
In Vitro Insights into the Dietary Role of Glucoraphanin and Its Metabolite Sulforaphane in Celiac Disease

 , , , , ,
, , , , ,  , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Cell Viability
2.4. Evaluation of Chemokine Release
2.5. Measurement of Epithelial Integrity
2.6. Luciferase Assay and Transcription Factors
2.7. Expression and Activity of Catalase
2.8. Phosphorylation of Histone γ-H2AX
2.9. LC-MS/MS Analysis on Broccoli Sprouts Sample
2.10. Measurement of the Antioxidant Capacity through DPPH and FRAP Assays
2.11. Statistical Analysis
3. Results
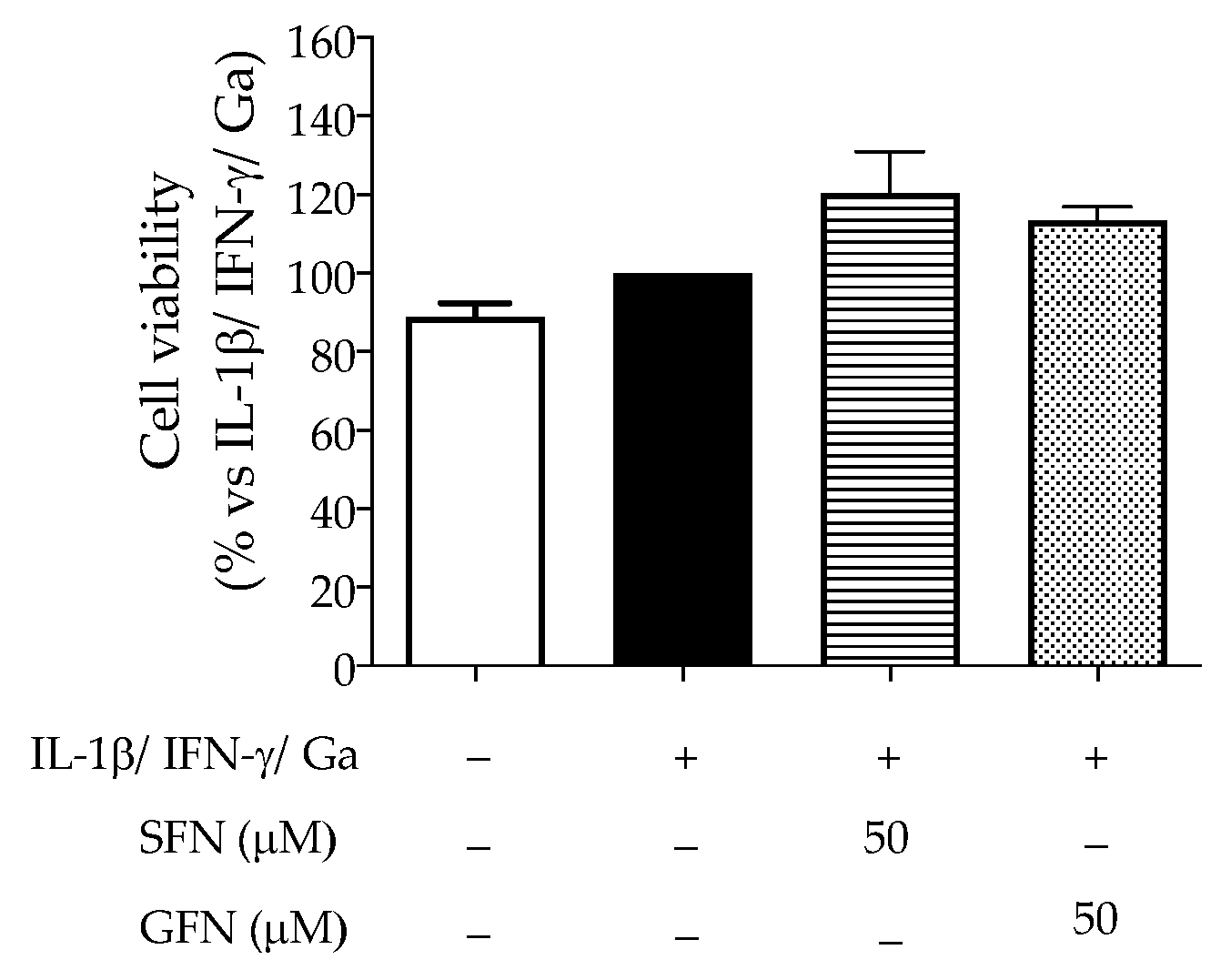
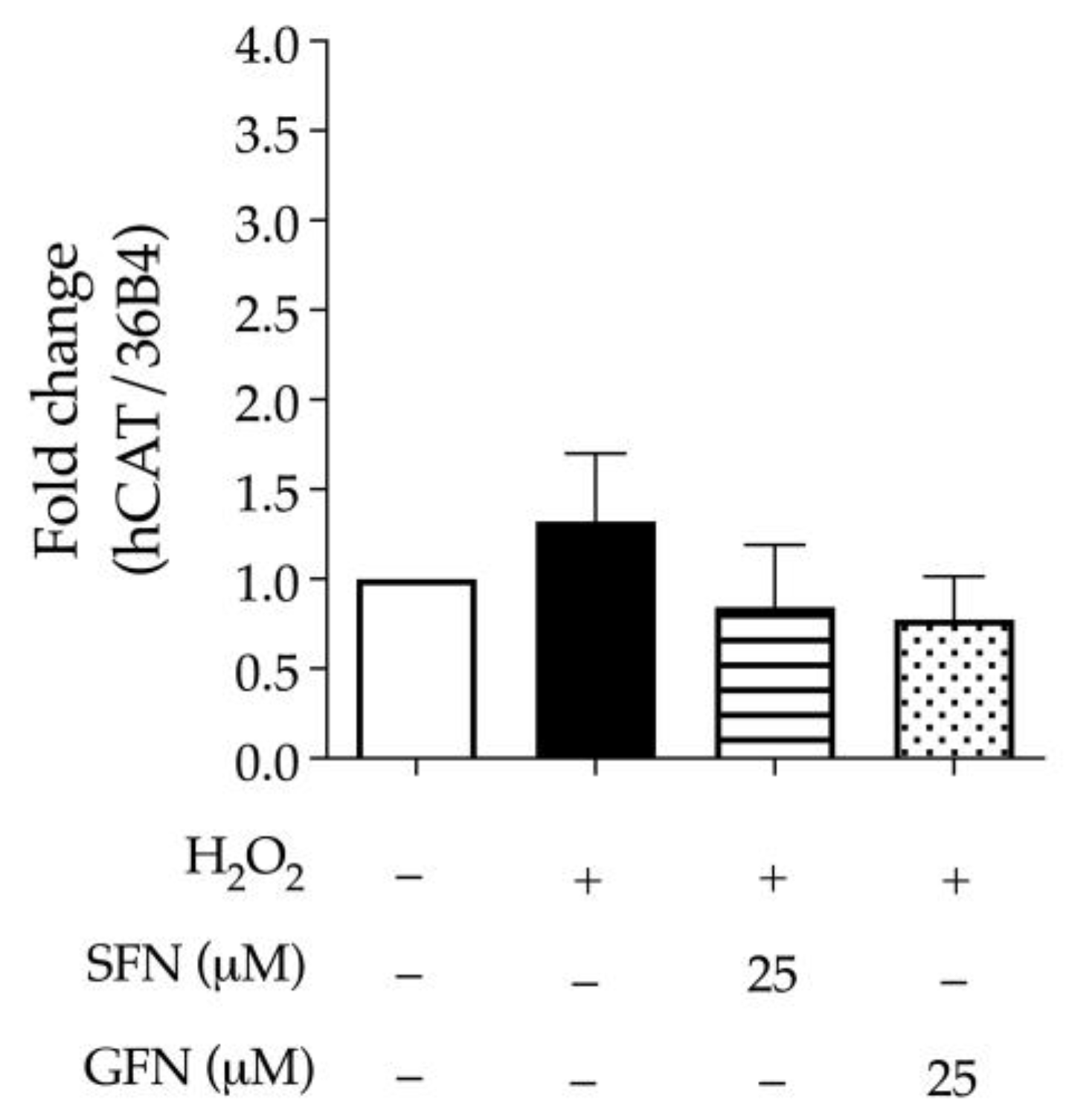
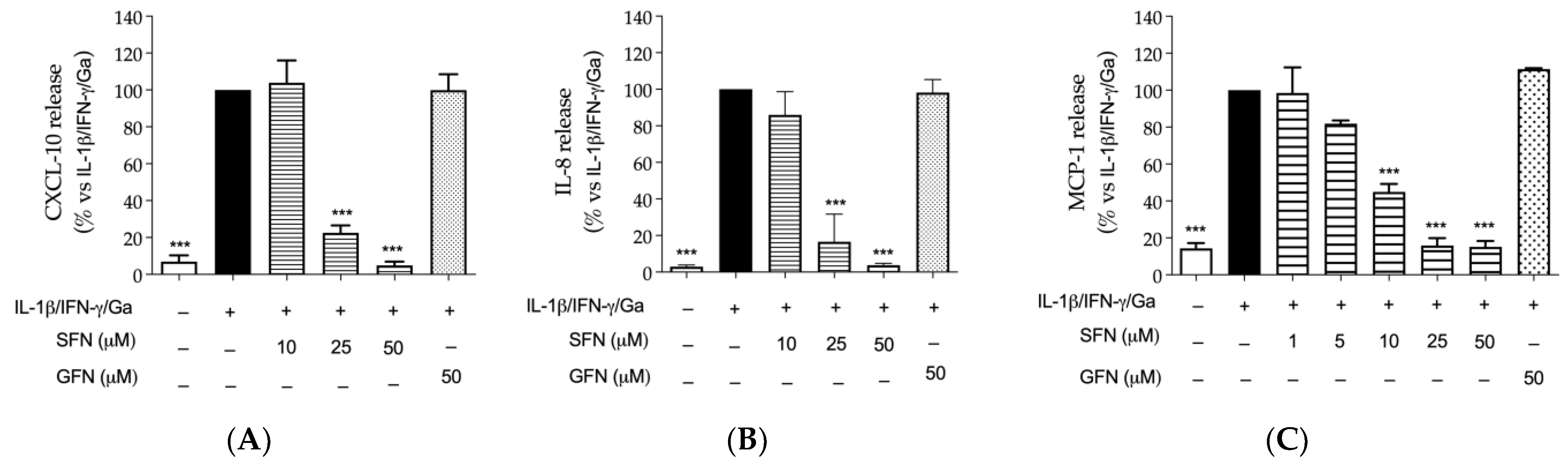
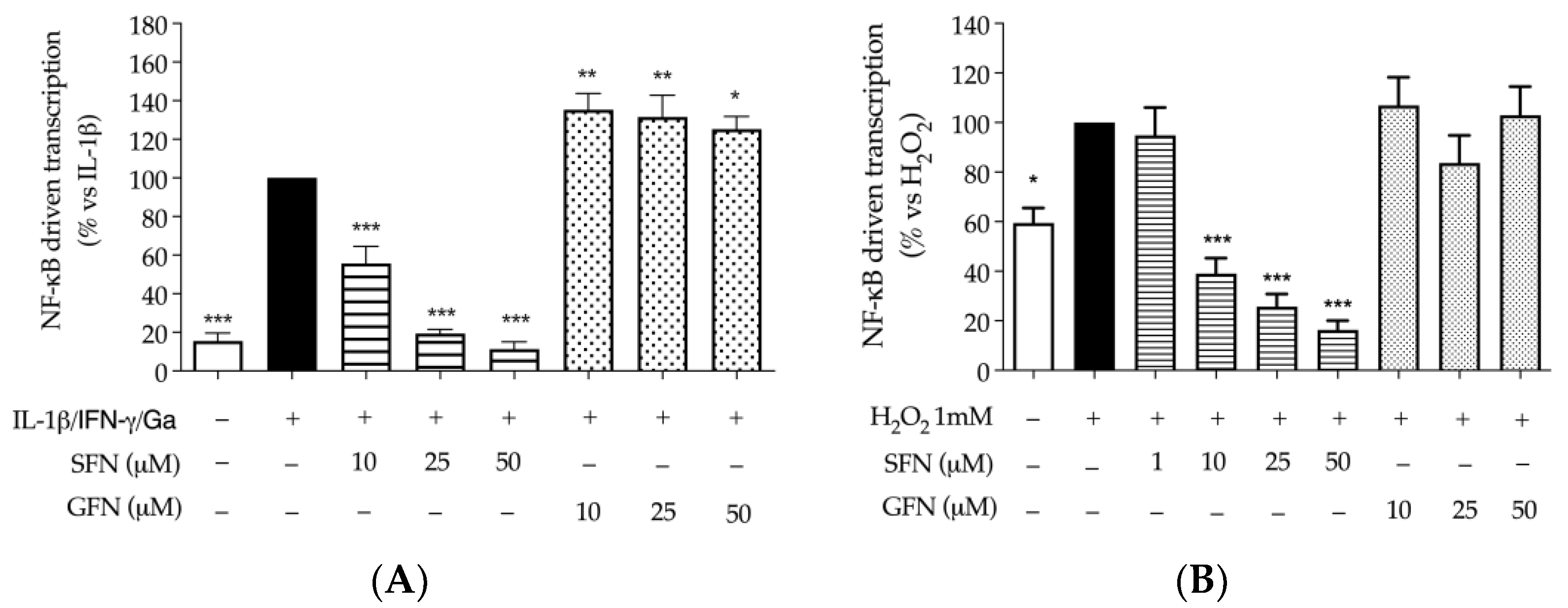
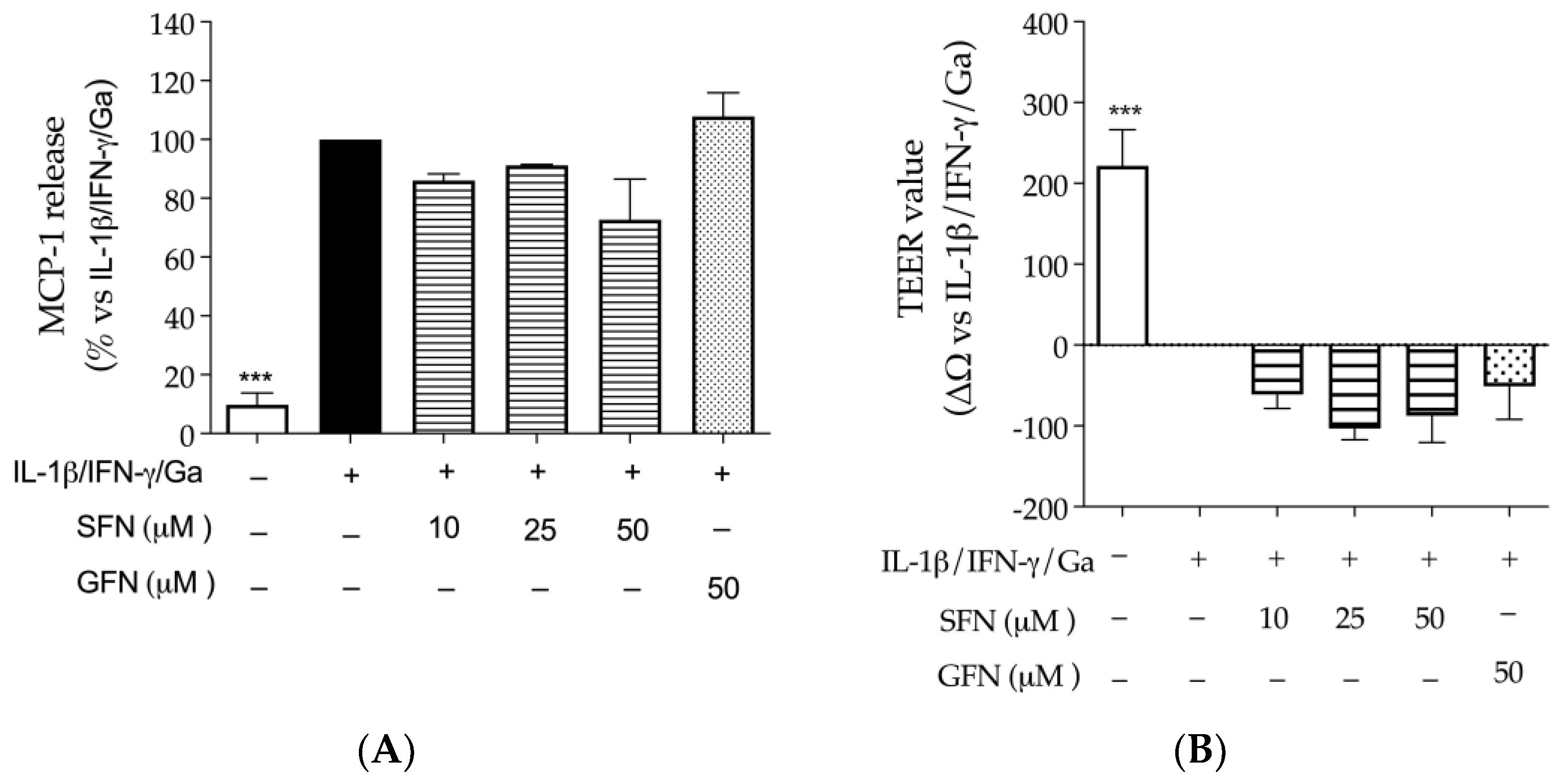
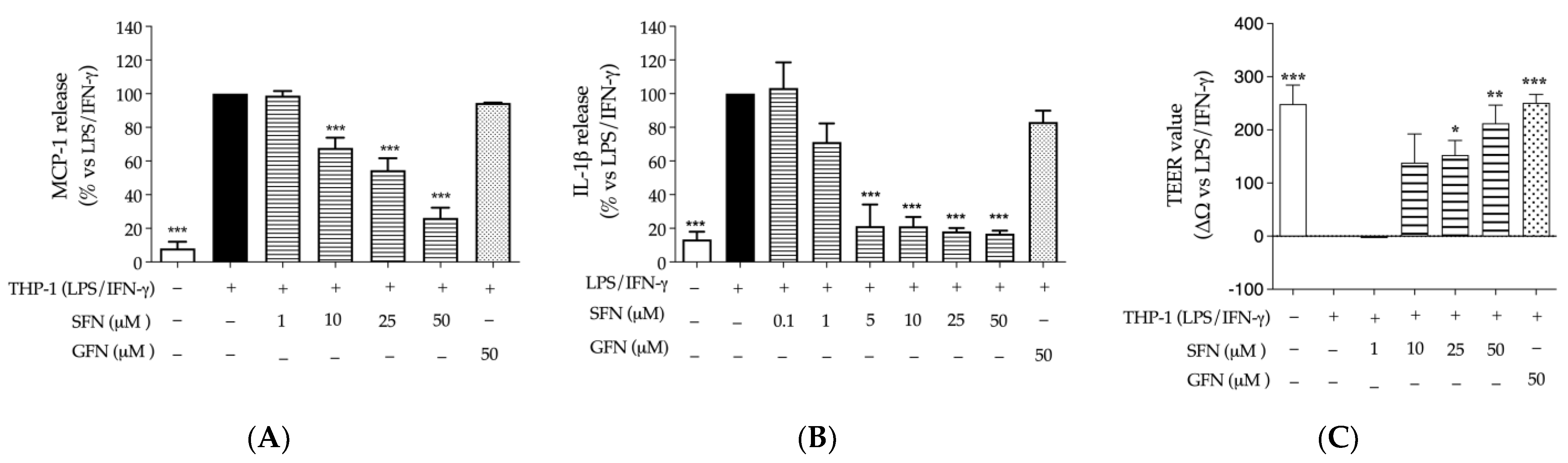
3.1. The Effect of Glucoraphanin and Sulforaphane on Inflammatory and Oxidative Markers in CaCo-2 Cells
3.2. Quantification of Sulforaphane before and after In Vitro Gastrointestinal Digestion of the Extract
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
References
- Prieto, M.A.; Lopez, C.J.; Simal-Gandara, J. Glucosinolates: Molecular structure, breakdown, genetic, bioavailability, properties and healthy and adverse effects. Adv. Food Nutr. Res. 2019, 90, 305–350. [Google Scholar] [CrossRef] [PubMed]
- Yagishita, Y.; Fahey, J.W.; Dinkova-Kostova, A.T.; Kensler, T.W. Broccoli or Sulforaphane: Is It the Source or Dose That Matters? Molecules 2019, 24, 3593. [Google Scholar] [CrossRef] [PubMed]
