The Role of Vitamin D Deficiency in Hepatic Encephalopathy: A Review of Pathophysiology, Clinical Outcomes, and Therapeutic Potential

 ,
,
Abstract
1. Introduction
2. History, Etiology, Clinical Features, and Diagnosis of Hepatic Encephalopathy
3. Pathophysiology of Cirrhosis and Hepatic Encephalopathy and the Role of Oxidative Stress
4. Vitamin D and Its Association with Cirrhosis and Hepatic Encephalopathy
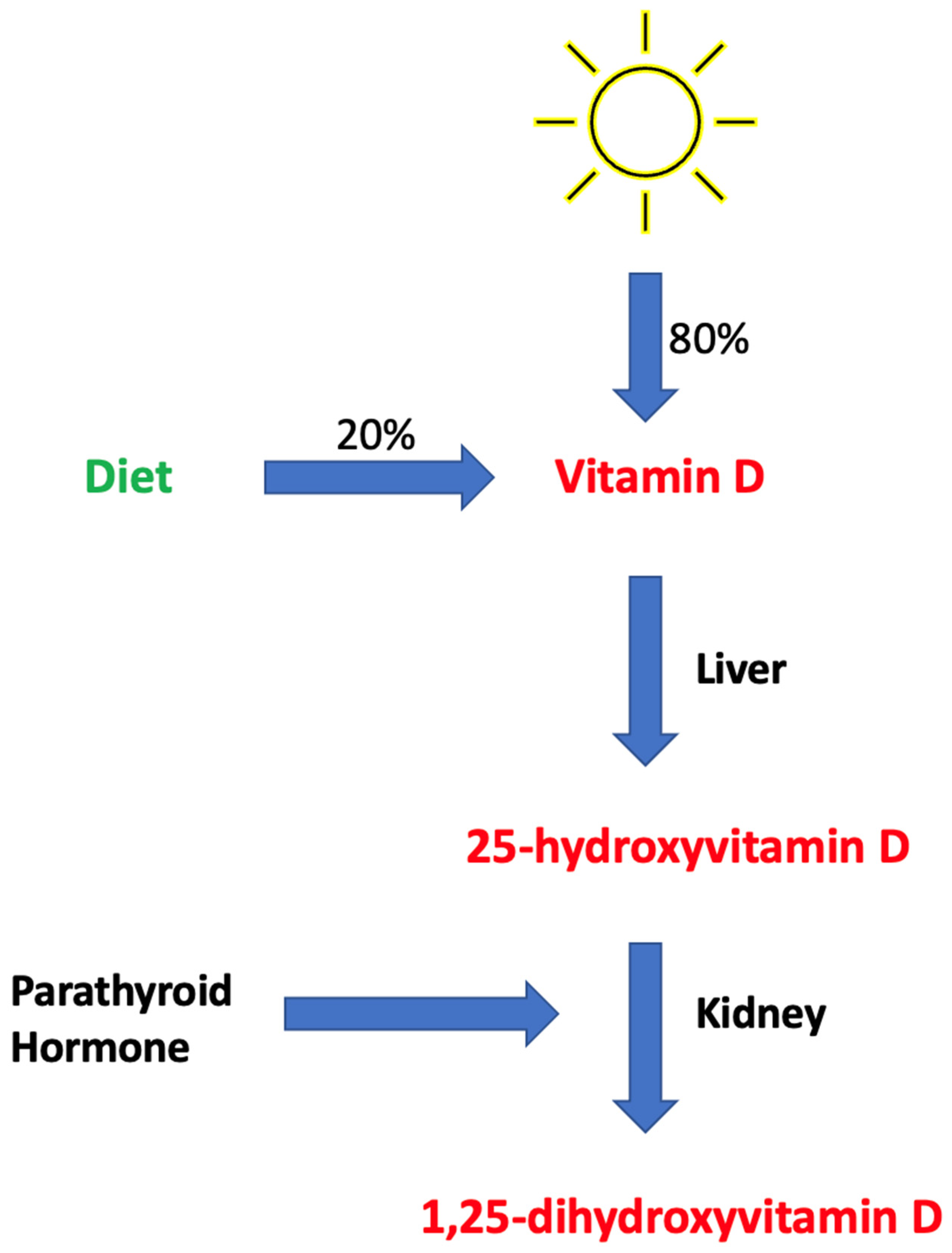
4.1. Brief Overview of Vitamin D Metabolism
4.2. Supplemental Forms of Vitamin D and the Definition of Deficiency
4.3. Vitamin D Levels in Association with Cirrhosis
4.4. Vitamin D Levels in Association with Hepatic Encephalopathy
4.5. Vitamin D Levels in Post-Operative Transjugular Intrahepatic Portosystemic Shunt (TIPS)
4.6. Vitamin D Has Been Shown to Suppress Oxidative Stress
5. Vitamin D Levels in Association with Survival
6. What Should Future Studies Explore?
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sahney, A.; Wadhawan, M. Encephalopathy in Cirrhosis: Prevention and Management. J. Clin. Exp. Hepatol. 2022, 12, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Mandiga, P.; Kommu, S.; Bollu, P.C. Hepatic Encephalopathy. In StatPearls; StatPearls: Treasure Island, FL, USA, 2024. [Google Scholar]
- Poordad, F.F. Review article: The burden of hepatic encephalopathy. Aliment. Pharmacol. Ther. 2007, 25 (Suppl. S1), 3–9. [Google Scholar] [CrossRef] [PubMed]
- Elsaid, M.I.; Rustgi, V.K. Epidemiology of Hepatic Encephalopathy. Clin. Liver Dis. 2020, 24, 157–174. [Google Scholar] [CrossRef] [PubMed]
- Mumit Sarkar, A.; Al Mukit, A.; Bari, T.; Islam, R.; Islam, S.; Sarker, K.; Chowdhury, M.; Rashid, M.H.O.; Alim, A. Association of low serum 25-Hydroxy vitamin D [25(OH)D] with hepatic encephalopathy in patients with decompensated liver cirrhosis. Arab. J. Gastroenterol. 2024, 25, 182–187. [Google Scholar] [CrossRef]
- Afifi, M.A.E.; Hussein, A.M.; Rizk, M. Low Serum 25-Hydroxy Vitamin D (25-OHD) and Hepatic Encephalopathy in HCV-Related Liver Cirrhosis. Int. J. Hepatol. 2021, 2021, 6669527. [Google Scholar] [CrossRef]
- Kalita, S.; Das, J.; Rajkakati, R.; Mili, C. Vitamin D in Patients of Chronic Liver Disease with Hepatic Encephalopathy. J. Assoc. Physicians India 2022, 70, 11–12. [Google Scholar]
- Harris, M.K.; Elliott, D.; Schwendimann, R.N.; Minagar, A.; Jaffe, S.L. Neurologic presentations of hepatic disease. Neurol. Clin. 2010, 28, 89–105. [Google Scholar] [CrossRef]
- Ferenci, P.; Lockwood, A.; Mullen, K.; Tarter, R.; Weissenborn, K.; Blei, A.T. Hepatic encephalopathy—Definition, nomenclature, diagnosis, and quantification: Final report of the working party at the 11th World Congresses of Gastroenterology, Vienna, 1998. Hepatology 2002, 35, 716–721. [Google Scholar] [CrossRef]
- Rudler, M.; Weiss, N.; Bouzbib, C.; Thabut, D. Diagnosis and Management of Hepatic Encephalopathy. Clin. Liver Dis. 2021, 25, 393–417. [Google Scholar] [CrossRef]
