
Arginine Dysregulation and Myocardial Dysfunction in a Mouse Model and Children with Chronic Kidney Disease

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Murine Experiments:
2.1.1. Mouse Model of CKD
2.1.2. Murine Echocardiography
2.1.3. Murine Non-Invasive Blood Pressure Monitoring
2.2. Human Experiments
2.2.1. Pediatric CKD Subjects
2.2.2. Pediatric Healthy Control Subjects
2.2.3. Pediatric Echocardiography
2.3. Tissue and Plasma Analyses
2.3.1. Measurement of NO Metabolites (NOx), Amino Acids, and ADMA in Plasma
2.3.2. Measurement of Amino Acids, eNOS and ADMA in Myocardial Tissue
2.3.3. Measurement of Arginase Concentration and Activity
2.3.4. Statistical Analyses
3. Results
3.1. Mice with CKD Demonstrate Altered Amino Acid Metabolism Compared to Healthy Controls
3.2. Myocardial Dysfunction Correlates with Dysregulated Amino Acid Metabolism in Mice with and without CKD
3.3. Plasma Arginase Activity Increases in Mice with Longer Duration of CKD
3.4. Arginase Inhibition Improves Global Longitudinal Strain in Mice with CKD
3.5. Amino Acid Metabolism and Arginase Activity Are Dysregulated in Children with CKD/ESRD
3.6. ADMA Accumulates in Mice and Children with CKD/ESRD
3.7. Total Nitrate/Nitrite Accumulates in Children with CKD
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parekh, R.S.; Carroll, C.E.; Wolfe, R.A.; Port, F.K. Cardiovascular mortality in children and young adults with end-stage kidney disease. J. Pediatr. 2002, 141, 191–197. [Google Scholar] [CrossRef] [PubMed]
- McDonald, S.P.; Craig, J.C. Long-term survival of children with end-stage renal disease. N. Engl. J. Med. 2004, 350, 2654–2662. [Google Scholar] [CrossRef] [PubMed]
- Mitsnefes, M.M. Cardiovascular disease in children with chronic kidney disease. J. Am. Soc. Nephrol. JASN 2012, 23, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Mitsnefes, M.M.; Kimball, T.R.; Border, W.L.; Witt, S.A.; Glascock, B.J.; Khoury, P.R.; Daniels, S.R. Impaired left ventricular diastolic function in children with chronic renal failure. Kidney Int. 2004, 65, 1461–1466. [Google Scholar] [CrossRef] [PubMed]
- Chinali, M.; Matteucci, M.C.; Franceschini, A.; Doyon, A.; Pongiglione, G.; Rinelli, G.; Schaefer, F. Advanced Parameters of Cardiac Mechanics in Children with CKD: The 4C Study. Clin. J. Am. Soc. Nephrol. CJASN 2015, 10, 1357–1363. [Google Scholar] [CrossRef]
- Malatesta-Muncher, R.; Wansapura, J.; Taylor, M.; Lindquist, D.; Hor, K.; Mitsnefes, M. Early cardiac dysfunction in pediatric patients on maintenance dialysis and post kidney transplant. Pediatr. Nephrol. 2012, 27, 1157–1164. [Google Scholar] [CrossRef]
- Liu, Y.W.; Su, C.T.; Sung, J.M.; Wang, S.P.; Su, Y.R.; Yang, C.S.; Tsai, L.M.; Chen, J.H.; Tsai, W.C. Association of left ventricular longitudinal strain with mortality among stable hemodialysis patients with preserved left ventricular ejection fraction. Clin. J. Am. Soc. Nephrol. CJASN 2013, 8, 1564–1574. [Google Scholar] [CrossRef]
- Krishnasamy, R.; Isbel, N.M.; Hawley, C.M.; Pascoe, E.M.; Leano, R.; Haluska, B.A.; Stanton, T. The association between left ventricular global longitudinal strain, renal impairment and all-cause mortality. Nephrol. Dial. Transpl. 2014, 29, 1218–1225. [Google Scholar] [CrossRef]
- Poulikakos, D.; Ross, L.; Recio-Mayoral, A.; Cole, D.; Andoh, J.; Chitalia, N.; Sharma, R.; Kaski, J.C.; Banerjee, D. Left ventricular hypertrophy and endothelial dysfunction in chronic kidney disease. Eur. Heart J. Cardiovasc. Imaging 2014, 15, 56–61. [Google Scholar] [CrossRef]
- Malyszko, J. Mechanism of endothelial dysfunction in chronic kidney disease. Clin. Chim. Acta Int. J. Clin. Chem. 2010, 411, 1412–1420. [Google Scholar] [CrossRef]