- Traka, M.; Gasper, A.V.; Melchini, A.; Bacon, J.R.; Needs, P.W.; Frost, V.; Chantry, A.; Jones, A.M.; Ortori, C.A.; Barrett, D.A.; et al. Broccoli consumption interacts with GSTM1 to perturb oncogenic signalling pathways in the prostate. PLoS ONE 2008, 3, e2568. [Google Scholar] [CrossRef] [PubMed]
- Gasmi, A.; Gasmi Benahmed, A.; Shanaida, M.; Chirumbolo, S.; Menzel, A.; Anzar, W.; Arshad, M.; Cruz-Martins, N.; Lysiuk, R.; Beley, N.; et al. Anticancer activity of broccoli, its organosulfur and polyphenolic compounds. Crit. Rev. Food Sci. Nutr. 2023, 2, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Mthembu, S.X.H.; Mazibuko-Mbeje, S.E.; Moetlediwa, M.T.; Muvhulawa, N.; Silvestri, S.; Orlando, P.; Nkambule, B.B.; Muller, C.J.F.; Ndwandwe, D.; Basson, A.K.; et al. Sulforaphane: A nutraceutical against diabetes-related complications. Pharmacol. Res. 2023, 196, 106918. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.; Dayal, H.; Swanson, M.; Mittelman, A.; Wilkinson, G. Diet in the epidemiology of cancer of the colon and rectum. J. Natl. Cancer Inst. 1978, 61, 709–714. [Google Scholar] [PubMed]
- Tse, G.; Eslick, G.D. Cruciferous vegetables and risk of colorectal neoplasms: A systematic review and meta-analysis. Nutr. Cancer 2014, 66, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Lippmann, D.; Lehmann, C.; Florian, S.; Barknowitz, G.; Haack, M.; Mewis, I.; Wiesner, M.; Schreiner, M.; Glatt, H.; Brigelius-Flohe, R.; et al. Glucosinolates from pak choi and broccoli induce enzymes and inhibit inflammation and colon cancer differently. Food Funct. 2014, 5, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Bessler, H.; Djaldetti, M. Broccoli and human health: Immunomodulatory effect of sulforaphane in a model of colon cancer. Int. J. Food Sci. Nutr. 2018, 69, 946–953. [Google Scholar] [CrossRef]
