
Vitamin D, Cellular Senescence and Chronic Kidney Diseases: What Is Missing in the Equation?
 ,
,  , , , and
, , , and 
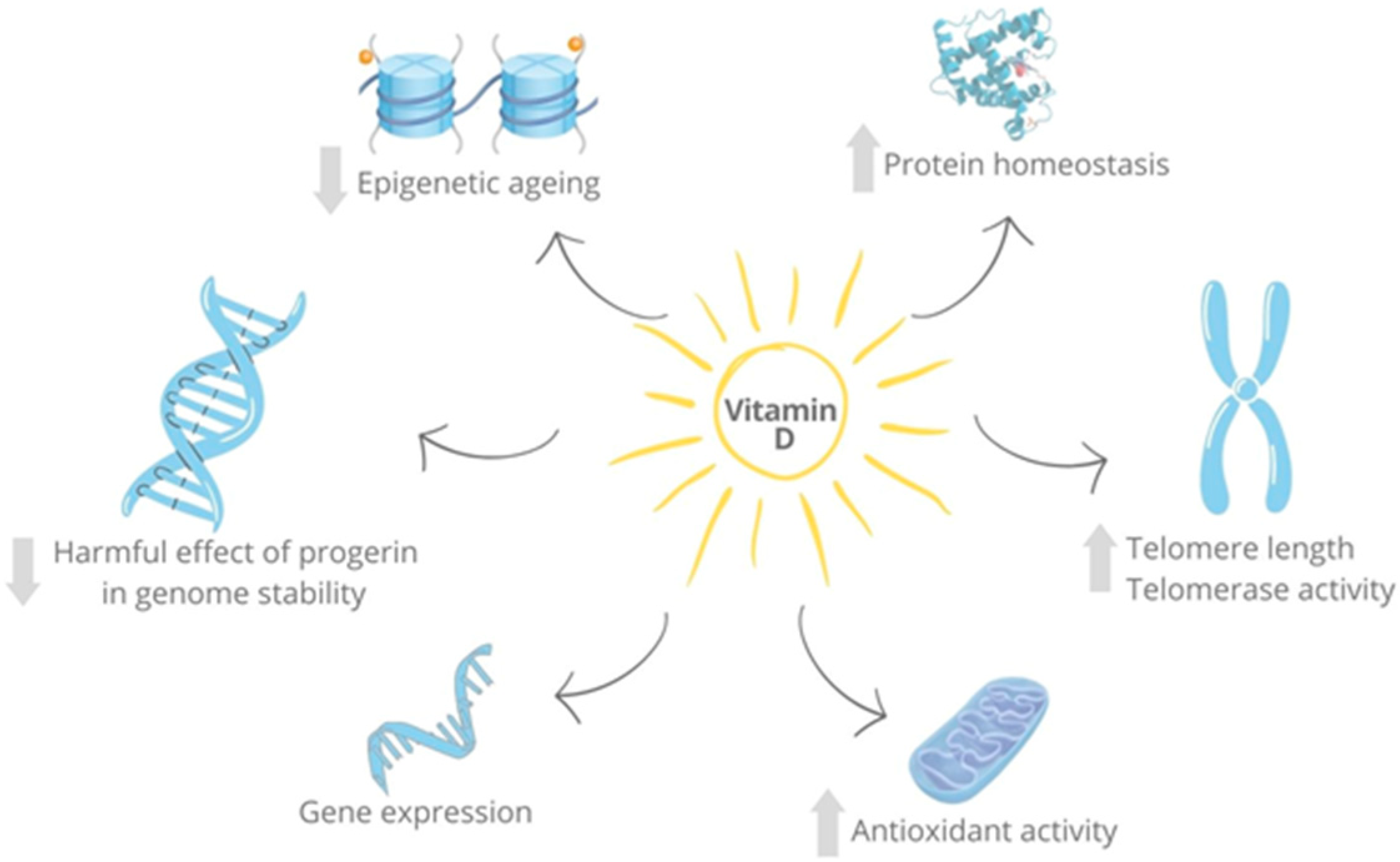{kind=link}
{kind=link}
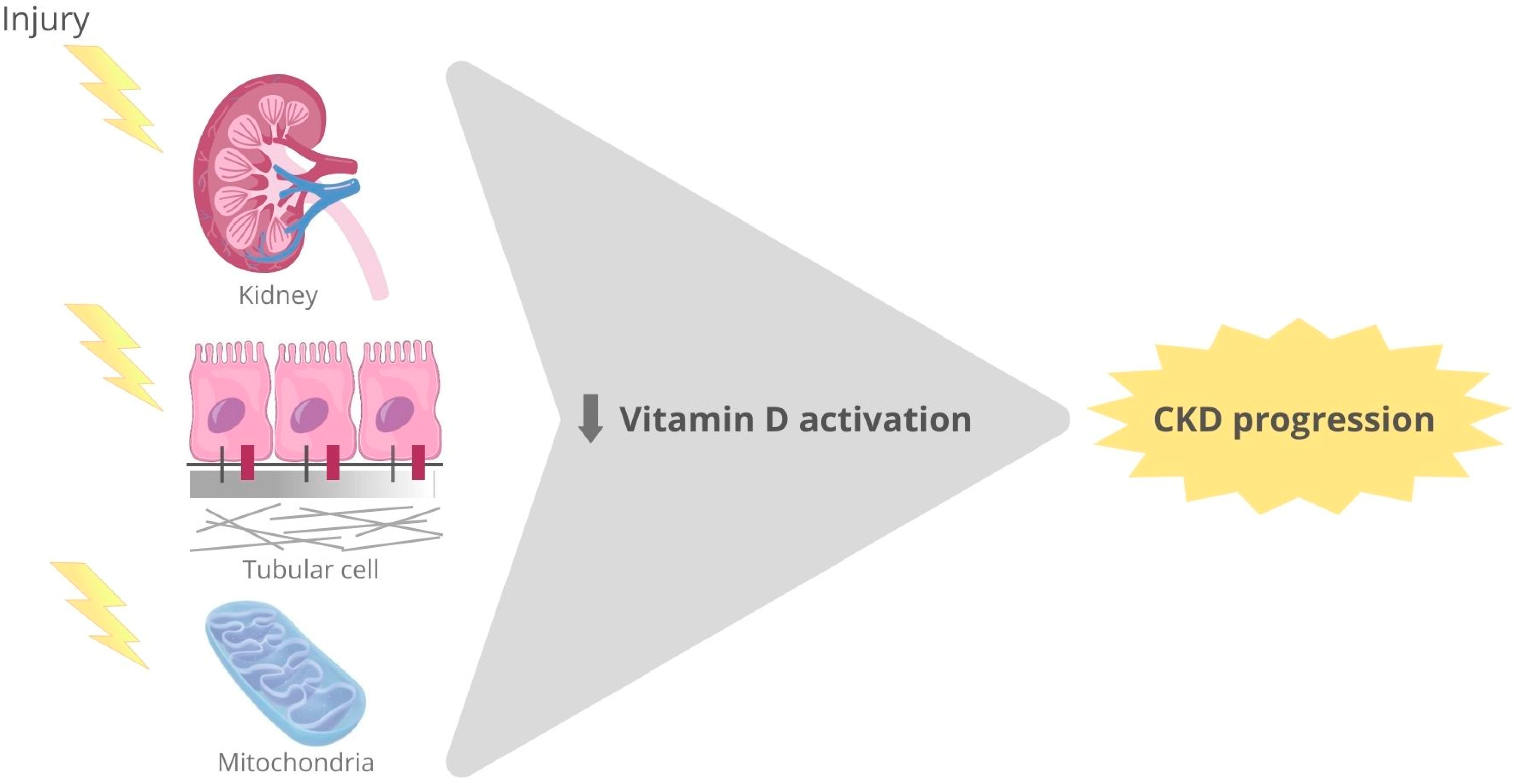{kind=link}
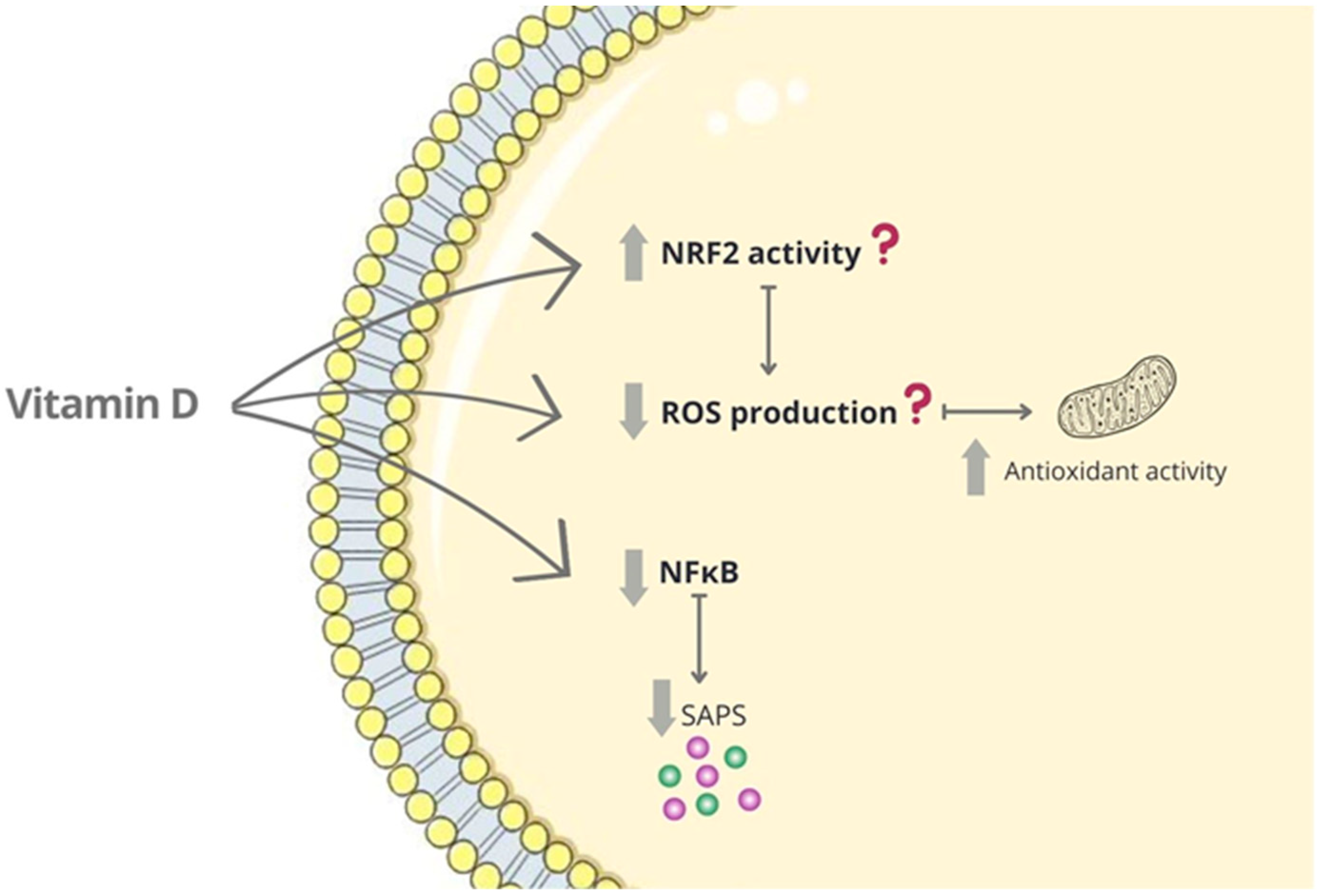{kind=link}
Abstract
1. Introduction
2. Cellular Senescence in Biological Aging and Age-Related Disorders
Senescence: The Molecular View
3. CKD and Cellular Senescence
4. Vitamin D, Aging and CKD
4.1. Vitamin D Synthesis and Mechanisms of Action
4.2. Vitamin D as an Anti-Aging Factor
4.3. Vitamin D and the Immune System
4.4. Other Senescence-Related Targets of Vitamin D
4.5. Vitamin D and CKD
4.5.1. Vitamin D Receptor Activation and Experimental Kidney Damage
4.5.2. Vitamin D Receptor Activation and Human Kidney Diseases
5. Conclusions
6. Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ortiz, A.; Asociación Información Enfermedades Renales Genéticas (AIRG-E); European Kidney Patients’ Federation (EKPF); Federación Nacional de Asociaciones para la Lucha Contra las Enfermedades del Riñón (ALCER); Fundación Renal Íñigo Álvarez de Toledo (FRIAT); Red de Investigación Renal (REDINREN); Resultados en Salud 2040 (RICORS2040); Sociedad Española de Nefrología (SENEFRO) Council; Sociedad Española de Trasplante (SET) Council; Organización Nacional de Trasplantes (ONT). RICORS2040: The need for collaborative research in chronic kidney disease. Clin. Kidney J. 2021, 15, 372–387. [Google Scholar] [CrossRef] [PubMed]
- Rayego-Mateos, S.; Marquez-Expósito, L.; Rodrigues-Diez, R.; Sanz, A.B.; Guiteras, R.; Doladé, N.; Rubio-Soto, I.; Manonelles, A.; Codina, S.; Ortiz, A.; et al. Molecular Mechanisms of Kidney Injury and Repair. Int. J. Mol. Sci. 2022, 23, 1542. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ortega, M.; Rayego-Mateos, S.; Lamas, S.; Ortiz, A.; Rodrigues-Diez, R.R. Targeting the progression of chronic kidney disease. Nat. Rev. Nephrol. 2020, 16, 269–288. [Google Scholar] [CrossRef] [PubMed]
- Rayego-Mateos, S.; Rodrigues-Diez, R.R.; Fernandez-Fernandez, B.; Mora-Fernández, C.; Marchant, V.; Donate-Correa, J.; Navarro-González, J.F.; Ortiz, A.; Ruiz-Ortega, M. Targeting inflammation to treat diabetic kidney disease: The road to 2030. Kidney Int. 2022, 103, 282–296. [Google Scholar] [CrossRef]
- Ruiz-Ortega, M.; Lamas, S.; Ortiz, A. Antifibrotic Agents for the Management of CKD: A Review. Am. J. Kidney Dis. 2022, 80, 251–263. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- López-Otín, C.; Pietrocola, F.; Roiz-Valle, D.; Galluzzi, L.; Kroemer, G. Meta-hallmarks of aging and cancer. Cell Metab. 2023, 35, 12–35. [Google Scholar] [CrossRef]
