRef]
- Bettelli, E.; Oukka, M.; Kuchroo, V.K. T(H)-17 cells in the circle of immunity and autoimmunity. Nat. Immunol. 2007, 8, 345–350. [Google Scholar] [CrossRef]
- Codarri, L.; Gyulveszi, G.; Tosevski, V.; Hesske, L.; Fontana, A.; Magnenat, L.; Suter, T.; Becher, B. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat. Immunol. 2011, 12, 560–567. [Google Scholar] [CrossRef]
- Lassmann, H.; van Horssen, J.; Mahad, D. Progressive multiple sclerosis: Pathology and pathogenesis. Nat. Rev. Neurol. 2012, 8, 647–656. [Google Scholar] [CrossRef]
- Ludwin, S.K.; Rao, V.; Moore, C.S.; Antel, J.P. Astrocytes in multiple sclerosis. Mult. Scler. 2016, 22, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Buron, M.D.; Chalmer, T.A.; Sellebjerg, F.; Barzinji, I.; Danny, B.; Christensen, J.R.; Christensen, M.K.; Hansen, V.; Illes, Z.; Jensen, H.B.; et al. Initial high-efficacy disease-modifying therapy in multiple sclerosis: A nationwide cohort study. Neurology 2020, 95, e1041–e1051. [Google Scholar] [CrossRef] [PubMed]
- van der Star, B.J.; Vogel, D.Y.; Kipp, M.; Puentes, F.; Baker, D.; Amor, S. In vitro and in vivo models of multiple sclerosis. CNS Neurol. Disord. Drug Targets 2012, 11, 570–588. [Google Scholar] [CrossRef] [PubMed]
- Pachner, A.R. Experimental models of multiple sclerosis. Curr. Opin. Neurol. 2011, 24, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Bittner, S.; Afzali, A.M.; Wiendl, H.; Meuth, S.G. Myelin oligodendrocyte glycoprotein (MOG35-55) induced experimental autoimmune encephalomyelitis (EAE) in C57BL/6 mice. J. Vis. Exp. JoVE 2014, 86, e51275. [Google Scholar] [CrossRef]
- Bore, L.; Honda, T.; Gribble, G.W. Partial synthesis of 23-hydroxyursolic acid isolated from medicinal plants of the Rubiaceae family. Nat. Prod. Lett. 2002, 16, 273–276. [Google Scholar] [CrossRef]
- Nguyen, H.N.; Ahn, Y.J.; Medina, E.A.; Asmis, R. Dietary 23-hydroxy ursolic acid protects against atherosclerosis and obesity by preventing dyslipidemia-induced monocyte priming and dysfunction. Atherosclerosis 2018, 275, 333–341. [Google Scholar] [CrossRef]
- Raphael, I.; Gomez-Rivera, F.; Raphael, R.A.; Robinson, R.R.; Nalawade, S.; Forsthuber, T.G. TNFR2 limits proinflammatory astrocyte functions during EAE induced by pathogenic DR2b-restricted T cells. JCI Insight 2019, 4, e132527. [Google Scholar] [CrossRef] [PubMed]
- Bar-Or, A.; Li, R. Cellular immunology of relapsing multiple sclerosis: Interactions, checks, and balances. Lancet Neurol. 2021, 20, 470–483. [Google Scholar] [CrossRef]
- Dong, Y.; Yong, V.W. When encephalitogenic T cells collaborate with microglia in multiple sclerosis. Nat. Rev. Neurol. 2019, 15, 704–717. [Google Scholar] [CrossRef] [PubMed]
- Hohlfeld, R.; Dornmair, K.; Meinl, E.; Wekerle, H. The search for the target antigens of multiple sclerosis, part 1: Autoreactive CD4+ T lymphocytes as pathogenic effectors and therapeutic targets. Lancet Neurol. 2016, 15, 198–209. [Google Scholar] [CrossRef]
- Ahn, Y.J.; Wang, L.; Foster, S.; Asmis, R. Dietary 23-hydroxy ursolic acid protects against diet-induced weight gain and hyperglycemia by protecting monocytes and macrophages against nutrient stress-triggered reprogramming and dysfunction and preventing adipose tissue inflammation. J. Nutr. Biochem. 2020, 86, 108483. [Google Scholar] [CrossRef] [PubMed]