- Holman, J.; Hurd, M.; Moses, P.L.; Mawe, G.M.; Zhang, T.; Ishaq, S.L.; Li, Y. Interplay of broccoli/broccoli sprout bioactives with gut microbiota in reducing inflammation in inflammatory bowel diseases. J. Nutr. Biochem. 2023, 113, 109238. [Google Scholar] [CrossRef]
- Wei, L.Y.; Zhang, J.K.; Zheng, L.; Chen, Y. The functional role of sulforaphane in intestinal inflammation: A review. Food Funct. 2022, 13, 514–529. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.H.; Miller, M.J.; Jeffery, E. Glucoraphanin hydrolysis by microbiota in the rat cecum results in sulforaphane absorption. Food Funct. 2010, 1, 161–166. [Google Scholar] [CrossRef]
- Vancamelbeke, M.; Vermeire, S. The intestinal barrier: A fundamental role in health and disease. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Caio, G.; Lungaro, L.; Segata, N.; Guarino, M.; Zoli, G.; Volta, U.; De Giorgio, R. Effect of Gluten-Free Diet on Gut Microbiota Composition in Patients with Celiac Disease and Non-Celiac Gluten/Wheat Sensitivity. Nutrients 2020, 12, 1832. [Google Scholar] [CrossRef]
- Ramirez-Sanchez, A.D.; Tan, I.L.; Gonera-de Jong, B.C.; Visschedijk, M.C.; Jonkers, I.; Withoff, S. Molecular Biomarkers for Celiac Disease: Past, Present and Future. Int. J. Mol. Sci. 2020, 21, 8528. [Google Scholar] [CrossRef]
- Capozzi, A.; Vincentini, O.; Gizzi, P.; Porzia, A.; Longo, A.; Felli, C.; Mattei, V.; Mainiero, F.; Silano, M.; Sorice, M.; et al. Modulatory Effect of Gliadin Peptide 10-mer on Epithelial Intestinal CACO-2 Cell Inflammatory Response. PLoS ONE 2013, 8, e66561. [Google Scholar] [CrossRef]
