
GHSR Deletion in β-Cells of Male Mice: Ineffective in Obesity, but Effective in Protecting against Streptozotocin-Induced β-Cell Injury in Aging

 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. High-Fat-Diet (HFD) Feeding and Streptozotocin-Treated Regular Diet (RD)-Fed Mice
2.3. Glucose Tolerance Test and In Vivo Glucose-Stimulated Insulin Secretion
2.4. Ex Vivo Glucose-Stimulated Insulin Secretion
2.5. Metabolic Hormone Measurement
2.6. RNA Isolation, RT-PCR, and Real-Time PCR
2.7. Statistical Analysis
3. Results
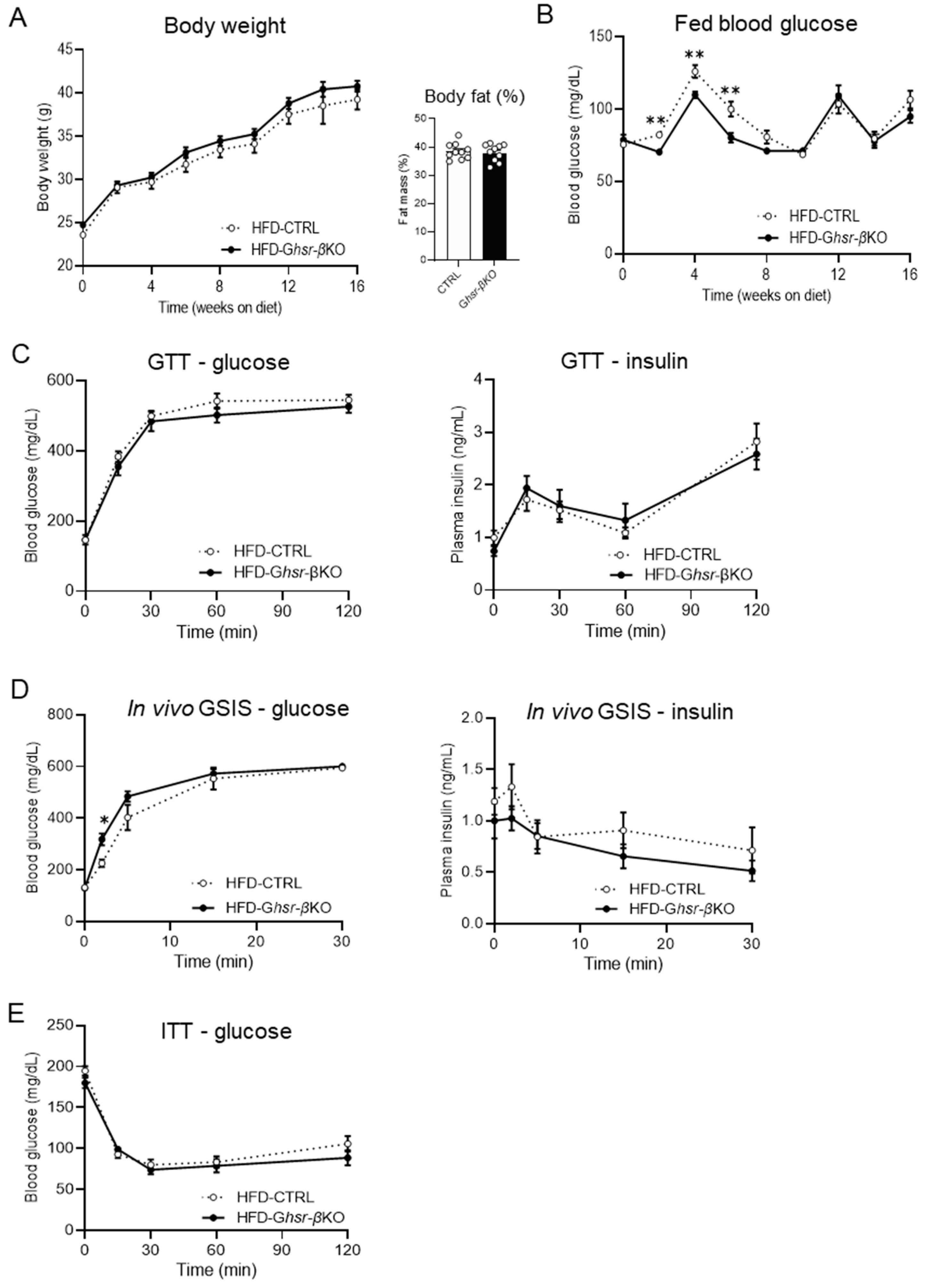
3.1. β-Cell Ghsr Deficiency Had no Discernable Effect on HFD-Induced Obesity and Glucose Dysregulation
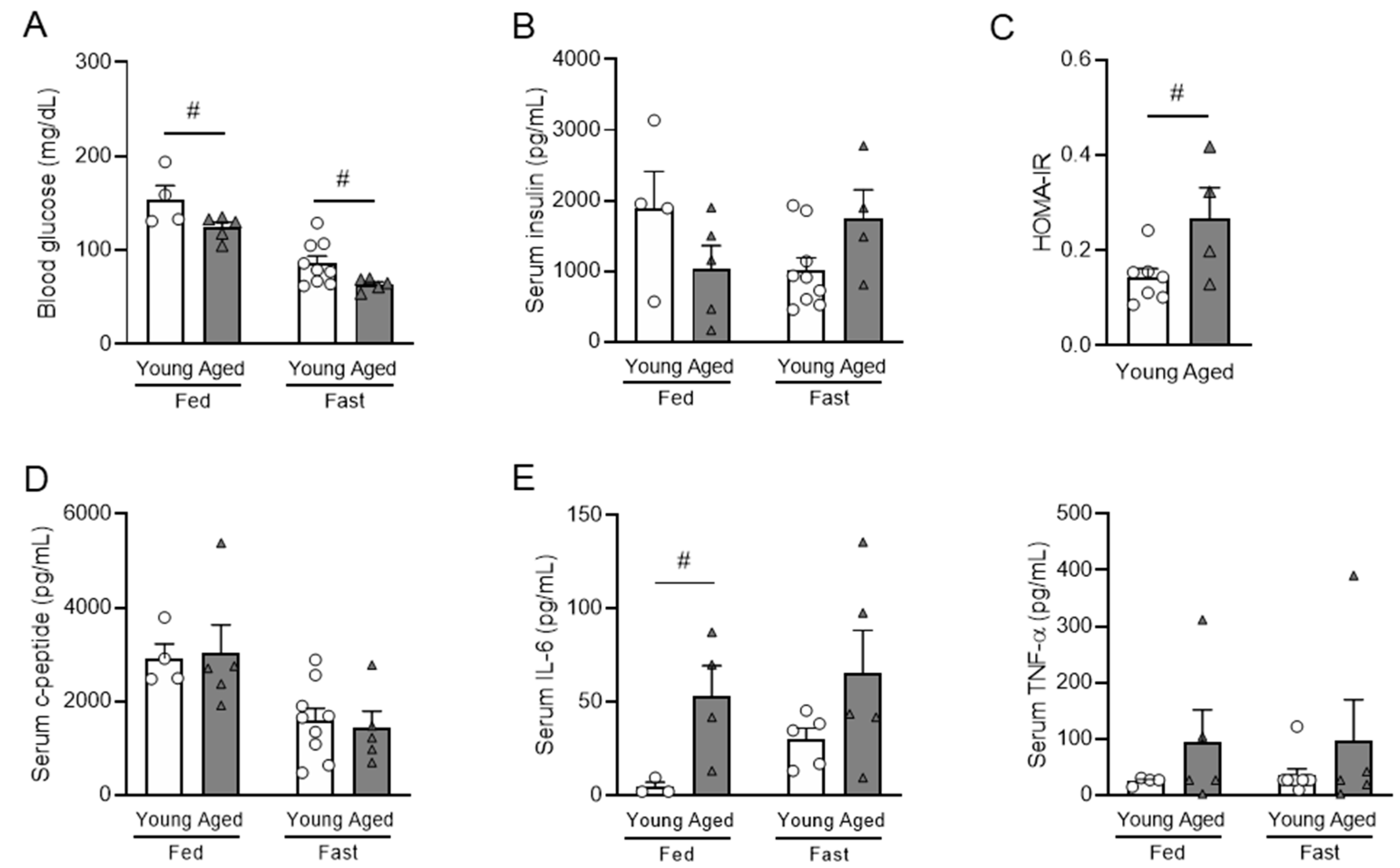
3.2. Aging Is Correlated with Impaired Insulin Sensitivity and Elevated Inflammatory Cytokines
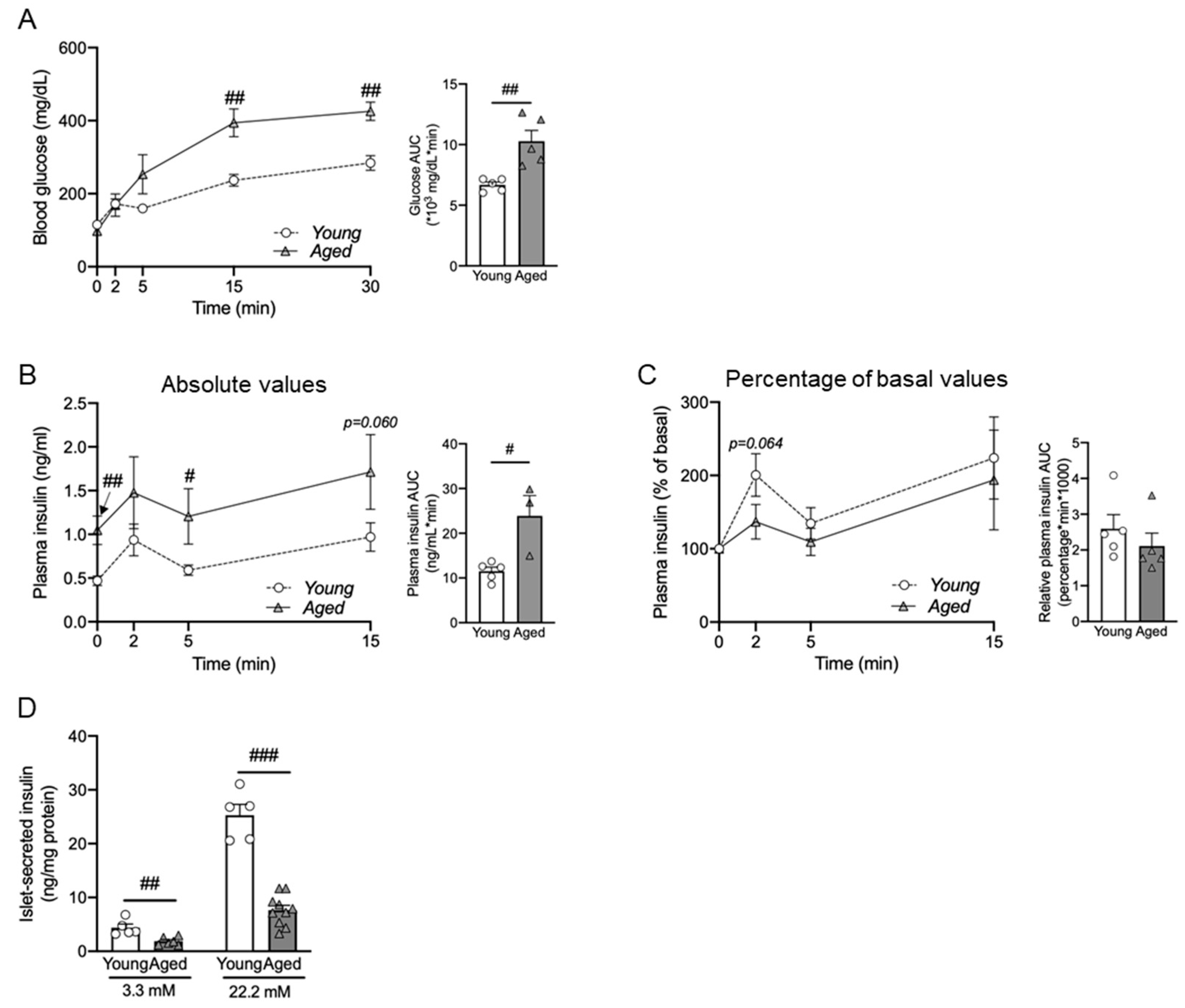
3.3. Aging Impairs Glucose-Stimulated Insulin Secretion (GSIS) Both In Vivo and Ex Vivo
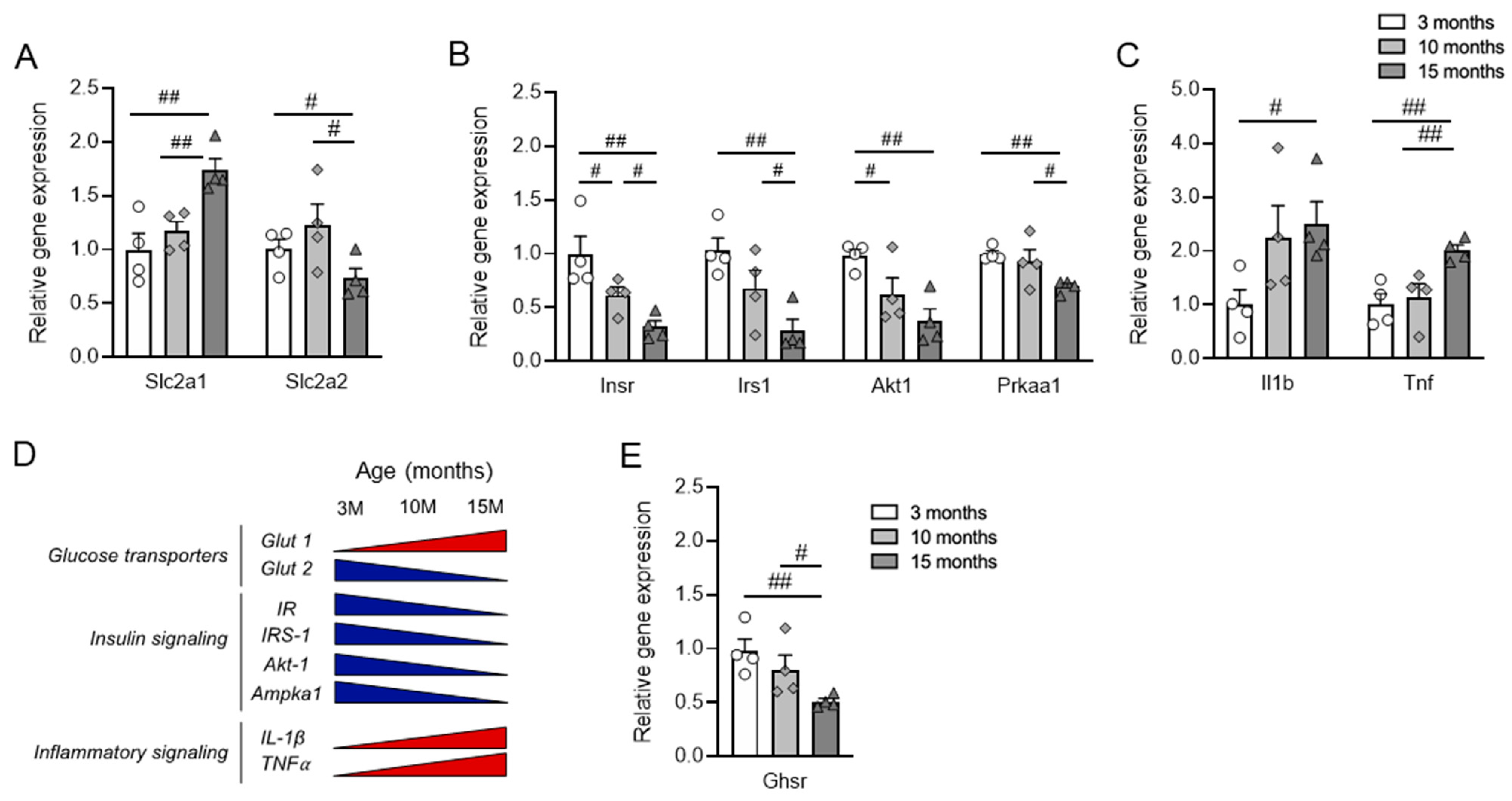
3.4. Pancreatic Islets Show Differential Changes in Gene Expression during Aging
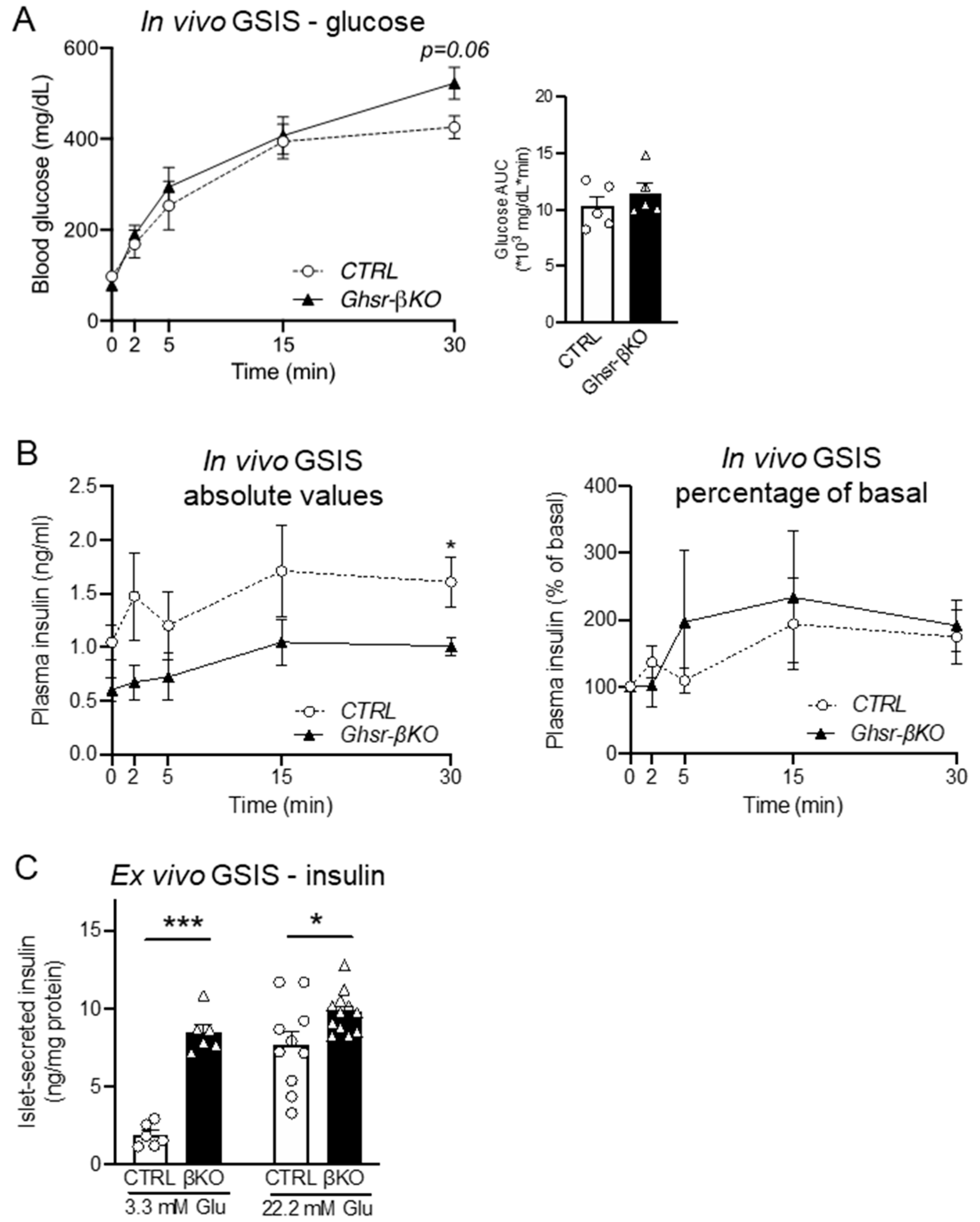
3.5. β-Cell Ghsr Deficiency has Little Effect on In Vivo GSIS but Improves Ex Vivo GSIS in Normal Aged Mice
3.6. β-Cell Ghsr Deficiency Protects against STZ-Induced β-Cell Damage in Aged Mice
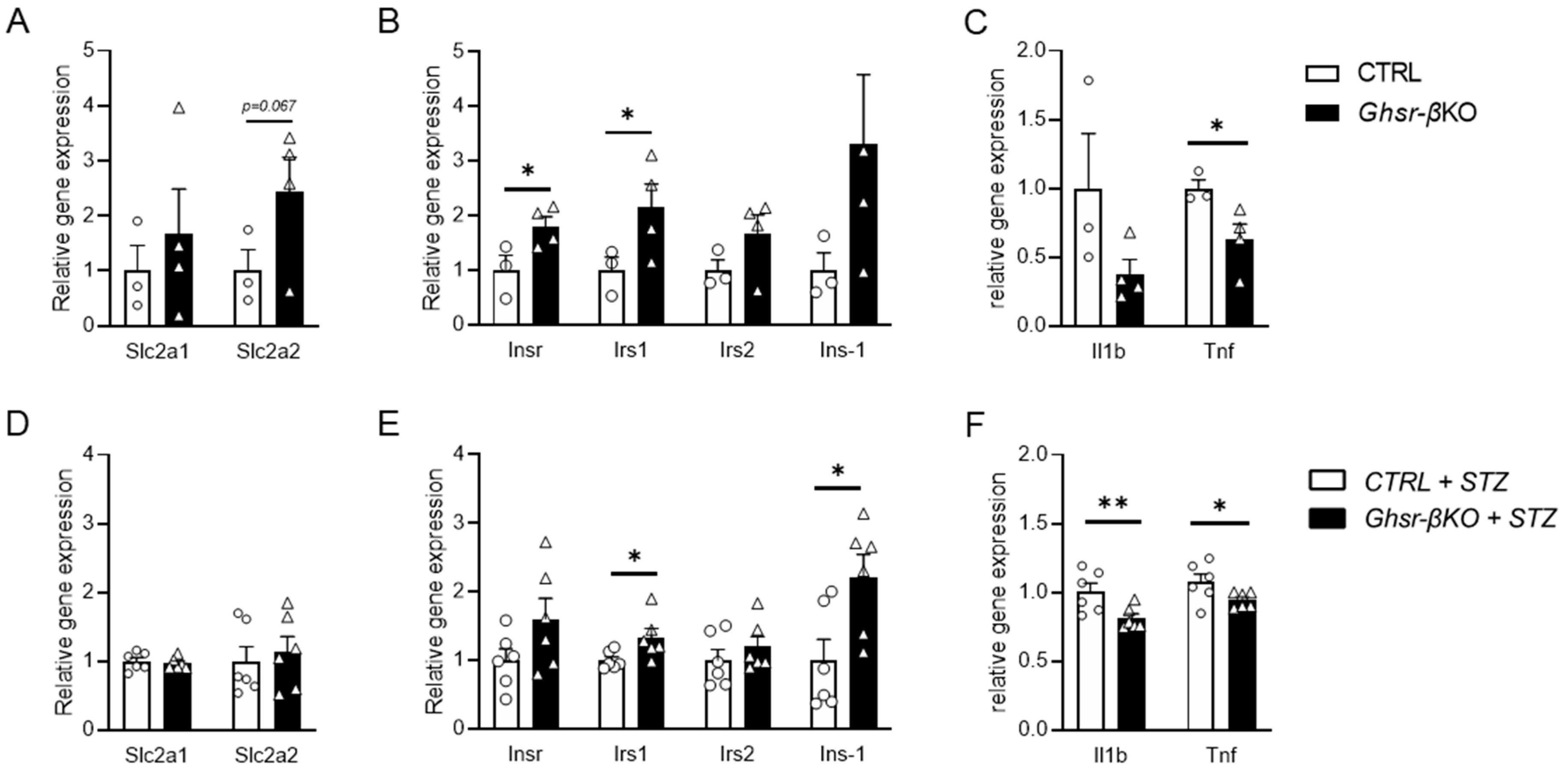
3.7. Islets from β-Cell Ghsr-Deficient Mice Exhibit Differential Gene Expressions of Insulin Signaling and Pro-Inflammatory Cytokines
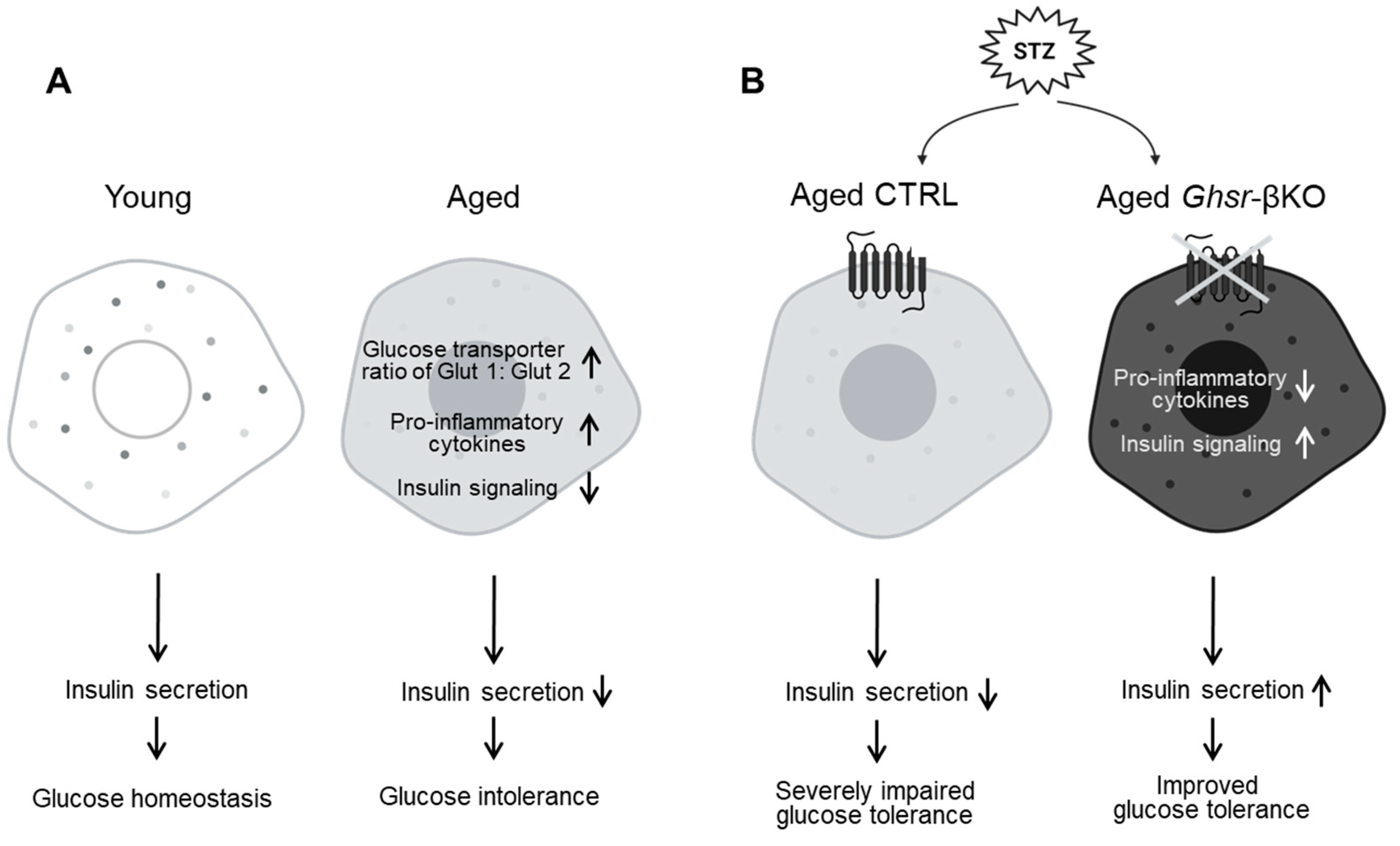
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xu, G.; Liu, B.; Sun, Y.; Du, Y.; Snetselaar, L.G.; Hu, F.B.; Bao, W. Prevalence of diagnosed type 1 and type 2 diabetes among us adults in 2016 and 2017: Population based study. BMJ 2018, 362, k1497. Available online: https://www.bmj.com/content/bmj/362/bmj.k1497.full.pdf (accessed on 1 May 2024). [CrossRef]
- Frasca, D.; Blomberg, B.B.; Paganelli, R. Aging, obesity, and inflammatory age-related diseases. Front. Immunol. 2017, 8, 1745. [Google Scholar] [CrossRef] [PubMed]
- Dludla, P.V.; Mabhida, S.E.; Ziqubu, K.; Nkambule, B.B.; Mazibuko-Mbeje, S.E.; Hanser, S.; Basson, A.K.; Pheiffer, C.; Kengne, A.P. Pancreatic β-cell dysfunction in type 2 diabetes: Implications of inflammation and oxidative stress. World J. Diabetes 2023, 14, 130. [Google Scholar] [CrossRef] [PubMed]
- Cerf, M.E. Beta cell physiological dynamics and dysfunctional transitions in response to islet inflammation in obesity and diabetes. Metabolites 2020, 10, 452. [Google Scholar] [CrossRef] [PubMed]
- Khanna, D.; Khanna, S.; Khanna, P.; Kahar, P.; Patel, B.M. Obesity: A chronic low-grade inflammation and its markers. Cureus 2022, 14, e22711. [Google Scholar] [CrossRef] [PubMed]
- Febbraio, M.A. Role of interleukins in obesity: Implications for metabolic disease. Trends Endocrinol. Metab. 2014, 25, 312–319. [Google Scholar] [CrossRef]