- Peng, T.; Hu, Z.; Wu, L.; Li, D.; Yang, X. Correlation between endothelial dysfunction and left ventricular remodeling in patients with chronic kidney disease. Kidney Blood Press. Res. 2014, 39, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Wever, R.; Boer, P.; Hijmering, M.; Stroes, E.; Verhaar, M.; Kastelein, J.; Versluis, K.; Lagerwerf, F.; van Rijn, H.; Koomans, H.; et al. Nitric oxide production is reduced in patients with chronic renal failure. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1168–1172. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D. Effect of chronic renal failure on nitric oxide metabolism. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2001, 38 (Suppl. S1), S74–S79. [Google Scholar] [CrossRef] [PubMed]
- Baylis, C. Arginine, arginine analogs and nitric oxide production in chronic kidney disease. Nat. Clin. Pract. Nephrol. 2006, 2, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Reddy, Y.S.; Kiranmayi, V.S.; Bitla, A.R.; Krishna, G.S.; Rao, P.V.; Sivakumar, V. Nitric oxide status in patients with chronic kidney disease. Indian J. Nephrol. 2015, 25, 287–2891. [Google Scholar] [CrossRef] [PubMed]
- Shukla, V.; Dey, R.; Chandra, A.; Karoli, R.; Khanduri, S. Endothelial Dysfunction by Flow-Mediated Vasodilatation in Chronic Kidney Disease. J. Assoc. Physicians India 2015, 63, 30–33. [Google Scholar]
- Endemann, D.H.; Touyz, R.M.; Iglarz, M.; Savoia, C.; Schiffrin, E.L. Eplerenone prevents salt-induced vascular remodeling and cardiac fibrosis in stroke-prone spontaneously hypertensive rats. Hypertension 2004, 43, 1252–1257. [Google Scholar] [CrossRef]
- Stam, F.; van Guldener, C.; Becker, A.; Dekker, J.M.; Heine, R.J.; Bouter, L.M.; Stegouwer, C.D.A. Endothelial dysfunction contributes to renal function-associated cardiovascular mortality in a population with mild renal insufficiency: The Hoorn study. J. Am. Soc. Nephrol. JASN 2006, 17, 537–545. [Google Scholar] [CrossRef]
- Wagner, L.; Klein, J.D.; Sands, J.M.; Baylis, C. Urea transporters are distributed in endothelial cells and mediate inhibition of L-arginine transport. Am. J. Physiol. Ren. Physiol. 2002, 283, F578–F582. [Google Scholar] [CrossRef]
- Xiao, S.; Wagner, L.; Mahaney, J.; Baylis, C. Uremic levels of urea inhibit L-arginine transport in cultured endothelial cells. Am. J. Physiol. Ren. Physiol. 2011, 280, F989–F995. [Google Scholar] [CrossRef]
- Brosnan, M.E.; Brosnan, J.T. Renal arginine metabolism. J. Nutr. 2004, 134, 2791S–2795S; discussion 2796S–2797S. [Google Scholar] [CrossRef] [PubMed]
- Pernow, J.; Jung, C. Arginase as a potential target in the treatment of cardiovascular disease: Reversal of arginine steal? Cardiovasc. Res. 2013, 98, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Quitter, F.; Figulla, H.R.; Ferrari, M.; Pernow, J.; Jung, C. Increased arginase levels in heart failure represent a therapeutic target to rescue microvascular perfusion. Clin. Hemorheol. Microcirc. 2013, 54, 75–85. [Google Scholar] [CrossRef]
- Pernow, J.; Jung, C. The Emerging Role of Arginase in Endothelial Dysfunction in Diabetes. Curr. Vasc. Pharmacol. 2016, 14, 155–162. [Google Scholar] [CrossRef]
- Morris, C.R.; Kato, G.J.; Poljakovic, M.; Wang, X.; Blackwelder, W.C.; Sachdev, V.; Hazen, S.L.; Vichinsky, E.P.; Morris, S.M., Jr.; Gladwin, M.T. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA 2005, 294, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Shemyakin, A.; Kovamees, O.; Rafnsson, A.; Bohm, F.; Svenarud, P.; Settergren, M.; Jung, C.; Pernow, J. Arginase inhibition improves endothelial function in patients with coronary artery disease and type 2 diabetes mellitus. Circulation 2012, 126, 2943–2950. [Google Scholar] [CrossRef] [PubMed]