- De Stefano, D.; Maiuri, M.C.; Iovine, B.; Ialenti, A.; Bevilacqua, M.A.; Carnuccio, R. The role of NF-kappaB, IRF-1, and STAT-1alpha transcription factors in the iNOS gene induction by gliadin and IFN-gamma in RAW 264.7 macrophages. J. Mol. Med. 2006, 84, 65–74. [Google Scholar] [CrossRef]
- Lammers, K.M.; Lu, R.; Brownley, J.; Lu, B.; Gerard, C.; Thomas, K.; Rallabhandi, P.; Shea-Donohue, T.; Tamiz, A.; Alkan, S.; et al. Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3. Gastroenterology 2008, 135, 194–204.e193. [Google Scholar] [CrossRef]
- Monguzzi, E.; Marabini, L.; Elli, L.; Vaira, V.; Ferrero, S.; Ferretti, F.; Branchi, F.; Gaudioso, G.; Scricciolo, A.; Lombardo, V.; et al. Gliadin effect on the oxidative balance and DNA damage: An in-vitro, ex-vivo study. Dig. Liver Dis. 2019, 51, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Odetti, P.; Valentini, S.; Aragno, I.; Garibaldi, S.; Pronzato, M.A.; Rolandi, E.; Barreca, T. Oxidative stress in subjects affected by celiac disease. Free. Radic. Res. 1998, 29, 17–24. [Google Scholar] [CrossRef]
- Szaflarska-Poplawska, A.; Siomek, A.; Czerwionka-Szaflarska, M.; Gackowski, D.; Rozalski, R.; Guz, J.; Szpila, A.; Zarakowska, E.; Olinski, R. Oxidatively damaged DNA/oxidative stress in children with celiac disease. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1960–1965. [Google Scholar] [CrossRef] [PubMed]
- Piazza, S.; Colombo, F.; Bani, C.; Fumagalli, M.; Vincentini, O.; Sangiovanni, E.; Martinelli, G.; Biella, S.; Silano, M.; Restani, P.; et al. Evaluation of the Potential Anti-Inflammatory Activity of Black Rice in the Framework of Celiac Disease. Foods 2022, 12, 63. [Google Scholar] [CrossRef] [PubMed]