- Montoliu, C.; Cauli, O.; Urios, A.; ElMlili, N.; Serra, M.A.; Giner-Duran, R.; González-Lopez, O.; Del Olmo, J.A.; Wassel, A.; Rodrigo, J.M.; et al. 3-nitro-tyrosine as a peripheral biomarker of minimal hepatic encephalopathy in patients with liver cirrhosis. Am. J. Gastroenterol. 2011, 106, 1629–1637. [Google Scholar] [CrossRef]
- Sharma, B.; John, S. Hepatic Cirrhosis. In StatPearls; StatPearls: Treasure Island, FL, USA, 2024. [Google Scholar]
- Shojaie, L.; Iorga, A.; Dara, L. Cell Death in Liver Diseases: A Review. Int. J. Mol. Sci. 2020, 21, 9682. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Premkumar, M.; Anand, A.C. Overview of Complications in Cirrhosis. J. Clin. Exp. Hepatol. 2022, 12, 1150–1174. [Google Scholar] [CrossRef] [PubMed]
- Gressner, O.A.; Lahme, B.; Gressner, A.M. Gc-globulin (vitamin D binding protein) is synthesized and secreted by hepatocytes and internalized by hepatic stellate cells through Ca(2+)-dependent interaction with the megalin/gp330 receptor. Clin. Chim. Acta 2008, 390, 28–37. [Google Scholar] [CrossRef]
- Nardelli, S.; Riggio, O.; Gioia, S.; Puzzono, M.; Pelle, G.; Ridola, L. Spontaneous porto-systemic shunts in liver cirrhosis: Clinical and therapeutical aspects. World J. Gastroenterol. 2020, 26, 1726–1732. [Google Scholar] [CrossRef]
- Lu, K. Cellular Pathogenesis of Hepatic Encephalopathy: An Update. Biomolecules 2023, 13, 396. [Google Scholar] [CrossRef]
- Bai, Y.; Li, K.; Li, X.; Chen, X.; Zheng, J.; Wu, F.; Chen, J.; Li, Z.; Zhang, S.; Wu, K.; et al. Effects of oxidative stress on hepatic encephalopathy pathogenesis in mice. Nat. Commun. 2023, 14, 4456. [Google Scholar] [CrossRef]
- Simicic, D.; Cudalbu, C.; Pierzchala, K. Overview of oxidative stress findings in hepatic encephalopathy: From cellular and ammonium-based animal models to human data. Anal. Biochem. 2022, 654, 114795. [Google Scholar] [CrossRef]
- de la Puente Yague, M.; Yurrita, L.C.; Cabañas, M.J.C.; Cenzual, M.A.C. Role of Vitamin D in Athletes and Their Performance: Current Concepts and New Trends. Nutrients 2020, 12, 579. [Google Scholar] [CrossRef]
- Sizar, O.; Khare, S.; Goyal, A.; Givler, A. Vitamin D Deficiency. In StatPearls; StatPearls: Treasure Island, FL, USA, 2024. [Google Scholar]
- Stevens, C.M.; Jain, S.K. Vitamin D/Bone Mineral Density and Triglyceride Paradoxes Seen in African Americans: A Cross-Sectional Study and Review of the Literature. Int. J. Mol. Sci. 2024, 25, 1305. [Google Scholar] [CrossRef]
- Pludowski, P. Supplementing Vitamin D in Different Patient Groups to Reduce Deficiency. Nutrients 2023, 15, 3725. [Google Scholar] [CrossRef] [PubMed]
- Amrein, K.; Scherkl, M.; Hoffmann, M.; Neuwersch-Sommeregger, S.; Köstenberger, M.; Berisha, A.T.; Martucci, G.; Pilz, S.; Malle, O. Vitamin D deficiency 2.0: An update on the current status worldwide. Eur. J. Clin. Nutr. 2020, 74, 1498–1513. [Google Scholar] [CrossRef]
- Holick, M.F.; Binkley, N.C.; Bischoff-Ferrari, H.A.; Gordon, C.M.; Hanley, D.A.; Heaney, R.P.; Murad, M.H.; Weaver, C.M. Evaluation, treatment, and prevention of vitamin D deficiency: An Endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 2011, 96, 1911–1930. [Google Scholar] [CrossRef] [PubMed]