- Donath, M.Y.; Størling, J.; Berchtold, L.A.; Billestrup, N.; Mandrup-Poulsen, T. Cytokines and β-cell biology: From concept to clinical translation. Endocr. Rev. 2008, 29, 334–350. [Google Scholar] [CrossRef] [PubMed]
- Khodabandehloo, H.; Gorgani-Firuzjaee, S.; Panahi, G.; Meshkani, R. Molecular and cellular mechanisms linking inflammation to insulin resistance and β-cell dysfunction. Transl. Res. 2016, 167, 228–256. [Google Scholar] [CrossRef] [PubMed]
- Dahllöf, M.S.; Christensen, D.P.; Lundh, M.; Dinarello, C.A.; Mascagni, P.; Grunnet, L.G.; Mandrup-Poulsen, T. The lysine deacetylase inhibitor givinostat inhibits β-cell il-1β induced il-1β transcription and processing. Islets 2012, 4, 417–422. [Google Scholar] [CrossRef]
- Sinclair, A.; Saeedi, P.; Kaundal, A.; Karuranga, S.; Malanda, B.; Williams, R. Diabetes and global ageing among 65–99-year-old adults: Findings from the international diabetes federation diabetes atlas. Diabetes Res. Clin. Pract. 2020, 162, 108078. [Google Scholar] [CrossRef]
- Longo, M.; Bellastella, G.; Maiorino, M.I.; Meier, J.J.; Esposito, K.; Giugliano, D. Diabetes and aging: From treatment goals to pharmacologic therapy. Front. Endocrinol. 2019, 10, 45. [Google Scholar] [CrossRef] [PubMed]
- Chentli, F.; Azzoug, S.; Mahgoun, S. Diabetes mellitus in elderly. Indian J. Endocrinol. Metab. 2015, 19, 744. [Google Scholar] [CrossRef] [PubMed]
- Gunasekaran, U.; Gannon, M. Type 2 diabetes and the aging pancreatic beta cell. Aging 2011, 3, 565. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.M.; Halter, J.B. Aging and insulin secretion. Am. J. Physiol.-Endocrinol. Metab. 2003, 284, E7–E12. [Google Scholar] [CrossRef]
- Aguayo-Mazzucato, C.; Andle, J.; Lee, T.B., Jr.; Midha, A.; Talemal, L.; Chipashvili, V.; Hollister-Lock, J.; van Deursen, J.; Weir, G.; Bonner-Weir, S. Acceleration of β cell aging determines diabetes and senolysis improves disease outcomes. Cell Metab. 2019, 30, 129–142.e4. [Google Scholar] [CrossRef]
- Aguayo-Mazzucato, C. Functional changes in beta cells during ageing and senescence. Diabetologia 2020, 63, 2022–2029. [Google Scholar] [CrossRef]
- Cerf, M.E. Beta cell dysfunction and insulin resistance. Front. Endocrinol. 2013, 4, 37. [Google Scholar] [CrossRef]
- Kojima, M.; Kangawa, K. Ghrelin: More than endogenous growth hormone secretagogue. Ann. N. Y. Acad. Sci. 2010, 1200, 140–148. [Google Scholar] [CrossRef]
- Pradhan, G.; Samson, S.L.; Sun, Y. Ghrelin: Much more than a hunger hormone. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 619. [Google Scholar] [CrossRef]
- Yin, Y.; Li, Y.; Zhang, W. The growth hormone secretagogue receptor: Its intracellular signaling and regulation. Int. J. Mol. Sci. 2014, 15, 4837–4855. [Google Scholar] [CrossRef]
- Cornejo, M.P.; Mustafá, E.R.; Cassano, D.; Banères, J.; Raingo, J.; Perello, M. The ups and downs of growth hormone secretagogue receptor signaling. FEBS J. 2021, 288, 7213–7229. [Google Scholar] [CrossRef] [PubMed]