- Piazza, S.; Bani, C.; Colombo, F.; Mercogliano, F.; Pozzoli, C.; Martinelli, G.; Petroni, K.; Roberto Pilu, S.; Sonzogni, E.; Fumagalli, M.; et al. Pigmented corn as a gluten-free source of polyphenols with anti-inflammatory and antioxidant properties in CaCo-2 cells. Food Res. Int. 2024, 191, 114640. [Google Scholar] [CrossRef] [PubMed]
- Le, N.P.K.; Altenburger, M.J.; Lamy, E. Development of an Inflammation-Triggered In Vitro “Leaky Gut” Model Using Caco-2/HT29-MTX-E12 Combined with Macrophage-like THP-1 Cells or Primary Human-Derived Macrophages. Int. J. Mol. Sci. 2023, 24, 7427. [Google Scholar] [CrossRef] [PubMed]
- Hoensch, H.P.; Weigmann, B. Regulation of the intestinal immune system by flavonoids and its utility in chronic inflammatory bowel disease. World J. Gastroenterol. 2018, 24, 877–881. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Oshima, T.; Tomita, T.; Fukui, H.; Miwa, H. Butyrate Alleviates Cytokine-Induced Barrier Dysfunction by Modifying Claudin-2 Levels. Biology 2021, 10, 205. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, D.J.; Kolde, R.; Sooknah, M.; Graham, D.B.; Sundberg, T.B.; Latorre, I.; Mikkelsen, T.S.; Xavier, R.J. Simultaneous Pathway Activity Inference and Gene Expression Analysis Using RNA Sequencing. Cell Syst. 2016, 2, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Piazza, S.; Martinelli, G.; Vrhovsek, U.; Masuero, D.; Fumagalli, M.; Magnavacca, A.; Pozzoli, C.; Canilli, L.; Terno, M.; Angarano, M.; et al. Anti-Inflammatory and Anti-Acne Effects of Hamamelis virginiana Bark in Human Keratinocytes. Antioxidants 2022, 11, 1119. [Google Scholar] [CrossRef] [PubMed]