- Kövamees, O.; Shemyakin, A.; Checa, A.; Wheelcok, C.E.; Lundberg, J.O.; Ostenson, C.G.; Pernow, J. Arginase Inhibition Improves Microvascular Endothelial Function in Patients With Type 2 Diabetes Mellitus. J. Clin. Endocrinol. Metab. 2016, 101, 3952–3958. [Google Scholar] [CrossRef]
- Boger, R.H.; Zoccali, C. ADMA: A novel risk factor that explains excess cardiovascular event rate in patients with end-stage renal disease. Atheroscler. Suppl. 2003, 4, 23–28. [Google Scholar] [CrossRef]
- Tizianello, A.; De Ferrari, G.; Garibotto, G.; Gurreri, G.; Robaudo, C. Renal metabolism of amino acids and ammonia in subjects with normal renal function and in patients with chronic renal insufficiency. J. Clin. Investig. 1980, 65, 1162–1173. [Google Scholar] [CrossRef]
- Bouby, N.; Hassler, C.; Parvy, P.; Bankir, L. Renal synthesis of arginine in chronic renal failure: In vivo and in vitro studies in rats with 5/6 nephrectomy. Kidney Int. 1993, 44, 676–683. [Google Scholar] [CrossRef]
- Chen, G.F.; Baylis, C. In vivo renal arginine release is impaired throughout development of chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2010, 298, F95–F102. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Wang, Z.; Cho, L.; Brennan, D.M.; Hazen, S.L. Diminished global arginine bioavailability and increased arginine catabolism as metabolic profile of increased cardiovascular risk. J. Am. Coll. Cardiol. 2009, 53, 2061–2067. [Google Scholar] [CrossRef] [PubMed]
- Onalo, R.; Cooper, P.; Cilliers, A.; Vorster, B.C.; Uche, N.A.; Oluseyi, O.O.; Onalo, V.D.; Zubairu, Y.; Ayodele-Kehinde, A.U.; Damilare, O.M.; et al. Randomized control trial of oral arginine therapy for children with sickle cell anemia hospitalized for pain in Nigeria. Am. J. Hematol. 2021, 96, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Onalo, R.; Cilliers, A.; Cooper, P.; Morris, C.R. Arginine Therapy and Cardiopulmonary Hemodynamics in Hospitalized Children with Sickle Cell Anemia: A Prospective, Double-blinded, Randomized Placebo-controlled Clinical Trial. Am. J. Respir. Crit. Care Med. 2022, 206, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.R.; Kim, H.Y.; Klings, E.S.; Wood, J.; Porter, J.B.; Trachtenberg, F.; Sweeters, N.; Olivieri, N.F.; Kwiatkowski, J.L.; Virzi, L.; et al. Dysregulated arginine metabolism and cardiopulmonary dysfunction in patients with thalassaemia. Br. J. Haematol. 2015, 169, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Cox, S.E.; Makani, J.; Komba, A.N.; Soka, D.; Newton, C.R.; Kirkham, F.J.; Prentice, A.M. Global arginine bioavailability in Tanzanian sickle cell anaemia patients at steady-state: A nested case control study of deaths versus survivors. Br. J. Haematol. 2011, 155, 522–524. [Google Scholar] [CrossRef] [PubMed]
- Sourij, H.; Meinitzer, A.; Pilz, S.; Grammer, T.B.; Winkelmann, B.R.; Boehm, B.O.; Marz, W. Arginine bioavailability ratios are associated with cardiovascular mortality in patients referred to coronary angiography. Atherosclerosis 2011, 218, 220–225. [Google Scholar] [CrossRef]
- Ishinoda, Y.; Masaki, N.; Hitomi, Y.; Taruoka, A.; Kawai, A.; Iwashita, M.; Yumita, Y.; Kagami, K.; Yasuda, R.; Ido, Y.; et al. A Low Arginine/Ornithine Ratio is Associated with Long-Term Cardiovascular Mortality. J. Atheroscler. Thromb. 2023, 63779, Online ahead of print. [Google Scholar] [CrossRef]
- Winterberg, P.D.; Jiang, R.; Maxwell, J.T.; Wang, B.; Wagner, M.B. Myocardial dysfunction occurs prior to changes in ventricular geometry in mice with chronic kidney disease (CKD). Physiol. Rep. 2016, 4, e12732. [Google Scholar] [CrossRef]
- Stockelman, M.G.; Lorenz, J.N.; Smith, F.N.; Boivin, G.P.; Sahota, A.; Tischfield, J.A.; Stambrook, P.J. Chronic renal failure in a mouse model of human adenine phosphoribosyltransferase deficiency. Am. J. Physiol. 1998, 275 Pt 2, F154–F163. [Google Scholar] [CrossRef]