- Camina, J. Cell biology of the ghrelin receptor. J. Neuroendocrinol. 2006, 18, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.D.; Nogueiras, R.; Andermann, M.L.; Andrews, Z.B.; Anker, S.D.; Argente, J.; Batterham, R.L.; Benoit, S.; Bowers, C.Y.; Broglio, F. Ghrelin. Mol. Metab. 2015, 4, 437–460. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Z.-T.; Luo, Q. Molecular mechanisms and health benefits of ghrelin: A narrative review. Nutrients 2022, 14, 4191. [Google Scholar] [CrossRef] [PubMed]
- Granata, R.; Baragli, A.; Settanni, F.; Scarlatti, F.; Ghigo, E. Unraveling the role of the ghrelin gene peptides in the endocrine pancreas. J. Mol. Endocrinol. 2010, 45, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Gnanapavan, S.; Kola, B.; Bustin, S.A.; Morris, D.G.; McGee, P.; Fairclough, P.; Bhattacharya, S.; Carpenter, R.; Grossman, A.B.; Korbonits, M. The tissue distribution of the mrna of ghrelin and subtypes of its receptor, ghs-r, in humans. J. Clin. Endocrinol. Metab. 2002, 87, 2988–2991. [Google Scholar] [CrossRef] [PubMed]
- DiGruccio, M.R.; Mawla, A.M.; Donaldson, C.J.; Noguchi, G.M.; Vaughan, J.; Cowing-Zitron, C.; van der Meulen, T.; Huising, M.O. Comprehensive alpha, beta and delta cell transcriptomes reveal that ghrelin selectively activates delta cells and promotes somatostatin release from pancreatic islets. Mol. Metab. 2016, 5, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, A.E.; Svendsen, B.; Lam, B.Y.; Yeo, G.S.; Holst, J.J.; Reimann, F.; Gribble, M.F. Transcriptomic profiling of pancreatic alpha, beta and delta cell populations identifies delta cells as a principal target for ghrelin in mouse islets. Diabetologia 2016, 59, 2156–2165. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, G.; Wu, C.-S.; Villarreal, D.; Lee, J.H.; Han, H.W.; Gaharwar, A.; Tian, Y.; Fu, W.; Guo, S.; Smith, R.G. Β cell ghs-r regulates insulin secretion and sensitivity. Int. J. Mol. Sci. 2021, 22, 3950. [Google Scholar] [CrossRef]
- Pradhan, G.; Lee, J.H.; Wu, C.-S.; Wang, H.; Lin, L.; Donti, T.; Graham, B.H.; Rajan, A.S.; Balasubramanyam, A.; Samson, S.L.; et al. Balasubramanyam and Samson, S.L. Mechanistic investigation of ghs-r mediated glucose-stimulated insulin secretion in pancreatic islets. Biomolecules 2022, 12, 407. [Google Scholar] [CrossRef]
- Gupta, D.; Dowsett, G.K.C.; Mani, B.K.; Shankar, K.; Osborne-Lawrence, S.; Metzger, N.P.; Lam, B.Y.H.; Yeo, G.S.H.; Zigman, J.M. High coexpression of the ghrelin and leap2 receptor ghsr with pancreatic polypeptide in mouse and human islets. Endocrinology 2021, 162, bqab148. [Google Scholar] [CrossRef] [PubMed]
- MacKay, H.; Charbonneau, V.R.; St-Onge, V.; Murray, E.; Watts, A.; Wellman, M.K.; Abizaid, A. Rats with a truncated ghrelin receptor (ghsr) do not respond to ghrelin, and show reduced intake of palatable, high-calorie food. Physiol. Behav. 2016, 163, 88–96. [Google Scholar] [CrossRef]