- Sun, Y.; Li, Q.; Kirkland, J.L. Targeting senescent cells for a healthier longevity: The roadmap for an era of global aging. Life Med. 2022, 1, 103–119. [Google Scholar] [CrossRef]
- Sharma, A.; Chabloz, S.; Lapides, R.A.; Roider, E.; Ewald, C.Y. Potential Synergistic Supplementation of NAD+ Promoting Compounds as a Strategy for Increasing Healthspan. Nutrients 2023, 15, 445. [Google Scholar] [CrossRef]
- Ao, T.; Kikuta, J.; Ishii, M. The Effects of Vitamin D on Immune System and Inflammatory Diseases. Biomolecules 2021, 11, 1624. [Google Scholar] [CrossRef]
- Martens, P.-J.; Gysemans, C.; Verstuyf, A.; Mathieu, C. Vitamin D’s Effect on Immune Function. Nutrients 2020, 12, 1248. [Google Scholar] [CrossRef] [PubMed]
- Holick, M.F. Vitamin D Deficiency. N. Engl. J. Med. 2007, 357, 266–281. [Google Scholar] [CrossRef]
- Prietl, B.; Treiber, G.; Pieber, T.R.; Amrein, K. Vitamin D and immune function. Nutrients 2013, 5, 2502–2521. [Google Scholar] [CrossRef] [PubMed]
- Bellan, M.; Andreoli, L.; Mele, C.; Sainaghi, P.P.; Rigamonti, C.; Piantoni, S.; De Benedittis, C.; Aimaretti, G.; Pirisi, M.; Marzullo, P. Pathophysiological Role and Therapeutic Implications of Vitamin D in Autoimmunity: Focus on Chronic Autoimmune Diseases. Nutrients 2020, 12, 789. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Warnakula, S.; Kunutsor, S.; Crowe, F.; Ward, H.A.; Johnson, L.; Franco, O.; Butterworth, A.S.; Forouhi, N.; Thompson, S.G.; et al. Association of dietary, circulating, and supplement fatty acids with coronary risk: A systematic review and meta-analysis. Ann. Intern. Med. 2014, 160, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Mitri, J.; Muraru, M.D.; Pittas, A.G. Vitamin D and type 2 diabetes: A systematic review. Eur. J. Clin. Nutr. 2011, 65, 1005–1015. [Google Scholar] [CrossRef]
- Sommer, I.; Griebler, U.; Kien, C.; Auer, S.; Klerings, I.; Hammer, R.; Holzer, P.; Gartlehner, G. Vitamin D deficiency as a risk factor for dementia: A systematic review and meta-analysis. BMC Geriatr. 2017, 17, 16. [Google Scholar] [CrossRef]
- Wu, X.; Hu, W.; Lu, L.; Zhao, Y.; Zhou, Y.; Xiao, Z.; Zhang, L.; Zhang, H.; Li, X.; Li, W.; et al. Repurposing vitamin D for treatment of human malignancies via targeting tumor microenvironment. Acta Pharm. Sin. B 2018, 9, 203–219. [Google Scholar] [CrossRef]
- Nowak, J.; Hudzik, B.; Jagielski, P.; Kulik-Kupka, K.; Danikiewicz, A.; Zubelewicz-Szkodzińska, B. Lack of Seasonal Variations in Vitamin D Concentrations among Hospitalized Elderly Patients. Int. J. Environ. Res. Public Health 2021, 18, 1676. [Google Scholar] [CrossRef]