- Baylis, C. Sexual dimorphism in the aging kidney: Differences in the nitric oxide system. Nat. Rev. Nephrol. 2009, 5, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Meyer, M.; Turk, T.R.; Wilde, B.; Feldkamp, T.; Assert, R.; Wu, K.; Kribben, A.; Witzke, O. Serum cystatin C in mouse models: A reliable and precise marker for renal function and superior to serum creatinine. Nephrol. Dial. Transpl. 2009, 24, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- George, R.P.; Mehta, A.K.; Perez, S.D.; Winterberg, P.D.; Cheeseman, J.; Johnson, B.; Kwun, J.; Monday, S.; Stempora, L.; Warshaw, B.; et al. Premature T Cell Senescence in Pediatric CKD. J. Am. Soc. Nephrol. JASN 2017, 28, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Devereux, R.B.; Alonsa, D.R.; Lutas, E.M.; Gottlieb, G.J.; Campo, E.; Sachs, I.; Reichek, N. Echocardiographic assessment of left ventricular hypertrophy: Comparison to necropsy findings. Am. J. Cardiol. 1986, 57, 450–458. [Google Scholar] [CrossRef]
- De Simone, G.; Daniels, S.R.; Devereux, R.B.; Meyer, R.A.; Roman, M.J.; de Divitiis, O.; Alderman, M.H. Left ventricular mass and body size in normotensive children and adults: Assessment of allometric relations and impact of overweight. J. Am. Coll. Cardiol. 1992, 20, 1251–1260. [Google Scholar] [CrossRef]
- Khoury, P.R.; Mitsnefes, M.; Daniels, S.R.; Kimball, T.R. Age-specific reference intervals for indexed left ventricular mass in children. J. Am. Soc. Echocardiogr. 2009, 22, 709–714. [Google Scholar] [CrossRef]
- Morris, C.R.; Kuypers, F.A.; Larkin, S.; Sweeters, N.; Simon, J.; Vichinsky, E.P.; Styles, A. Arginine therapy: A novel strategy to induce nitric oxide production in sickle cell disease. Br. J. Haematol. 2000, 111, 498–500. [Google Scholar] [CrossRef]
- Morris, C.R.; Kuypers, F.A.; Larkin, S.; Vichinsky, E.P.; Styles, L.A. Patterns of arginine and nitric oxide in patients with sickle cell disease with vaso-occlusive crisis and acute chest syndrome. J. Pediatr. Hematol. Oncol. 2000, 22, 515–520. [Google Scholar] [CrossRef]
- Hsu, C.N.; Lu, P.C.; Lo, M.H.; Lin, I.C.; Tain, Y.L. The Association between Nitric Oxide Pathway, Blood Pressure Abnormalities, and Cardiovascular Risk Profile in Pediatric Chronic Kidney Disease. Int. J. Mol. Sci. 2019, 20, 5301. [Google Scholar] [CrossRef]
- Liu, X.; Xu, X.; Shang, R.; Chen, Y. Asymmetric dimethylarginine (ADMA) as an important risk factor for the increased cardiovascular diseases and heart failure in chronic kidney disease. Nitric Oxide 2018, 78, 113–120. [Google Scholar] [CrossRef]
- Holowatz, L.A.; Kenney, W.L. Up-regulation of arginase activity contributes to attenuated reflex cutaneous vasodilatation in hypertensive humans. J. Physiol. 2007, 581 Pt 2, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, D.E.; White, R.; Li, D.; Minhas, K.M.; Cernetich, A.; Kim, S.; Burke, S.; Shoukas, A.A.; Nyhan, D.; Champion, H.C.; et al. Arginase reciprocally regulates nitric oxide synthase activity and contributes to endothelial dysfunction in aging blood vessels. Circulation 2003, 108, 2000–2006. [Google Scholar] [CrossRef] [PubMed]
- Durante, W.; Johnson, F.K.; Johnson, R.A. Arginase: A critical regulator of nitric oxide synthesis and vascular function. Clin. Exp. Pharmacol. Physiol. 2007, 34, 906–911. [Google Scholar] [CrossRef]