- Sun, Y.; Butte, N.F.; Garcia, J.M.; Smith, R.G. Characterization of adult ghrelin and ghrelin receptor knockout mice under positive and negative energy balance. Endocrinology 2008, 149, 843–850. [Google Scholar] [CrossRef]
- Yuan, F.; Ma, J.; Xiang, X.; Lan, H.; Xu, Y.; Zhao, J.; Li, Y.; Zhang, W. Improvement of adipose macrophage polarization in high fat diet-induced obese ghsr knockout mice. BioMed Res. Int. 2018, 2018, 4924325. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Lee, J.H.; Buras, E.D.; Yu, K.; Wang, R.; Smith, C.W.; Wu, H.; Sheikh-Hamad, D.; Sun, Y. Ghrelin receptor regulates adipose tissue inflammation in aging. Aging 2016, 8, 178. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Fang, C.; Li, X.; Wu, C.S.; Noh, J.Y.; Ye, X.; Chapkin, R.S.; Sun, K.; Sun, Y. Ghs-r suppression in adipose tissues protects against obesity and insulin resistance by regulating adipose angiogenesis and fibrosis. Int. J. Obes. 2021, 45, 1565–1575. [Google Scholar] [CrossRef]
- Kim, D.M.; Lee, J.H.; Pan, Q.; Han, H.W.; Shen, Z.; Eshghjoo, S.; Wu, C.-S.; Yang, W.; Noh, J.Y.; Threadgill, D.W.; et al. Nutrient-sensing growth hormone secretagogue receptor in macrophage programming and meta-inflammation. Mol. Metab. 2024, 79, 101852. [Google Scholar] [CrossRef]
- Lin, L.; Saha, P.K.; Ma, X.; Henshaw, I.O.; Shao, L.; Chang, B.H.J.; Buras, E.D.; Tong, Q.; Chan, L.; McGuinness, O.P.; et al. Ablation of ghrelin receptor reduces adiposity and improves insulin sensitivity during aging by regulating fat metabolism in white and brown adipose tissues. Aging Cell 2011, 10, 996–1010. [Google Scholar] [CrossRef]
- Ma, X.; Lin, L.; Qin, G.; Lu, X.; Fiorotto, M.; Dixit, V.D.; Sun, Y. Ablations of ghrelin and ghrelin receptor exhibit differential metabolic phenotypes and thermogenic capacity during aging. PLoS ONE 2011, 6, e16391. [Google Scholar] [CrossRef]
- Lin, L.; Lee, J.H.; Bongmba, O.Y.N.; Ma, X.; Zhu, X.; Sheikh-Hamad, D.; Sun, Y. The suppression of ghrelin signaling mitigates age-associated thermogenic impairment. Aging 2014, 6, 1019. [Google Scholar] [CrossRef]
- Villarreal, D.; Pradhan, G.; Wu, C.-S.; Allred, C.D.; Guo, S.; Sun, Y. A simple high efficiency protocol for pancreatic islet isolation from mice. JoVE (J. Vis. Exp.) 2019, e57048. [Google Scholar] [CrossRef]
- Maggio, M.; Guralnik, J.M.; Longo, D.L.; Ferrucci, L. Interleukin-6 in aging and chronic disease: A magnificent pathway. J. Gerontol. Ser. A 2006, 61, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Morley, J.E.; Baumgartner, R.N. Cytokine-related aging process. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2004, 59, M924–M929. [Google Scholar] [CrossRef]
- Berger, C.; Zdzieblo, D. Glucose transporters in pancreatic islets. Pflügers Arch.-Eur. J. Physiol. 2020, 472, 1249–1272. [Google Scholar] [CrossRef]