- Dobnig, H.; Pilz, S.; Scharnagl, H.; Renner, W.; Seelhorst, U.; Wellnitz, B.; Kinkeldei, J.; Boehm, B.O.; Weihrauch, G.; Maerz, W. Independent association of low serum 25-hydroxyvitamin D and 1,25-dihydroxyvitamin D levels with all-cause and cardiovascular mortality. Arch. Intern. Med. 2008, 168, 1340–1349. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef] [PubMed]
- Mas-Bargues, C.; Alique, M.; Barrús-Ortiz, M.T.; Borrás, C.; Rodrigues-Díez, R. Exploring New Kingdoms: The Role of Extracellular Vesicles in Oxi-Inflamm-Aging Related to Cardiorenal Syndrome. Antioxidants 2021, 11, 78. [Google Scholar] [CrossRef] [PubMed]
- Vernot, J.P. Senescence-Associated Pro-inflammatory Cytokines and Tumor Cell Plasticity. Front. Mol. Biosci. 2020, 7, 63. [Google Scholar] [CrossRef] [PubMed]
- Estévez-Souto, V.; Da Silva-Álvarez, S.; Collado, M. The role of extracellular vesicles in cellular senescence. FEBS J. 2022, 290, 1203–1211. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.; Koh, D.; Bin Jeon, H.; Kim, K.M. The Role of Extracellular Vesicles in Senescence. Mol. Cells 2022, 45, 603–609. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging: An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Pawelec, G. The human immunosenescence phenotype: Does it exist? Semin. Immunopathol. 2020, 42, 537–544. [Google Scholar] [CrossRef]
- Pawelec, G. Age and immunity: What is “immunosenescence”? Exp. Gerontol. 2017, 105, 4–9. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Alique, M.; Ruíz-Torres, M.P.; Bodega, G.; Noci, M.V.; Troyano, N.; Bohórquez, L.; Luna, C.; Luque, R.; Carmona, A.; Carracedo, J.; et al. Microvesicles from the plasma of elderly subjects and from senescent endothelial cells promote vascular calcification. Aging 2017, 9, 778–789. [Google Scholar] [CrossRef]
- Fumagalli, M.; Rossiello, F.; Mondello, C.; Di Fagagna, F.D. Stable cellular senescence is associated with persistent DDR activation. PLoS ONE 2014, 9, e110969. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, T.; Malik, R.; Thomas, S.; Sage, J.; Johnson, P.F. C/EBPβ cooperates with RB:E2F to implement RasV12-induced cellular senescence. EMBO J. 2005, 24, 3301–3312. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Salotti, J.; Johnson, P.F. Regulation of senescence and the SASP by the transcription factor C/EBPβ. Exp. Gerontol. 2019, 128, 110752. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; DeMaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660.e4. [Google Scholar] [CrossRef] [PubMed]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef]