- Roca, P.; Di Lorenzo, C.; Bani, C.; Mercogliano, F.; Bosso, A.; Restani, P. Valorization of wine industry by-products: Characterization of phenolic profile and investigation of potential healthy properties. BIO Web Conf. 2023, 68, 04016. [Google Scholar] [CrossRef]
- Sjakste, N.; Djelic, N.; Dzintare, M.; Zivkovic, L. DNA-BINDING and DNA-protecting activities of small natural organic molecules and food extracts. Chem. Biol. Interact. 2020, 323, 109030. [Google Scholar] [CrossRef]
- Navrkalova, V.; Kafkova, L.R.; Divoky, V.; Pospisilova, S. Oxidative stress as a therapeutic perspective for ATM-deficient chronic lymphocytic leukemia patients. Haematologica 2015, 100, 994–996. [Google Scholar] [CrossRef] [PubMed]
- Bollati, C.; Cruz-Chamorro, I.; Aiello, G.; Li, J.; Bartolomei, M.; Santos-Sanchez, G.; Ranaldi, G.; Ferruzza, S.; Sambuy, Y.; Arnoldi, A.; et al. Investigation of the intestinal trans-epithelial transport and antioxidant activity of two hempseed peptides WVSPLAGRT (H2) and IGFLIIWV (H3). Food Res. Int. 2022, 152, 110720. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.W.; Talalay, P. Antioxidant functions of sulforaphane: A potent inducer of Phase II detoxication enzymes. Food Chem. Toxicol. 1999, 37, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Demoulin, B.; Hermant, M.; Castrogiovanni, C.; Staudt, C.; Dumont, P. Resveratrol induces DNA damage in colon cancer cells by poisoning topoisomerase II and activates the ATM kinase to trigger p53-dependent apoptosis. Toxicol. Vitr. 2015, 29, 1156–1165. [Google Scholar] [CrossRef] [PubMed]