- Ying, W.; Fu, W.; Lee, Y.S.; Olefsky, J.M. The role of macrophages in obesity-associated islet inflammation and β-cell abnormalities. Nat. Rev. Endocrinol. 2020, 16, 81–90. [Google Scholar] [CrossRef]
- Amitani, M.; Amitani, H.; Cheng, K.C.; Kairupan, T.S.; Sameshima, N.; Shimoshikiryo, I.; Mizuma, K.; Rokot, N.T.; Nerome, Y.; Owaki, T.; et al. The role of ghrelin and ghrelin signaling in aging. Int. J. Mol. Sci. 2017, 18, 1511. [Google Scholar] [CrossRef]
- Deschaine, S.L.; Leggio, L. From “hunger hormone” to “it’s complicated”: Ghrelin beyond feeding control. Physiology 2022, 37, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Yada, T.; Damdindorj, B.; Rita, R.S.; Kurashina, T.; Ando, A.; Taguchi, M.; Koizumi, M.; Sone, H.; Nakata, M.; Kakei, M. Ghrelin signalling in β-cells regulates insulin secretion and blood glucose. Diabetes Obes. Metab. 2014, 16, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Alamri, B.N.; Shin, K.; Chappe, V.; Anini, Y. The role of ghrelin in the regulation of glucose homeostasis. Horm. Mol. Biol. Clin. Investig. 2016, 26, 3–11. [Google Scholar] [CrossRef]
- Sun, Y.; Asnicar, M.; Saha, P.K.; Chan, L.; Smith, R.G. Ablation of ghrelin improves the diabetic but not obese phenotype of ob/ob mice. Cell Metab. 2006, 3, 379–386. [Google Scholar] [CrossRef]
- Poher, A.-L.; Tschöp, M.H.; Müller, T.D. Ghrelin regulation of glucose metabolism. Peptides 2018, 100, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Chabot, F.; Caron, A.; Laplante, M.; St-Pierre, D.H. Interrelationships between ghrelin, insulin and glucose homeostasis: Physiological relevance. World J. Diabetes 2014, 5, 328. [Google Scholar] [CrossRef]
- Kurashina, T.; Dezaki, K.; Yoshida, M.; Rita, R.S.; Ito, K.; Taguchi, M.; Miura, R.; Tominaga, M.; Ishibashi, S.; Kakei, M.; et al. The β-cell ghsr and downstream camp/trpm2 signaling account for insulinostatic and glycemic effects of ghrelin. Sci. Rep. 2015, 5, 14041. [Google Scholar] [CrossRef] [PubMed]
- Dezaki, K.; Sone, H.; Koizumi, M.; Nakata, M.; Kakei, M.; Nagai, H.; Hosoda, H.; Kangawa, K.; Yada, T. Blockade of pancreatic islet–derived ghrelin enhances insulin secretion to prevent high-fat diet–induced glucose intolerance. Diabetes 2006, 55, 3486–3493. [Google Scholar] [CrossRef]
- Zhao, T.-J.; Liang, G.; Li, R.L.; Xie, X.; Sleeman, M.W.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Goldstein, J.L.; Brown, M.S. Ghrelin o-acyltransferase (goat) is essential for growth hormone-mediated survival of calorie-restricted mice. Proc. Natl. Acad. Sci. USA 2010, 107, 7467–7472. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Burstein, A.W.; Schwalbe, D.C.; Shankar, K.; Varshney, S.; Singh, O.; Paul, S.; Ogden, S.B.; Osborne-Lawrence, S.; Metzger, N.P.; et al. Ghrelin deletion and conditional ghrelin cell ablation increase pancreatic islet size in mice. J. Clin. Investig. 2023, 133, e169349. [Google Scholar] [CrossRef]