- Valentijn, F.A.; Knoppert, S.N.; Marquez-Exposito, L.; Rodrigues-Diez, R.R.; Pissas, G.; Tang, J.; Tejedor-Santamaria, L.; Broekhuizen, R.; Samarakoon, R.; Eleftheriadis, T.; et al. Cellular communication network 2 (connective tissue growth factor) aggravates acute DNA damage and subsequent DNA damage response-senescence-fibrosis following kidney ischemia reperfusion injury. Kidney Int. 2022, 102, 1305–1319. [Google Scholar] [CrossRef]
- Marquez-Exposito, L.; Tejedor-Santamaria, L.; Valentijn, F.A.; Tejera-Muñoz, A.; Rayego-Mateos, S.; Marchant, V.; Rodrigues-Diez, R.R.; Rubio-Soto, I.; Knoppert, S.N.; Ortiz, A.; et al. Oxidative Stress and Cellular Senescence Are Involved in the Aging Kidney. Antioxidants 2022, 11, 301. [Google Scholar] [CrossRef]
- Valentijn, F.A.; Knoppert, S.N.; Pissas, G.; Rodrigues-Diez, R.R.; Marquez-Exposito, L.; Broekhuizen, R.; Mokry, M.; Kester, L.A.; Falke, L.L.; Goldschmeding, R.; et al. CCN2 Aggravates the Immediate Oxidative Stress–DNA Damage Response following Renal Ischemia–Reperfusion Injury. Antioxidants 2021, 10, 2020. [Google Scholar] [CrossRef]
- Marquez-Exposito, L.; Tejedor-Santamaria, L.; Santos-Sanchez, L.; Valentijn, F.A.; Cantero-Navarro, E.; Rayego-Mateos, S.; Rodrigues-Diez, R.R.; Tejera-Muñoz, A.; Marchant, V.; Sanz, A.B.; et al. Acute Kidney Injury is Aggravated in Aged Mice by the Exacerbation of Proinflammatory Processes. Front. Pharmacol. 2021, 12, 662020. [Google Scholar] [CrossRef] [PubMed]
- Shilovsky, G.A.; Ashapkin, V.V. Transcription Factor Nrf2 and Mitochondria—Friends or Foes in the Regulation of Aging Rate. Biochemistry 2022, 87, 1477–1486. [Google Scholar] [CrossRef] [PubMed]
- Knoppert, S.N.; Valentijn, F.A.; Nguyen, T.Q.; Goldschmeding, R.; Falke, L.L. Cellular Senescence and the Kidney: Potential Therapeutic Targets and Tools. Front. Pharmacol. 2019, 10, 770. [Google Scholar] [CrossRef]
- Dusso, A.S. Kidney disease and vitamin D levels: 25-hydroxyvitamin D, 1,25-dihydroxyvitamin D, and VDR activation. Kidney Int. Suppl. 2011, 1, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Dusso, A.S.; Brown, A.J.; Slatopolsky, E. Vitamin D. Am. J. Physiol. Ren. Physiol. 2005, 289, 8–28. [Google Scholar] [CrossRef]
- Harrison, S.R.; Li, D.; Jeffery, L.E.; Raza, K.; Hewison, M. Vitamin D, Autoimmune Disease and Rheumatoid Arthritis. Calcif. Tissue Int. 2019, 106, 58–75. [Google Scholar] [CrossRef] [PubMed]
- Akeno, N.; Saikatsu, S.; Kawane, T.; Horiuchi, N. Mouse vitamin D-24-hydroxylase: Molecular cloning, tissue distribution, and transcriptional regulation by 1α,25-dihydroxyvitamin D3. Endocrinology 1997, 138, 2233–2240. [Google Scholar] [CrossRef]