- Daroux, M.; Shenouda, M.; Bacri, J.-L.; Lemaitre, V.; Vanhille, P.; Bataille, P. Vitamin D2 versus vitamin D3 supplementation in hemodialysis patients: A comparative pilot study. J. Nephrol. 2013, 26, 152–157. [Google Scholar] [CrossRef]
- Cesareo, R.; Falchetti, A.; Attanasio, R.; Tabacco, G.; Naciu, A.M.; Palermo, A. Hypovitaminosis D: Is It Time to Consider the Use of Calcifediol? Nutrients 2019, 11, 1016. [Google Scholar] [CrossRef]
- Quesada-Gomez, J.M.; Bouillon, R. Is calcifediol better than cholecalciferol for vitamin D supplementation? Osteoporos. Int. 2018, 29, 1697–1711. [Google Scholar] [CrossRef] [PubMed]
- Jodar, E.; Campusano, C.; de Jongh, R.T.; Holick, M.F. Calcifediol: A review of its pharmacological characteristics and clinical use in correcting vitamin D deficiency. Eur. J. Nutr. 2023, 62, 1579–1597. [Google Scholar] [CrossRef]
- Arteh, J.; Narra, S.; Nair, S. Prevalence of vitamin D deficiency in chronic liver disease. Dig. Dis. Sci. 2010, 55, 2624–2628. [Google Scholar] [CrossRef]
- Clements, M.R.; Davies, M.; Hayes, M.E.; Hlckey, C.D.; Lumb, G.A.; Mawer, E.B.; Adams, P.H. The role of 1,25-dihydroxyvitamin D in the mechanism of acquired vitamin D deficiency. Clin. Endocrinol. 1992, 37, 17–27. [Google Scholar] [CrossRef]
- Sitrin, M.D.; Bengoa, J.M. Intestinal absorption of cholecalciferol and 25-hydroxycholecalciferol in chronic cholestatic liver disease. Am. J. Clin. Nutr. 1987, 46, 1011–1015. [Google Scholar] [CrossRef]
- Putz-Bankuti, C.; Pilz, S.; Stojakovic, T.; Scharnagl, H.; Pieber, T.R.; Trauner, M.; Obermayer-Pietsch, B.; Stauber, R.E. Association of 25-hydroxyvitamin D levels with liver dysfunction and mortality in chronic liver disease. Liver Int. 2012, 32, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Stokes, C.S.; Krawczyk, M.; Reichel, C.; Lammert, F.; Grünhage, F. Vitamin D deficiency is associated with mortality in patients with advanced liver cirrhosis. Eur. J. Clin. Investig. 2014, 44, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Anty, R.; Tonohouan, M.; Ferrari-Panaia, P.; Piche, T.; Pariente, A.; Anstee, Q.M.; Gual, P.; Tran, A. Low Levels of 25-Hydroxy Vitamin D are Independently Associated with the Risk of Bacterial Infection in Cirrhotic Patients. Clin. Transl. Gastroenterol. 2014, 5, e56. [Google Scholar] [CrossRef]
- Koop, A.H.; Mousa, O.Y.; Pham, L.E.; Corral-Hurtado, J.E.; Pungpapong, S.; Keaveny, A.P. An Argument for Vitamin D, A, and Zinc Monitoring in Cirrhosis. Ann. Hepatol. 2018, 17, 920–932. [Google Scholar] [CrossRef]
- Kumar, P.; Chaudhry, S.; Dev, N.; Kumar, R.; Singh, G. Serum 25-hydroxyvitamin D level in patients with chronic liver disease and its correlation with hepatic encephalopathy: A cross-sectional study. J. Fam. Med. Prim. Care 2020, 9, 798–803. [Google Scholar]
- Kubesch, A.; Quenstedt, L.; Saleh, M.; Rüschenbaum, S.; Schwarzkopf, K.; Martinez, Y.; Welsch, C.; Zeuzem, S.; Welzel, T.M.; Lange, C.M. Vitamin D deficiency is associated with hepatic decompensation and inflammation in patients with liver cirrhosis: A prospective cohort study. PLoS ONE 2018, 13, e0207162. [Google Scholar] [CrossRef] [PubMed]