- Demougeot, C.; Prigent-Tessier, A.; Marie, C.; Berthelot, A. Arginase inhibition reduces endothelial dysfunction and blood pressure rising in spontaneously hypertensive rats. J. Hypertens. 2005, 23, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Steppan, J.; Tran, H.T.; Bead, V.R.; Oh, Y.J.; Sikka, G.; Bivalacqua, T.J.; Burnett, A.L.; Berkowitz, D.E.; Santhanam, L. Arginase Inhibition Reverses Endothelial Dysfunction, Pulmonary Hypertension, and Vascular Stiffness in Transgenic Sickle Cell Mice. Anesth. Analg. 2016, 123, 652–658. [Google Scholar] [CrossRef]
- Prati, C.; Berthelot, A.; Kantelip, B.; Wendling, D.; Demougeot, C. Treatment with the arginase inhibitor Nw-hydroxy-nor-L-arginine restores endothelial function in rat adjuvant-induced arthritis. Arthritis Res. Ther. 2012, 14, R130. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekharan, U.M.; Wang, Z.; Wu, Y.; Tang, W.H.W.; Hazen, S.L.; Wang, S.; Husni, M.E. Elevated levels of plasma symmetric dimethylarginine and increased arginase activity as potential indicators of cardiovascular comorbidity in rheumatoid arthritis. Arthritis Res. Ther. 2018, 20, 123. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.R.; Morris Jr, S.M.; Hagar, W.; Van Warmerdam, J.; Claster, S.; Kepka-Lenhart, D.; Machado, L.; Kuypers, F.A.; Vichinsky, E.P. Arginine therapy: A new treatment for pulmonary hypertension in sickle cell disease? Am. J. Respir. Crit. Care Med. 2003, 168, 63–69. [Google Scholar] [CrossRef]
- Rees, C.A.; Rostad, C.A.; Mantus, G.; Anderson, E.J.; Chahroudi, A.; Jaggi, P.; Wrammert, P.; Ochoa, J.B.; Ochoa, A.; Basu, R.K.; et al. Altered amino acid profile in patients with SARS-CoV-2 infection. Proc. Natl. Acad. Sci. USA 2021, 118, e2101708118. [Google Scholar] [CrossRef]
- Dean, M.J.; Ochoa, J.B.; Sanchez-Pino, M.D.; Zabaleta, J.; Garai, J.; Del Valle, L.; Wyczechowska, D.; Baiamonte, L.B.; Philbrook, P.; Majumder, R.; et al. Severe COVID-19 Is Characterized by an Impaired Type I Interferon Response and Elevated Levels of Arginase Producing Granulocytic Myeloid Derived Suppressor Cells. Front. Immunol. 2021, 12, 695972. [Google Scholar] [CrossRef]
- Caldwell, R.W.; Rodriguez, P.C.; Toque, H.A.; Narayanan, S.P.; Caldwell, R.B. Arginase: A Multifaceted Enzyme Important in Health and Disease. Physiol. Rev. 2018, 98, 641–665. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Shrestha, K.; Wang, Z.; Troughton, R.W.; Klein, A.L.; Hazen, S.L. Diminished global arginine bioavailability as a metabolic defect in chronic systolic heart failure. J. Card. Fail. 2013, 19, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.F.; Moningka, N.C.; Sasser, J.M.; Zharikov, S.; Cunningham Jr, M.; Tain, Y.L.; Schwartz, I.F.; Baylis, C. Arginine and asymmetric dimethylarginine in puromycin aminonucleoside-induced chronic kidney disease in the rat. Am. J. Nephrol. 2012, 35, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Sabbatini, M.; Pisani, A.; Uccello, F.; Fuiano, G.; Alfieri, R.; Cesaro, A.; Cianciaruso, B.; Andreucci, V.E. Arginase inhibition slows the progression of renal failure in rats with renal ablation. Am. J. Physiol. Ren. Physiol. 2003, 284, F680–F687. [Google Scholar] [CrossRef]
- Martens, C.R.; Kuczmarski, J.M.; Lennon-Edwards, S.; Edwards, D.G. Impaired L-arginine uptake but not arginase contributes to endothelial dysfunction in rats with chronic kidney disease. J. Cardiovasc. Pharmacol. 2014, 63, 40–48. [Google Scholar] [CrossRef]
- Ashish, K.; Faisaluddin, M.; Bandyopadhyay, D.; Hajra, A.; Herzog, E. Prognostic value of global longitudinal strain in heart failure subjects: A recent prototype. Int. J. Cardiol. Heart Vasc. 2019, 22, 48–49. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ma, L.; Song, M.; Li, X.; He, F.; Wang, C.; Chen, M.; Zhou, J.; Mei, C. The role of the complement factor B-arginase-polyamine molecular axis in uremia-induced cardiac remodeling in mice. Eur. J. Immunol. 2020, 50, 220–233. [Google Scholar] [CrossRef]