- Rosen, C.J.; Abrams, S.A.; Aloia, J.F.; Brannon, P.M.; Clinton, S.K.; Durazo-Arvizu, R.A.; Gallagher, J.C.; Gallo, R.L.; Jones, G.; Kovacs, C.S.; et al. IOM committee members respond to endocrine society vitamin D guideline. J. Clin. Endocrinol. Metab. 2012, 97, 1146–1152. [Google Scholar] [CrossRef]
- Carlberg, C. Vitamin D Genomics: From In Vitro to In Vivo. Front. Endocrinol. 2018, 9, 250. [Google Scholar] [CrossRef]
- Campbell, M.J. Vitamin D and the RNA transcriptome: More than mRNA regulation. Front. Physiol. 2014, 5, 181. [Google Scholar] [CrossRef]
- Hanel, A.; Carlberg, C. Time-Resolved Gene Expression Analysis Monitors the Regulation of Inflammatory Mediators and Attenuation of Adaptive Immune Response by Vitamin D. Int. J. Mol. Sci. 2022, 23, 911. [Google Scholar] [CrossRef] [PubMed]
- Gil, Á.; Plaza-Diaz, J.; Mesa, M.D. Vitamin D: Classic and Novel Actions. Ann. Nutr. Metab. 2018, 72, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Cortes, M.; Chen, M.J.; Stachura, D.L.; Liu, S.Y.; Kwan, W.; Wright, F.; Vo, L.T.; Theodore, L.N.; Esain, V.; Frost, I.M.; et al. Developmental Vitamin D Availability Impacts Hematopoietic Stem Cell Production. Cell Rep. 2016, 17, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Müller, V.; de Boer, R.J.; Bonhoeffer, S.; Szathmáry, E. An evolutionary perspective on the systems of adaptive immunity. Biol. Rev. 2018, 93, 505–528. [Google Scholar] [CrossRef] [PubMed]
- Vanherwegen, A.-S.; Gysemans, C.; Mathieu, C. Vitamin D endocrinology on the cross-road between immunity and metabolism. Mol. Cell. Endocrinol. 2017, 453, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Nemere, I.; Safford, S.E.; Rohe, B.; DeSouza, M.M.; Farach-Carson, M.C. Identification and characterization of 1,25D3-membrane-associated rapid response, steroid (1,25D3-MARRS) binding protein. J. Steroid Biochem. Mol. Biol. 2004, 89–90, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Khanal, R.C.; Nemere, I. The ERp57/GRp58/1,25D3-MARRS receptor: Multiple functional roles in diverse cell systems. Curr. Med. Chem. 2007, 14, 1087–1093. [Google Scholar] [CrossRef]
- Nemere, I. The 1,25D3-MARRS protein: Contribution to steroid stimulated calcium uptake in chicks and rats. Steroids 2005, 70, 455–457. [Google Scholar] [CrossRef]
- Nemere, I.; Dormanen, M.C.; Hammond, M.W.; Okamura, W.H.; Norman, A.W. Identification of a specific binding protein for 1 alpha,25-dihydroxyvitamin D3 in basal-lateral membranes of chick intestinal epithelium and relationship to transcaltachia. J. Biol. Chem. 1994, 269, 23750–23756. [Google Scholar] [CrossRef]