- Zhao, X.; An, X.; Yang, C.; Sun, W.; Ji, H.; Lian, F. The crucial role and mechanism of insulin resistance in metabolic disease. Front. Endocrinol. 2023, 14, 1149239. [Google Scholar] [CrossRef]
- Boeni-Schnetzler, M.; Mereau, H.; Rachid, L.; Wiedemann, S.J.; Schulze, F.; Trimigliozzi, K.; Meier, D.T.; Donath, M.Y. Il-1beta promotes the age-associated decline of beta cell function. Iscience 2021, 24, 103250. [Google Scholar] [CrossRef]
- Shrestha, S.; Erikson, G.; Lyon, J.; Spigelman, A.F.; Bautista, A.; Fox, J.E.M.; dos Santos, C.; Shokhirev, M.; Cartailler, J.-P.; Hetzer, M.W.; et al. Aging compromises human islet beta cell function and identity by decreasing transcription factor activity and inducing er stress. Sci. Adv. 2022, 8, eabo3932. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.S.; Seo, E.-H.; Lee, Y.-S.; Cho, S.C.; Jung, H.S.; Park, S.C.; Jun, H.-S. Increase of calcium sensing receptor expression is related to compensatory insulin secretion during aging in mice. PLoS ONE 2016, 11, e0159689. [Google Scholar] [CrossRef]
- Gregg, T.; Poudel, C.; Schmidt, B.A.; Dhillon, R.S.; Sdao, S.M.; Truchan, N.A.; Baar, E.L.; Fernandez, L.A.; Denu, J.M.; Eliceiri, K.W.; et al. Pancreatic β-cells from mice offset age-associated mitochondrial deficiency with reduced katp channel activity. Diabetes 2016, 65, 2700–2710. [Google Scholar] [CrossRef] [PubMed]
- Avrahami, D.; Li, C.; Zhang, J.; Schug, J.; Avrahami, R.; Rao, S.; Stadler, M.B.; Burger, L.; Schübeler, D.; Glaser, B.; et al. Aging-dependent demethylation of regulatory elements correlates with chromatin state and improved β cell function. Cell Metab. 2015, 22, 619–632. [Google Scholar] [CrossRef] [PubMed]
- De Leon, E.R.; Brinkman, J.A.; Fenske, R.J.; Gregg, T.; Schmidt, B.A.; Sherman, D.S.; Cummings, N.E.; Peter, D.C.; Kimple, M.E.; Lamming, D.W.; et al. Age-dependent protection of insulin secretion in diet induced obese mice. Sci. Rep. 2018, 8, 17814. [Google Scholar] [CrossRef] [PubMed]
- Pacini, G.; Beccaro, F.; Valerio, A.; Nosadini, R.; Crepaldi, G. Reduced beta-cell secretion and insulin hepatic extraction in healthy elderly subjects. J. Am. Geriatr. Soc. 1990, 38, 1283–1289. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Guan, Y.; Yang, J. Cytokines in the progression of pancreatic β-cell dysfunction. Int. J. Endocrinol. 2010, 2010, 515136. [Google Scholar] [CrossRef] [PubMed]
- Citro, A.; Campo, F.; Dugnani, E.; Piemonti, L. Innate immunity mediated inflammation and beta cell function: Neighbors or enemies? Front. Endocrinol. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ren, A.; Hu, S.; Mo, W.; Xin, X.; Jia, W. The significance of tumor necrosis factor-α in newly diagnosed type 2 diabetic patients by transient intensive insulin treatment. Diabetes Res. Clin. Pract. 2007, 75, 327–332. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, C.M.; Lu, C.; Corbin, K.L.; Sharma, P.R.; Dula, S.B.; Carter, J.D.; Ramadan, J.W.; Xin, W.; Lee, J.K.; Nunemaker, C.S. Circulating levels of il-1b+ il-6 cause er stress and dysfunction in islets from prediabetic male mice. Endocrinology 2013, 154, 3077–3088. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Vitale, G.; Capri, M.; Salvioli, S. Inflammaging and ‘garb-aging’. Trends Endocrinol. Metab. 2017, 28, 199–212. [Google Scholar] [CrossRef]