- Morris, C.R.; Brown, L.A.S.; Reynolds, M.; Dampier, C.D.; Lane, P.A.; Watt, A.; Kumari, P.; Harris, F.; Manoranjithan, S.; Mendis, R.; et al. Impact of arginine therapy on mitochondrial function in children with sickle cell disease during vaso-occlusive pain. Blood 2020, 136, 1402–1406. [Google Scholar] [CrossRef]
- Morris, C.R.; Kuypers, F.A.; Lavrisha, L.; Ansari, M.; Sweeters, N.; Stewart, M.; Gildengorin, G.; Naumayr, L.; Vichinsky, E.P. A randomized, placebo-controlled trial of arginine therapy for the treatment of children with sickle cell disease hospitalized with vaso-occlusive pain episodes. Haematologica 2013, 98, 1375–1382. [Google Scholar] [CrossRef]
- Reyes, L.Z.; Figueroa, J.; Leake, D.; Khemani, K.; Kumari, P.; Bakshi, N.; Lane, P.A.; Dampier, C.; Morris, C.R. Safety of intravenous arginine therapy in children with sickle cell disease hospitalized for vaso-occlusive pain: A randomized placebo-controlled trial in progress. Am. J. Hematol. 2022, 97, e21–e24. [Google Scholar] [CrossRef]
- Zhou, S.; Zhu, Q.; Li, X.; Chen, C.; Liu, J.; Ye, Y.; Ruan, Y.; Hei, Z. Asymmetric dimethylarginine and all-cause mortality: A systematic review and meta-analysis. Sci. Rep. 2017, 7, 44692. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Yamagishi, S.I.; Matsumoto, Y.; Fukami, K.; Okuda, S. Asymmetric dimethylarginine (ADMA) is a novel emerging risk factor for cardiovascular disease and the development of renal injury in chronic kidney disease. Clin. Exp. Nephrol. 2007, 11, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, N.; Androulakis, E.; Papaioannou, S.; Antoniades, C.; Tousoulis, D. Homoarginine in the shadow of asymmetric dimethylarginine: From nitric oxide to cardiovascular disease. Amino Acids 2015, 47, 1741–1750. [Google Scholar] [CrossRef]
- Lu, T.M.; Ding, Y.A.; Lin, S.J.; Lee, W.S.; Tai, H.C. Plasma levels of asymmetrical dimethylarginine and adverse cardiovascular events after percutaneous coronary intervention. Eur. Heart J. 2003, 24, 1912–1919. [Google Scholar] [CrossRef] [PubMed]
- Valkonen, V.P.; Paiva, H.; Salonen, J.T.; Lakka, T.A.; Lehtimaki, T.; Laasko, J.; Laaksonen, R. Risk of acute coronary events and serum concentration of asymmetrical dimethylarginine. Lancet 2001, 358, 2127–2128. [Google Scholar] [CrossRef] [PubMed]
- Achan, V.; Broadhead, M.; Malaki, M.; Whitley, G.; Leiper, J.; MacAllister, R.; Vallance, P. Asymmetric dimethylarginine causes hypertension and cardiac dysfunction in humans and is actively metabolized by dimethylarginine dimethylaminohydrolase. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1455–1459. [Google Scholar] [CrossRef]
- Hu, X.; Xu, X.; Zhu, G.; Atzler, D.; Kimoto, M.; Chen, J.; Schwedhelm, E.; Luneburg, N.; Boger, R.; Zhang, P.; et al. Vascular endothelial-specific dimethylarginine dimethylaminohydrolase-1-deficient mice reveal that vascular endothelium plays an important role in removing asymmetric dimethylarginine. Circulation 2009, 120, 2222–2229. [Google Scholar] [CrossRef]





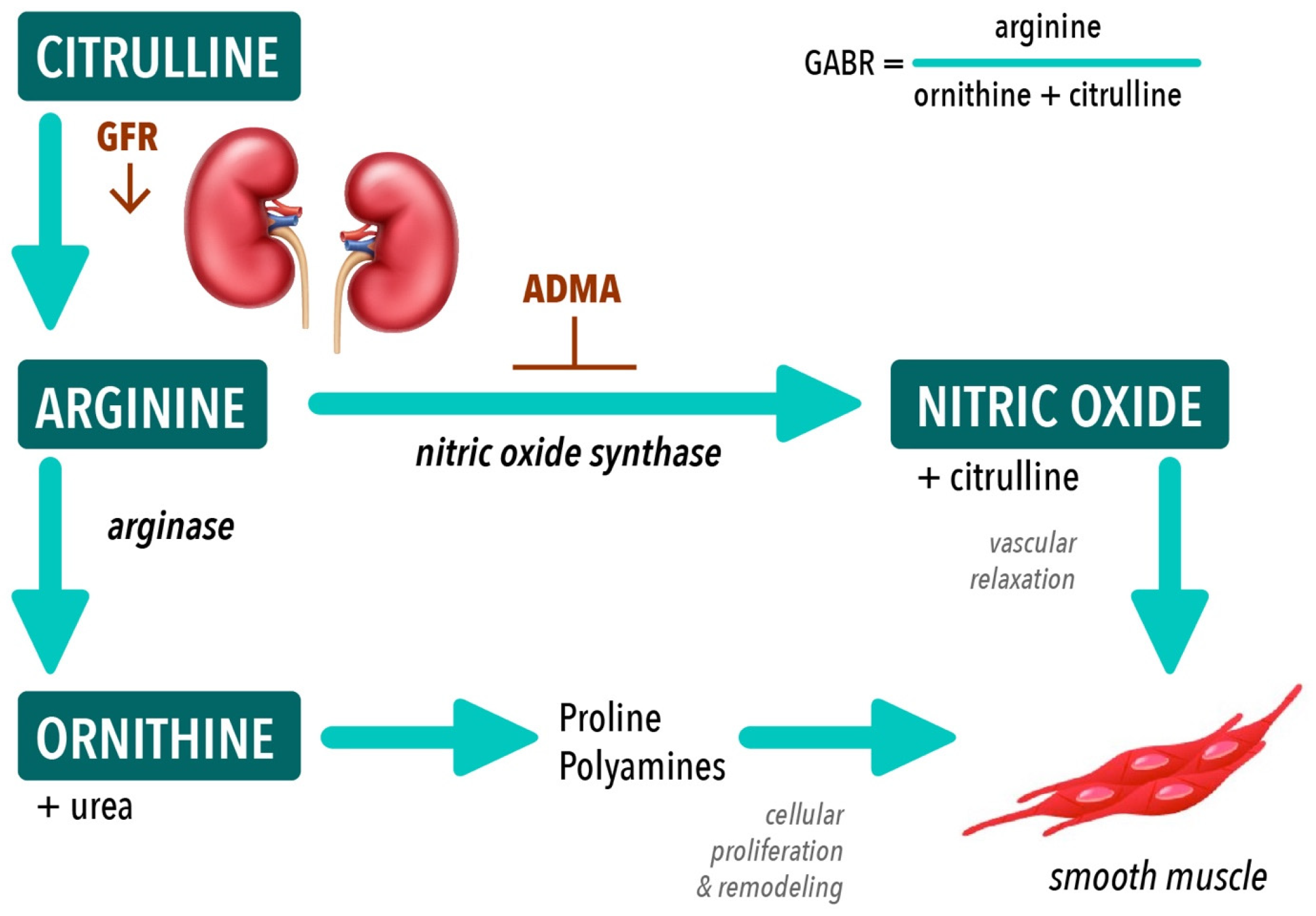{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Measure | 8 Weeks (n = 18) | 16 Weeks (n = 17) | ||||
|---|---|---|---|---|---|---|
| Control n = 9, 50.0% | CKD n = 9, 50.0% | p-Value | Control n = 7, 41.2% | CKD n = 10, 58.8% | p-Value | |
| Plasma BUN (mg/dL) | 53.7 (44.6–67.1) | 43.5 (25.0–58.0) | 0.224 | 26.7 (25.7–31.8) | 54.3 (47.4–66.6) | <0.001 |
| Plasma Cystatin C (ug/mL) | 0.293 (0.232–0.376) | 0.717 (0.677–0.772) | 0.002 | 0.327 (0.292–0.414) | 0.642 (0.582–0.865) | <0.001 |
| Plasma Arginine (µM/L) | 152.8 (125.8–158.9) | 128.0 (89.6–163.4) | 0.603 | 123.3 (54.3–134.7) | 102.1 (70.1–117.2) | 0.737 |
| Plasma Ornithine (µM/L) | 152.3 (106.7–157.6) | 85.9 (75.0–117.5) | 0.095 | 138.9 (112.3–235.5) | 229.4 (144.7–274) | 0.206 |