- Nemere, I. Apparent nonnuclear regulation of intestinal phosphate transport: Effects of 1,25-dihydroxyvitamin D3,24,25-dihydroxyvitamin D3, and 25-hydroxyvitamin D3. Endocrinology 1996, 137, 2254–2261. [Google Scholar] [CrossRef]
- Muhlenkamp, C.R.; Gill, S.S. A glucose-regulated protein, GRP58, is down-regulated in C57B6 mouse liver after diethylhexyl phthalate exposure. Toxicol. Appl. Pharmacol. 1998, 148, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Tourkova, I.L.; Shurin, G.V.; Chatta, G.S.; Perez, L.; Finke, J.; Whiteside, T.L.; Ferrone, S.; Shurin, M.R. Restoration by IL-15 of MHC class I antigen-processing machinery in human dendritic cells inhibited by tumor-derived gangliosides. J. Immunol. 2005, 175, 3045–3052. [Google Scholar] [CrossRef]
- Seliger, B.; Wollscheid, U.; Momburg, F.; Blankenstein, T.; Huber, C. Characterization of the major histocompatibility complex class I deficiencies in B16 melanoma cells. Cancer Res. 2001, 61, 1095–1099. [Google Scholar] [PubMed]
- Guo, G.G.; Patel, K.; Kumar, V.; Shah, M.; Fried, V.A.; Etlinger, J.D.; Sehgal, P.B. Association of the chaperone glucose-regulated protein 58 (GRP58/ER-60/ERp57) with Stat3 in cytosol and plasma membrane complexes. J. Interf. Cytokine Res. 2002, 22, 555–563. [Google Scholar] [CrossRef]
- Dihazi, H.; Dihazi, G.H.; Bibi, A.; Eltoweissy, M.; Mueller, C.A.; Asif, A.R.; Rubel, D.; Vasko, R.; Mueller, G.A. ERP57 secretion is important for extracellular matrix accumulation and renal fibrosis progression and is an earlier sign of disease onset. J. Cell Sci. 2013, 126, 3649–3663. [Google Scholar] [CrossRef] [PubMed]
- Rayego-Mateos, S.; Morgado-Pascual, J.L.; Valdivielso, J.M.; Sanz, A.B.; Bosch-Panadero, E.; Rodrigues-Díez, R.R.; Egido, J.; Ortiz, A.; González-Parra, E.; Ruiz-Ortega, M. TRAF3 Modulation: Novel Mechanism for the Anti-inflammatory Effects of the Vitamin D Receptor Agonist Paricalcitol in Renal Disease. J. Am. Soc. Nephrol. 2020, 31, 2026–2042. [Google Scholar] [CrossRef]
- Coll-Bonfill, N.; de Faria, R.C.; Bhoopatiraju, S.; Gonzalo, S. Calcitriol Prevents RAD51 Loss and cGAS-STING-IFN Response Triggered by Progerin. Proteomics 2019, 20, e1800406. [Google Scholar] [CrossRef]
- Zarei, M.; Zarezadeh, M.; Kalajahi, F.H.; Javanbakht, M.H. The Relationship Between Vitamin D and Telomere/Telomerase: A Comprehensive Review. J. Frailty Aging 2020, 10, 2–9. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115, Erratum in Genome Biol. 2015, 16, 96. [Google Scholar] [CrossRef]