- Savic, Z.; Damjanov, D.; Curic, N.; Kovacev-Zavisic, B.; Hadnadjev, L.; Novakovic-Paro, J.; Nikolic, S. Vitamin D status, bone metabolism and bone mass in patients with alcoholic liver cirrhosis. Bratisl. Lek. Listy 2014, 115, 573–578. [Google Scholar] [CrossRef]
- Vidot, H.; Potter, A.; Cheng, R.; Allman-Farinelli, M.; Shackel, N. Serum 25-hydroxyvitamin D deficiency and hepatic encephalopathy in chronic liver disease. World J. Hepatol. 2017, 9, 510–518. [Google Scholar] [CrossRef]
- Jha, A.K.; Jha, S.K.; Kumar, A.; Dayal, V.M.; Jha, S.K. Effect of replenishment of vitamin D on survival in patients with decompensated liver cirrhosis: A prospective study. World J. Gastrointest. Pathophysiol. 2017, 8, 133–141. [Google Scholar] [CrossRef]
- Khan, M.A.; Dar, H.A.; Baba, M.A.; Shah, A.H.; Singh, B.; Shiekh, N.A. Impact of Vitamin D Status in Chronic Liver Disease. J. Clin. Exp. Hepatol. 2019, 9, 574–580. [Google Scholar] [CrossRef]
- Yousif, M.M.; Sadek, A.M.E.M.; Farrag, H.A.; Selim, F.O.; Hamed, E.F.; Salama, R.I. Associated vitamin D deficiency is a risk factor for the complication of HCV-related liver cirrhosis including hepatic encephalopathy and spontaneous bacterial peritonitis. Intern. Emerg. Med. 2019, 14, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Narayanasamy, K.; Karthick, R.; Raj, A.K. High Prevalent Hypovitaminosis D Is Associated with Dysregulation of Calcium-parathyroid Hormone-vitamin D Axis in Patients with Chronic Liver Diseases. J. Clin. Transl. Hepatol. 2019, 7, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Simbrunner, B.; Semmler, G.; Stadlmann, A.; Scheiner, B.; Schwabl, P.; Paternostro, R.; Bucsics, T.; Bauer, D.; Eigenbauer, E.; Pinter, M.; et al. Vitamin A levels reflect disease severity and portal hypertension in patients with cirrhosis. Hepatol. Int. 2020, 14, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Casadaban, L.C.; Parvinian, A.; Minocha, J.; Lakhoo, J.; Grant, C.W.; Ray, C.E.; Knuttinen, M.G.; Bui, J.T.; Gaba, R.C. Clearing the Confusion over Hepatic Encephalopathy After TIPS Creation: Incidence, Prognostic Factors, and Clinical Outcomes. Dig. Dis. Sci. 2015, 60, 1059–1066. [Google Scholar] [CrossRef]
- Panchani, N.N.; Colvin, T.; Aryan, M.; Shoreibah, M.G. Nutritional Deficiencies and Clinical Practices in Decompensated Cirrhotics With Hepatic Encephalopathy. Cureus 2022, 14, e25352. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.K.; Stevens, C.M.; Margret, J.J.; Levine, S.N. Alzheimer’s Disease: A Review of Pathology, Current Treatments, and the Potential Therapeutic Effect of Decreasing Oxidative Stress by Combined Vitamin D and l-Cysteine Supplementation. Antioxid. Redox Signal. 2024, 40, 663–678. [Google Scholar] [CrossRef]
- Wenclewska, S.; Szymczak-Pajor, I.; Drzewoski, J.; Bunk, M.; Śliwińska, A. Vitamin D Supplementation Reduces Both Oxidative DNA Damage and Insulin Resistance in the Elderly with Metabolic Disorders. Int. J. Mol. Sci. 2019, 20, 2891. [Google Scholar] [CrossRef]