| Plasma Citrulline (µM/L) | 50.2 (37.2–53.2) | 87.6 (84.3–96.0) | <0.003 | 57.1 (50.8–73.1) | 106.0 (102.6–141.8) | 0.009 |
| GABR = Arg/(Orn+Cit) | 0.75 (0.62–0.87) | 0.61 (0.55–0.79) | 0.664 | 0.64 (0.22–0.70) | 0.30 (0.20–0.43) | 0.176 |
| Plasma Proline (µM/L) | 160.9 (129.3–166.9) | 152.3 (141.1–168.4) | 0.728 | 135.9 (102.8–161.5) | 127.0 (105.7–136.1) | 0.453 |
| Plasma Glutamate (µM/L) | 97.8 (84.6–168.2) | 49.0 (22.4–73.8) | 0.012 | 84.3 (68.9–151.2) | 78.5 (72.8–85.6) | 0.495 |
| Plasma Glutamine (µM/L) | 411.9 (397.7–475.2) | 322.3 (307.4–333.2) | <0.001 | 425.5 (319.5–440.1) | 359.1 (334.0–374.2) | 0.118 |
| Plasma Arginase Activity (U/L) | 7.3 (4.1–9.5) | 7.0 (3.7–7.6) | 0.438 | 5.5 (1.5–10.0) | 10.5 (8.4–11.7) | 0.052 |
| Arg/Orn ratio | 1.003 (0.641–2.343) | 1.56 (1.013–1.911) | 0.258 | 0.996 (0.269–1.168) | 0.53 (0.277–0.736) | 0.27 |
| Plasma ADMA (µM/L) | 7.135 (7.089–7.269) | 7.188 (7.094–7.236) | 0.081 | 7.092 (7.038–7.146) | 7.171 (7.091–7.285) | 0.036 |
| Myocardial Arginase I concentration (ng/mg protein) | 0.64 (0.49–0.71) | 0.14 (0.09–0.67) | 0.152 | 0.81 (0.18–1.10) | 0.45 (0.24–0.79) | 0.419 |
| Myocardial Arginase II concentration (ng/mg/protein) | 0.27 (0.25–0.31) | 0.31 (0.17–0.33) | 0.794 | 0.35 (0.29–0.40) | 0.29 (0.16–0.41) | 0.368 |
| Myocardial Arginase Activity (units/mg protein) | 16.47 (15.43–16.74) | 16.74 (15.41–17.27) | 0.827 | 14.39 (14.12–18.34) | 16.74 (15.43–17.54) | 0.222 |
| Myocardial eNOS concentration (ng/mg protein) | 3.22 (3.03–3.64) | 4.20 (2.78–4.52) | 0.728 | 3.47 (3.39–4.89) | 4.07 (2.90–5.97) | 1.000 |
| Measure | 8 Weeks (n = 18) | 16 Weeks (n = 17) | ||||
|---|---|---|---|---|---|---|
| Control n = 9, 50.0% | CKD n = 9, 50.0% | p-Value | Control n = 7, 41.2% | CKD n = 10, 58.8% | p-Value | |
| RWT | 0.69 (0.62–0.71) | 0.90 (0.81–1.11) | 0.020 | 0.89 (0.75–1.06) | 1.14 (1.05–1.26) | 0.036 |
| E/A ratio | 1.65 (1.49–1.85) | 1.20 (1.03–1.22) | 0.003 | 1.61 (1.27–1.85) | 0.96 (0.85–1.05) | 0.005 |
| GLS | 30.50 (29.50–32.50) | 25.10 (23.60–26.20) | 0.012 | 30.00 (28.96–33.70) | 23.85 (20.40–24.90) | 0.004 |
| Measures | CKD, n = 19 | Without CKD, n = 16 | ||||
|---|---|---|---|---|---|---|
| r | n | p-Value | r | n | p-Value | |
| Plasma Citrulline (µM/L) | ||||||
| RWT | 0.419 | 17 | 0.095 | 0.156 | 16 | 0.564 |
| E/A Ratio | −0.285 | 16 | 0.283 | 0.068 | 15 | 0.812 |
| GLS % | −0.058 | 17 | 0.826 | −0.076 | 16 | 0.780 |
| GABR | ||||||
| RWT | −0.495 | 19 | 0.033 | −0.134 | 14 | 0.649 |
| E/A Ratio | 0.577 | 18 | 0.014 | 0.225 | 13 | 0.459 |
| GLS % | 0.128 | 19 | 0.601 | 0.763 | 14 | 0.002 |
| Plasma Glutamine (µM/L) | ||||||
| RWT | −0.005 | 19 | 0.986 | −0.112 | 16 | 0.681 |
| E/A Ratio | −0.329 | 18 | 0.182 | 0.179 | 15 | 0.524 |
| GLS % | −0.218 | 19 | 0.371 | −0.132 | 16 | 0.625 |
| Plasma Arginase Activity (units/L) | ||||||
| RWT | 0.139 | 19 | 0.570 | −0.394 | 16 | 0.131 |
| E/A Ratio | −0.344 | 18 | 0.163 | −0.311 | 15 | 0.259 |
| GLS % | −0.129 | 19 | 0.599 | −0.459 | 16 | 0.074 |
| Measure | Normal n = 11 (19.0%) | CKD n = 15 (25.8%) | ESRD n = 32 (55.2%) | p-Value Nml vs. CKD | p-Value Nml vs. ESRD | p-Value CKD vs. ESRD |
|---|---|---|---|---|---|---|
| Demographics | ||||||
| Age at sample | 9.9 (4.5–12.6) | 11.9 (7.2–16.1) | 11.5 (5.0–14.8) | 0.476 | 0.797 | 0.746 |
| N (%) male | 5 (50.0%) | 12 (80.0%) | 22 (68.8%) | 0.194 | 0.451 | 0.503 |
| Race 1 | - | - | - | 0.515 | 0.791 | 0.056 |
| Black | 5 (50.0%) | 6 (40.0%) | 17 (53.1%) | - | - | - |
| White | 4 (40.0%) | 9 (60.0%) | 9 (28.1%) | - | - | - |
| Other | 1 (10.0%) | 0 (0.0%) | 6 (18.8%) | - | - | - |
| Ethnicity 2 | n/a | 0.399 | ||||