- Chen, L.; Dong, Y.; Bhagatwala, J.; Raed, A.; Huang, Y.; Zhu, H. Effects of Vitamin D3 Supplementation on Epigenetic Aging in Overweight and Obese African Americans With Suboptimal Vitamin D Status: A Randomized Clinical Trial. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 91–98. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, Y.; Schöttker, B.; Brenner, H. Vitamin D status and epigenetic-based mortality risk score: Strong independent and joint prediction of all-cause mortality in a population-based cohort study. Clin. Epigenet. 2018, 10, 84. [Google Scholar] [CrossRef] [PubMed]
- Mark, K.A.; Dumas, K.J.; Bhaumik, D.; Schilling, B.; Davis, S.; Oron, T.R.; Sorensen, D.J.; Lucanic, M.; Brem, R.B.; Melov, S.; et al. Vitamin D Promotes Protein Homeostasis and Longevity via the Stress Response Pathway Genes skn-1, ire-1, and xbp-1. Cell Rep. 2016, 17, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, R.; Qiao, W.; Zhang, W.; Chen, J.; Mao, L.; Goltzman, D.; Miao, D. 1,25-Dihydroxyvitamin D exerts an antiaging role by activation of Nrf2-antioxidant signaling and inactivation of p16/p53-senescence signaling. Aging Cell 2019, 18, e12951. [Google Scholar] [CrossRef] [PubMed]
- Keisala, T.; Minasyan, A.; Lou, Y.-R.; Zou, J.; Kalueff, A.V.; Pyykkö, I.; Tuohimaa, P. Premature aging in vitamin D receptor mutant mice. J. Steroid Biochem. Mol. Biol. 2009, 115, 91–97. [Google Scholar] [CrossRef]
- Gysemans, C.; van Etten, E.; Overbergh, L.; Giulietti, A.; Eelen, G.; Waer, M.; Verstuyf, A.; Bouillon, R.; Mathieu, C. Unaltered diabetes presentation in NOD mice lacking the vitamin D receptor. Diabetes 2008, 57, 269–275. [Google Scholar] [CrossRef]
- Simpson, R.U.; Hershey, S.H.; Nibbelink, K.A. Characterization of heart size and blood pressure in the vitamin D receptor knockout mouse. J. Steroid Biochem. Mol. Biol. 2007, 103, 521–524. [Google Scholar] [CrossRef]
- Burne, T.H.; McGrath, J.J.; Eyles, D.W.; Mackay-Sim, A. Behavioural characterization of Vitamin D receptor knockout mice. Behav. Brain Res. 2005, 157, 299–308. [Google Scholar] [CrossRef]
- Bonaccio, M.; Di Castelnuovo, A.; Pounis, G.; De Curtis, A.; Costanzo, S.; Persichillo, M.; Cerletti, C.; Donati, M.B.; de Gaetano, G.; Iacoviello, L. A score of low-grade inflammation and risk of mortality: Prospective findings from the Moli-sani study. Haematologica 2016, 101, 1434–1441. [Google Scholar] [CrossRef]
- De Gonzalo-Calvo, D.; De Luxán-Delgado, B.; Martínez-Camblor, P.; Rodríguez-González, S.; Garcia-Macia, M.; Suárez, F.M.; Solano, J.J.; Rodríguez-Colunga, M.J.; Coto-Montes, A. Chronic inflammation as predictor of 1-year hospitalization and mortality in elderly population. Eur. J. Clin. Investig. 2012, 42, 1037–1046. [Google Scholar] [CrossRef]