- Traka, M.; Gasper, A.V.; Smith, J.A.; Hawkey, C.J.; Bao, Y.; Mithen, R.F. Transcriptome analysis of human colon Caco-2 cells exposed to sulforaphane. J. Nutr. 2005, 135, 1865–1872. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Proll, M.; Neuhoff, C.; Zhang, R.; Cinar, M.U.; Hossain, M.M.; Tesfaye, D.; Grosse-Brinkhaus, C.; Salilew-Wondim, D.; Tholen, E.; et al. Sulforaphane epigenetically regulates innate immune responses of porcine monocyte-derived dendritic cells induced with lipopolysaccharide. PLoS ONE 2015, 10, e0121574. [Google Scholar] [CrossRef] [PubMed]
- Lucarini, E.; Micheli, L.; Trallori, E.; Citi, V.; Martelli, A.; Testai, L.; De Nicola, G.R.; Iori, R.; Calderone, V.; Ghelardini, C.; et al. Effect of glucoraphanin and sulforaphane against chemotherapy-induced neuropathic pain: Kv7 potassium channels modulation by H2S release in vivo. Phytother. Res. 2018, 32, 2226–2234. [Google Scholar] [CrossRef] [PubMed]
- Folkard, D.L.; Melchini, A.; Traka, M.H.; Al-Bakheit, A.; Saha, S.; Mulholland, F.; Watson, A.; Mithen, R.F. Suppression of LPS-induced transcription and cytokine secretion by the dietary isothiocyanate sulforaphane. Mol. Nutr. Food Res. 2014, 58, 2286–2296. [Google Scholar] [CrossRef]
- Krupa-Kozak, U.; Drabinska, N.; Baczek, N.; Simkova, K.; Starowicz, M.; Jelinski, T. Application of Broccoli Leaf Powder in Gluten-Free Bread: An Innovative Approach to Improve Its Bioactive Potential and Technological Quality. Foods 2021, 10, 819. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (min) | Phase A (%) | Phase B (%) |
|---|---|---|
| 0 | 90 | 10 |
| 1 | 90 | 10 |
| 6 | 0 | 100 |
| 8 | 0 | 100 |
| 8.10 | 90 | 10 |
| 10 | 90 | 10 |
| DPPH (μM GAE ± S.D.) | FRAP (μM FeSO4 ± S.D.) | |