- Frese, T.; Bazwinsky, I.; Mühlbauer, E.; Peschke, E. Circadian and age-dependent expression patterns of glut2 and glucokinase in the pancreatic β-cell of diabetic and nondiabetic rats. Horm. Metab. Res. 2007, 39, 567–574. [Google Scholar] [CrossRef]
- Baeeri, M.; Rahimifard, M.; Daghighi, S.M.; Khan, F.; Salami, S.A.; Moini-Nodeh, S.; Haghi-Aminjan, H.; Bayrami, Z.; Rezaee, F.; Abdollahi, M. Cannabinoids as anti-ros in aged pancreatic islet cells. Life Sci. 2020, 256, 117969. [Google Scholar] [CrossRef]
- Novosadová, Z.; Polidarová, L.; Sládek, M.; Sumová, A. Alteration in glucose homeostasis and persistence of the pancreatic clock in aged mper2 luc mice. Sci. Rep. 2018, 8, 11668. [Google Scholar] [CrossRef]
- Gorovits, N.; Charron, M.J. What we know about facilitative glucose transporters: Lessons from cultured cells, animal models, and human studies. Biochem. Mol. Biol. Educ. 2003, 31, 163–172. [Google Scholar] [CrossRef]
- Thorens, B.; Guillam, M.-T.; Beermann, F.; Burcelin, R.; Jaquet, M. Transgenic reexpression of glut1 or glut2 in pancreatic β cells rescues glut2-null mice from early death and restores normal glucose-stimulated insulin secretion. J. Biol. Chem. 2000, 275, 23751–23758. [Google Scholar] [CrossRef]
- Fernández-Díaz, C.M.; Merino, B.; López-Acosta, J.F.; Cidad, P.; De La Fuente, M.A.; Lobatón, C.D.; Moreno, A.; Leissring, M.A.; Perdomo, G.; Cózar-Castellano, I. Pancreatic β-cell-specific deletion of insulin-degrading enzyme leads to dysregulated insulin secretion and β-cell functional immaturity. Am. J. Physiol.-Endocrinol. Metab. 2019, 317, E805–E819. [Google Scholar] [CrossRef] [PubMed]
- Leibiger, I.B.; Leibiger, B.; Berggren, P.-O. Insulin signaling in the pancreatic β-cell. Annu. Rev. Nutr. 2008, 28, 233–251. [Google Scholar] [CrossRef]
- Furman, B.L. Streptozotocin-induced diabetic models in mice and rats. Curr. Protoc. 2021, 1, e78. [Google Scholar] [CrossRef]
- Like, A.A.; Rossini, A.A. Streptozotocin-induced pancreatic insulitis: New model of diabetes mellitus. Science 1976, 193, 415–417. [Google Scholar] [CrossRef] [PubMed]
- Lenzen, S. The mechanisms of alloxan-and streptozotocin-induced diabetes. Diabetologia 2008, 51, 216–226. [Google Scholar] [CrossRef]
- Hosokawa, M.; Dolci, W.; Thorens, B. Differential sensitivity of glut1-and glut2-expressing β cells to streptozotocin. Biochem. Biophys. Res. Commun. 2001, 289, 1114–1117. [Google Scholar] [CrossRef]