- Renke, G.; Starling-Soares, B.; Baesso, T.; Petronio, R.; Aguiar, D.; Paes, R. Effects of Vitamin D on Cardiovascular Risk and Oxidative Stress. Nutrients 2023, 15, 769. [Google Scholar] [CrossRef]
- Charoenngam, N.; Holick, M.F. Immunologic Effects of Vitamin D on Human Health and Disease. Nutrients 2020, 12, 2097. [Google Scholar] [CrossRef]
- Bouillon, R.; Antonio, L.; Olarte, O.R. Calcifediol (25OH Vitamin D(3)) Deficiency: A Risk Factor from Early to Old Age. Nutrients 2022, 14, 1168. [Google Scholar] [CrossRef]
- Wan, Z.; Guo, J.; Pan, A.; Chen, C.; Liu, L.; Liu, G. Association of Serum 25-Hydroxyvitamin D Concentrations With All-Cause and Cause-Specific Mortality Among Individuals With Diabetes. Diabetes Care 2021, 44, 350–357. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| References | Title | Disease (n) | Prevalence of VDD | Finding(s) |
|---|---|---|---|---|
| Putz-Bankuti et al., 2012 [34] | Association of 25-hydroxyvitamin D levels with liver dysfunction and mortality in chronic liver disease | CLD (n = 75) | VDD (<20 ng/mL) in 53(71%) of patients | 25-OHD levels were inversely correlated with CP and MELD scores |
| Savic et al., 2014 [40] | Vitamin D status, bone metabolism and bone mass in patients with alcoholic liver cirrhosis | ALD (n = 30) | VDD (<50 nmol/L) in 67% of patients | VDD had the highest prevalence in CP C class patients |
| Vidot et al., 2017 [41] | Serum 25-hydroxyvitamin D deficiency and hepatic encephalopathy in chronic liver disease | CLD (n = 165) | Moderate to severe 25-OHD deficiency was identified in 49 patients of whom 36 had grade 2–3 HE | 25-OHD deficiency is observed in many patients with CLD |
| Jha et al., 2017 [42] | Effect of replenishment of vitamin D on survival in patients with decompensated liver cirrhosis: A prospective study | Decompensated CLD (n = 153) | VDD (<20 ng/mL) in 129(84%) of patients | VD supplementation may increase survival probability of patients with decompensated liver cirrhosis with improvements in MELD and CP scores |
| Khan et al., 2019 [43] | Impact of Vitamin D Status in Chronic Liver Disease | CLD patients (n = 75) | VDD (<20 ng/dL) was found in 41% out of which 19% suffered from severe VDD (<10 ng/dL) | VDD was associated with CLD and an increased in CP score |
| Yousif et al., 2019 [44] | Associated vitamin D deficiency is a risk factor for the complication of HCV-related liver cirrhosis including hepatic encephalopathy and spontaneous bacterial peritonitis | HCV-related liver cirrhosis w/and w/o HE (n = 90) | 25-OHD level was on average 16.28 ng/mL in control and 6.81 ng/mL in HE group | Low serum levels of 25-OHD were associated with HE in cirrhotic patients |
| Narayanasamy et al., 2019 [45] | High Prevalent Hypovitaminosis D Is Associated with Dysregulation of Calcium-parathyroid Hormone-vitamin D Axis in Patients with Chronic Liver Diseases | CLD patients (n = 236) | VDD (<30 ng/dL) was found in 162 (69%) | VDD is associated with higher CP scores |