- Epstein, L.G.; Prineas, J.W.; Raine, C.S. Attachment of myelin to coated pits on macrophages in experimental allergic encephalomyelitis. J. Neurol. Sci. 1983, 61, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Bruck, W.; Sommermeier, N.; Bergmann, M.; Zettl, U.; Goebel, H.H.; Kretzschmar, H.A.; Lassmann, H. Macrophages in multiple sclerosis. Immunobiology 1996, 195, 588–600. [Google Scholar] [CrossRef] [PubMed]
- Prineas, J.W.; Parratt, J.D.E. Multiple Sclerosis: Microglia, Monocytes, and Macrophage-Mediated Demyelination. J. Neuropathol. Exp. Neurol. 2021, 80, 975–996. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, X.; Ciric, B.; Curtis, M.T.; Chen, W.J.; Rostami, A.; Zhang, G.X. A dual effect of ursolic acid to the treatment of multiple sclerosis through both immunomodulation and direct remyelination. Proc. Natl. Acad. Sci. USA 2020, 117, 9082–9093. [Google Scholar] [CrossRef] [PubMed]
- Correale, J.; Farez, M.F. The Role of Astrocytes in Multiple Sclerosis Progression. Front. Neurol. 2015, 6, 180. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Quintana, F.J. Control of autoimmune CNS inflammation by astrocytes. Semin. Immunopathol. 2015, 37, 625–638. [Google Scholar] [CrossRef]
- Rothhammer, V.; Kenison, J.E.; Tjon, E.; Takenaka, M.C.; de Lima, K.A.; Borucki, D.M.; Chao, C.C.; Wilz, A.; Blain, M.; Healy, L.; et al. Sphingosine 1-phosphate receptor modulation suppresses pathogenic astrocyte activation and chronic progressive CNS inflammation. Proc. Natl. Acad. Sci. USA 2017, 114, 2012–2017. [Google Scholar] [CrossRef]
- Li, T.; Chen, X.; Zhang, C.; Zhang, Y.; Yao, W. An update on reactive astrocytes in chronic pain. J. Neuroinflamm. 2019, 16, 140. [Google Scholar] [CrossRef]
- Kim, H.S.; Tavakoli, S.; Piefer, L.A.; Nguyen, H.N.; Asmis, R. Monocytic MKP-1 is a Sensor of the Metabolic Environment and Regulates Function and Phenotypic Fate of Monocyte-Derived Macrophages in Atherosclerosis. Sci. Rep. 2016, 6, 34223. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Ullevig, S.L.; Zamora, D.; Lee, C.F.; Asmis, R. Redox regulation of MAPK phosphatase 1 controls monocyte migration and macrophage recruitment. Proc. Natl. Acad. Sci. USA 2012, 109, E2803–E2812. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asmis, R.; Medrano, M.T.; Chase Huizar, C.; Griffith, W.P.; Forsthuber, T.G. Dietary Supplementation with 23-Hydroxy Ursolic Acid Reduces the Severity and Incidence of Acute Experimental Autoimmune Encephalomyelitis (EAE) in a Murine Model of Multiple Sclerosis. Nutrients 2024, 16, 348. https://doi.org/10.3390/nu16030348
Asmis R, Medrano MT, Chase Huizar C, Griffith WP, Forsthuber TG. Dietary Supplementation with 23-Hydroxy Ursolic Acid Reduces the Severity and Incidence of Acute Experimental Autoimmune Encephalomyelitis (EAE) in a Murine Model of Multiple Sclerosis. Nutrients. 2024; 16(3):348. https://doi.org/10.3390/nu16030348
Chicago/Turabian StyleAsmis, Reto, Megan T. Medrano, Carol Chase Huizar, Wendell P. Griffith, and Thomas G. Forsthuber. 2024. "Dietary Supplementation with 23-Hydroxy Ursolic Acid Reduces the Severity and Incidence of Acute Experimental Autoimmune Encephalomyelitis (EAE) in a Murine Model of Multiple Sclerosis" Nutrients 16, no. 3: 348. https://doi.org/10.3390/nu16030348
APA StyleAsmis, R., Medrano, M. T., Chase Huizar, C., Griffith, W. P., & Forsthuber, T. G. (2024). Dietary Supplementation with 23-Hydroxy Ursolic Acid Reduces the Severity and Incidence of Acute Experimental Autoimmune Encephalomyelitis (EAE) in a Murine Model of Multiple Sclerosis. Nutrients, 16(3), 348. https://doi.org/10.3390/nu16030348