|---|---|---|
| SFN (200 μM) | n.d. | n.d. |
| GFN (200 μM) | n.d. | n.d. |
| Resv. (100 μM) | 26.63 ± 2.37 | 333.98 ± 9.67 |
| Inflammatory Markers | IC50 (μM) of Sulforaphane |
|---|---|
| CXCL-10 | 23.55 |
| IL-8 | 15.74 |
| MCP-1 | 7.81 |
| NF-κB | 9.75 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sonzogni, E.; Martinelli, G.; Fumagalli, M.; Maranta, N.; Pozzoli, C.; Bani, C.; Marrari, L.A.; Di Lorenzo, C.; Sangiovanni, E.; Dell’Agli, M.; et al. In Vitro Insights into the Dietary Role of Glucoraphanin and Its Metabolite Sulforaphane in Celiac Disease. Nutrients 2024, 16, 2743. https://doi.org/10.3390/nu16162743
Sonzogni E, Martinelli G, Fumagalli M, Maranta N, Pozzoli C, Bani C, Marrari LA, Di Lorenzo C, Sangiovanni E, Dell’Agli M, et al. In Vitro Insights into the Dietary Role of Glucoraphanin and Its Metabolite Sulforaphane in Celiac Disease. Nutrients. 2024; 16(16):2743. https://doi.org/10.3390/nu16162743
Chicago/Turabian StyleSonzogni, Elisa, Giulia Martinelli, Marco Fumagalli, Nicole Maranta, Carola Pozzoli, Corinne Bani, Luigi Alberto Marrari, Chiara Di Lorenzo, Enrico Sangiovanni, Mario Dell’Agli, and et al. 2024. "In Vitro Insights into the Dietary Role of Glucoraphanin and Its Metabolite Sulforaphane in Celiac Disease" Nutrients 16, no. 16: 2743. https://doi.org/10.3390/nu16162743
APA StyleSonzogni, E., Martinelli, G., Fumagalli, M., Maranta, N., Pozzoli, C., Bani, C., Marrari, L. A., Di Lorenzo, C., Sangiovanni, E., Dell’Agli, M., & Piazza, S. (2024). In Vitro Insights into the Dietary Role of Glucoraphanin and Its Metabolite Sulforaphane in Celiac Disease. Nutrients, 16(16), 2743. https://doi.org/10.3390/nu16162743