- Jamali-Raeufy, N.; Baluchnejadmojarad, T.; Roghani, M. Isorhamnetin exerts neuroprotective effects in stz-induced diabetic rats via attenuation of oxidative stress, inflammation and apoptosis. J. Chem. Neuroanat. 2019, 102, 101709. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wu, R.; Kong, Y.; Yang, Y.; Gao, Y.; Sun, D.; Liu, Q.; Dai, D.; Lu, Z.; Wang, N.; et al. Tanshinone ii aattenuates renal damage in stz-induced diabetic rats via inhibiting oxidative stress and inflammation. Oncotarget 2017, 8, 31915. [Google Scholar] [CrossRef] [PubMed]
- Rad, M.G.; Sharifi, M.; Meamar, R.; Soltani, N. The role of pancreas to improve hyperglycemia in stz-induced diabetic rats by thiamine disulfide. Nutr. Diabetes 2022, 12, 32. [Google Scholar] [CrossRef] [PubMed]
- Riley, W.J.; McConnell, T.J.; McLaughlin, J.V.; Taylor, G. The diabetogenic effects of streptozotocin in mice are prolonged and inversely related to age. Diabetes 1981, 30, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Fève, B.; Bastard, J.-P. The role of interleukins in insulin resistance and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2009, 5, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Rehman, K.; Akash, M.S.H. Mechanisms of inflammatory responses and development of insulin resistance: How are they interlinked? J. Biomed. Sci. 2016, 23, 87. [Google Scholar] [CrossRef]
- Lindqvist, A.; Shcherbina, L.; Prasad, R.; Miskelly, M.; Abels, M.; Martínez-Lopéz, J.; Fred, R.; Nergård, B.; Hedenbro, J.; Groop, L.; et al. Ghrelin suppresses insulin secretion in human islets and type 2 diabetes patients have diminished islet ghrelin cell number and lower plasma ghrelin levels. Mol. Cell. Endocrinol. 2020, 511, 110835. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, H.W.; Pradhan, G.; Villarreal, D.; Kim, D.M.; Jain, A.; Gaharwar, A.; Tian, Y.; Guo, S.; Sun, Y. GHSR Deletion in β-Cells of Male Mice: Ineffective in Obesity, but Effective in Protecting against Streptozotocin-Induced β-Cell Injury in Aging. Nutrients 2024, 16, 1464. https://doi.org/10.3390/nu16101464
Han HW, Pradhan G, Villarreal D, Kim DM, Jain A, Gaharwar A, Tian Y, Guo S, Sun Y. GHSR Deletion in β-Cells of Male Mice: Ineffective in Obesity, but Effective in Protecting against Streptozotocin-Induced β-Cell Injury in Aging. Nutrients. 2024; 16(10):1464. https://doi.org/10.3390/nu16101464
Chicago/Turabian StyleHan, Hye Won, Geetali Pradhan, Daniel Villarreal, Da Mi Kim, Abhishek Jain, Akhilesh Gaharwar, Yanan Tian, Shaodong Guo, and Yuxiang Sun. 2024. "GHSR Deletion in β-Cells of Male Mice: Ineffective in Obesity, but Effective in Protecting against Streptozotocin-Induced β-Cell Injury in Aging" Nutrients 16, no. 10: 1464. https://doi.org/10.3390/nu16101464
APA StyleHan, H. W., Pradhan, G., Villarreal, D., Kim, D. M., Jain, A., Gaharwar, A., Tian, Y., Guo, S., & Sun, Y. (2024). GHSR Deletion in β-Cells of Male Mice: Ineffective in Obesity, but Effective in Protecting against Streptozotocin-Induced β-Cell Injury in Aging. Nutrients, 16(10), 1464. https://doi.org/10.3390/nu16101464