| Hispanic/Latino | n/a | 1 (9.1%) | 8 (27.6%) | - | - | - |
| Non-Hispanic/ Non-Latino | n/a | 10 (90.9%) | 21 (72.4%) | - | - | - |
| Clinical profile 3 | ||||||
| N (%) with SBP >95th %ile | n/a | 5 (62.5%) | 17 (65.4%) | n/a | n/a | 1.000 |
| N (%) with DBP >95th %ile | n/a | 4 (50.0%) | 14 (53.8%) | n/a | n/a | 1.000 |
| LVMI z-score | n/a | 1.38 (0.93–1.68) | 1.81 (−1.08–2.88) | n/a | n/a | 0.908 |
| Biochemical profile | ||||||
| Arginine (µM/L) | 18.3 (7.1–23.2) | 14.2 (9.2–20.7) | 20.9 (13.6–30.7) | 0.990 | 0.378 | 0.445 |
| Ornithine (µM/L) | 30.9 (25.2–37.9) | 38.5 (29.6–55.5) | 38.6 (29.5–51.4) | 0.191 | 0.349 | 0.989 |
| Citrulline (µM/L) | 6.5 (3.7–9.4) | 15.6 (11.5–19.3) | 29.1 (19.3–75.7) | 0.019 | <0.001 | 0.003 |
| GABR = Arg/(Orn + Cit) | 0.54 (0.28–0.63) | 0.23 (0.15–0.35) | 0.28 (0.17–0.48) | 0.338 | 0.134 | 0.951 |
| Proline (µM/L) | 47.4 (38.6–63.1) | 73.1 (62.5–97.1) | 124.8 (78.8–188.0) | 0.021 | <0.001 | 0.116 |
| Glutamate (µM/L) | 38.8 (33.6–55.2) | 42.1 (19.0–63.6) | 57.0 (36.6–93.9) | 0.9908 | 0.1937 | 0.2855 |
| Glutamine (µM/L) | 83.9 (69.4–110) | 98.3 (77.8–120.3) | 109.5 (81.8–144.5) | 0.6351 | 0.1665 | 0.9016 |
| Lysine (µM/L) | 161.1 (102.1–171) | 152.2 (97.7–195.2) | 149.1 (−10.9–190.4) | 0.9820 | 0.7917 | 0.9992 |
| Arginase concentration (ng/mL) | 18.1 (13.9–40.9) | 37.7 (20.4–46.2) | 11.5 (7.3–21.4) | 0.749 | 0.072 | 0.016 |
| Arginase activity (units/L) | 1.52 (1.23–2.15) | 3.65 (2.02–5.63) | 2.92 (2.04–5.34) | 0.047 | 0.040 | 0.799 |
| Arg/Orn ratio | 0.55 (0.17–0.79) | 0.32 (0.23–0.49) | 0.52 (0.37–0.86) | 0.819 | 0.840 | 0.111 |
| ADMA (µM/L) | 7.07 (7.05–7.07) | 7.5 (7.4–7.8) | 8.5 (7.6–8.7) | <0.001 | <0.001 | 0.004 |
| NOx (µM/L) | 92.9 (83.2–128.7) | 174.8 (145.1–257.9) | 163.9 (124.5–220.2) | <0.001 | <0.001 | 0.603 |
| Measure | Normal (n = 11) | HD (n = 20) | PD (n = 12) | p-Value (Nml vs. HD) | p-Value (Nml vs. PD) | p-Value (HD vs. PD) |
|---|---|---|---|---|---|---|
| Arginase concentration (ng/mL) | 18.1 (−13.9–40.9) | 9.4 (6.5–21.4) | 16.0 (11.2–25.3) | 0.040 | 0.444 | 0.220 |
| Arginase activity (units/L) | 1.52 (1.23–2.15) | 3.12 (2.10–5.34) | 2.28 (1.89–4.47) | 0.048 | 0.155 | 0.715 |
| Measures | LVMI z-Score < 2 (n = 24, 63.2%) | LVMI z-Score ≥ 2 (n = 14, 36.8%) | p-Value |
|---|---|---|---|
| Arginase concentration (ng/mL) | 15.8 (9.3–40.6) | 8.2 (6.4–17.4) | 0.084 |
| Arginase activity (units/L) | 2.57 (1.71–3.57) | 5.02 (2.44–6.67) | 0.083 |
| ADMA (µM/L) | 7.78 (7.36–8.51) | 8.45 (7.98–8.68) | 0.037 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reyes, L.Z.; Winterberg, P.D.; George, R.P.; Kelleman, M.; Harris, F.; Jo, H.; Brown, L.A.S.; Morris, C.R. Arginine Dysregulation and Myocardial Dysfunction in a Mouse Model and Children with Chronic Kidney Disease. Nutrients 2023, 15, 2162. https://doi.org/10.3390/nu15092162
Reyes LZ, Winterberg PD, George RP, Kelleman M, Harris F, Jo H, Brown LAS, Morris CR. Arginine Dysregulation and Myocardial Dysfunction in a Mouse Model and Children with Chronic Kidney Disease. Nutrients. 2023; 15(9):2162. https://doi.org/10.3390/nu15092162
Chicago/Turabian StyleReyes, Loretta Z., Pamela D. Winterberg, Roshan Punnoose George, Michael Kelleman, Frank Harris, Hanjoong Jo, Lou Ann S. Brown, and Claudia R. Morris. 2023. "Arginine Dysregulation and Myocardial Dysfunction in a Mouse Model and Children with Chronic Kidney Disease" Nutrients 15, no. 9: 2162. https://doi.org/10.3390/nu15092162
APA StyleReyes, L. Z., Winterberg, P. D., George, R. P., Kelleman, M., Harris, F., Jo, H., Brown, L. A. S., & Morris, C. R. (2023). Arginine Dysregulation and Myocardial Dysfunction in a Mouse Model and Children with Chronic Kidney Disease. Nutrients, 15(9), 2162. https://doi.org/10.3390/nu15092162