| Kumar et al., 2020 [38] | Serum 25-hydroxyvitamin D level in patients with chronic liver disease and its correlation with hepatic encephalopathy: A cross-sectional study | CLD w/and w/o HE (n = 100) | Severe 25-OHD deficiency was seen in 38% of HE group compared to 6% in control group | Serum 25-OHD deficiency was more prevalent in HE group compared to control |
| Simbrunner et al., 2020 [46] | Vitamin A levels reflect disease severity and portal hypertension in patients with cirrhosis | ACLD (n = 234) | VDD was found in 133 (57%) | VDD increased with increasing CP stages |
| Afifi et al., 2021 [6] | Low Serum 25-Hydroxy Vitamin D (25-OHD) and Hepatic Encephalopathy in HCV-Related Liver Cirrhosis | HCV-related liver cirrhosis w/and w/o HE (n = 100) | HE groups w/severe VDD was 16% compared to other group at 6%; HE group w/moderate VDD was 24% compared to other group at 10% | Lower levels of 25-OHD were associated with higher incidence of HE in cirrhotic HCV patients |
| Kalita et al., 2022 [7] | Vitamin D in Patients of Chronic Liver Disease with Hepatic Encephalopathy | CLD w/HE (n = 88) | Mean serum 25-OHD was 24.11, 13.61, 8.41, and 8.00 ng/mL in grades 1–4 HE, respectively | Mean serum 25-OHD deficiency became more severe as HE worsened |
| Sarkar et al., 2024 [5] | Association of low serum 25-Hydroxy vitamin D [25(OH)D] with hepatic encephalopathy in patients with decompensated liver cirrhosis | Decompensated cirrhosis of the liver w/and w/o HE (n = 70) | 91% of HE patients had moderate to severe 25-OHD deficiency compared 51% in control group | Significant association was found between low serum 25-OHD and HE |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, C.D.; Stevens, C.M.; Bennett, M.R.; Litch, A.B.; Rodrigue, E.M.; Quintanilla, M.D.; Wallace, E.; Allahyari, M. The Role of Vitamin D Deficiency in Hepatic Encephalopathy: A Review of Pathophysiology, Clinical Outcomes, and Therapeutic Potential. Nutrients 2024, 16, 4007. https://doi.org/10.3390/nu16234007
Johnson CD, Stevens CM, Bennett MR, Litch AB, Rodrigue EM, Quintanilla MD, Wallace E, Allahyari M. The Role of Vitamin D Deficiency in Hepatic Encephalopathy: A Review of Pathophysiology, Clinical Outcomes, and Therapeutic Potential. Nutrients. 2024; 16(23):4007. https://doi.org/10.3390/nu16234007
Chicago/Turabian StyleJohnson, Coplen D., Christopher M. Stevens, Matthew R. Bennett, Adam B. Litch, Eugenie M. Rodrigue, Maria D. Quintanilla, Eric Wallace, and Massoud Allahyari. 2024. "The Role of Vitamin D Deficiency in Hepatic Encephalopathy: A Review of Pathophysiology, Clinical Outcomes, and Therapeutic Potential" Nutrients 16, no. 23: 4007. https://doi.org/10.3390/nu16234007
APA StyleJohnson, C. D., Stevens, C. M., Bennett, M. R., Litch, A. B., Rodrigue, E. M., Quintanilla, M. D., Wallace, E., & Allahyari, M. (2024). The Role of Vitamin D Deficiency in Hepatic Encephalopathy: A Review of Pathophysiology, Clinical Outcomes, and Therapeutic Potential. Nutrients, 16(23), 4007. https://doi.org/10.3390/nu16234007