- Adams, J.S.; Ren, S.; Liu, P.T.; Chun, R.F.; Lagishetty, V.; Gombart, A.F.; Borregaard, N.; Modlin, R.L.; Hewison, M. Vitamin d-directed rheostatic regulation of monocyte antibacterial responses. J. Immunol. 2009, 182, 4289–4295. [Google Scholar] [CrossRef]
- Barlow, P.G.; Svoboda, P.; Mackellar, A.; Nash, A.A.; York, I.A.; Pohl, J.; Davidson, D.J.; Donis, R.O. Antiviral activity and increased host defense against influenza infection elicited by the human cathelicidin LL-37. PLoS ONE 2011, 6, e25333. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.T.; Stenger, S.; Li, H.; Wenzel, L.; Tan, B.H.; Krutzik, S.R.; Ochoa, M.T.; Schauber, J.; Wu, K.; Meinken, C.; et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 2006, 311, 1770–1773. [Google Scholar] [CrossRef] [PubMed]
- Shahmiri, M.; Enciso, M.; Adda, C.G.; Smith, B.J.; Perugini, M.A.; Mechler, A. Membrane Core-Specific Antimicrobial Action of Cathelicidin LL-37 Peptide Switches Between Pore and Nanofibre Formation. Sci. Rep. 2016, 6, 38184. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Leung, D.Y.M.; Richers, B.N.; Liu, Y.; Remigio, L.K.; Riches, D.W.; Goleva, E. Vitamin D inhibits monocyte/macrophage proinflammatory cytokine production by targeting MAPK phosphatase-1. J. Immunol. 2012, 188, 2127–2135. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.-Y.; Shi, G.-Y.; Shi, C.-S.; Kao, Y.-C.; Lin, S.-W.; Wu, H.-L. Monocytic thrombomodulin triggers LPS- and gram-negative bacteria-induced inflammatory response. J. Immunol. 2012, 188, 6328–6337. [Google Scholar] [CrossRef] [PubMed]
- Koivisto, O.; Hanel, A.; Carlberg, C. Key Vitamin D Target Genes with Functions in the Immune System. Nutrients 2020, 12, 1140. [Google Scholar] [CrossRef]
- Cantorna, M.T.; Snyder, L.; Lin, Y.-D.; Yang, L. Vitamin D and 1,25(OH)2D Regulation of T cells. Nutrients 2015, 7, 3011–3021. [Google Scholar] [CrossRef]
- Fisher, S.A.; Rahimzadeh, M.; Brierley, C.; Gration, B.; Doree, C.; Kimber, C.E.; Cajide, A.P.; Lamikanra, A.A.; Roberts, D.J. The role of vitamin D in increasing circulating T regulatory cell numbers and modulating T regulatory cell phenotypes in patients with inflammatory disease or in healthy volunteers: A systematic review. PLoS ONE 2019, 14, e0222313. [Google Scholar] [CrossRef]
- Zhou, Q.; Qin, S.; Zhang, J.; Zhon, L.; Pen, Z.; Xing, T. 1,25(OH) 2 D 3 induces regulatory T cell differentiation by influencing the VDR/PLC-γ1/TGF-β1/pathway. Mol. Immunol. 2017, 91, 156–164. [Google Scholar] [CrossRef]
- Duffy, M.M.; McNicholas, B.A.; Monaghan, D.A.; Hanley, S.A.; McMahon, J.M.; Pindjakova, J.; Alagesan, S.; Fearnhead, H.O.; Griffin, M.D. Mesenchymal stem cells and a vitamin D receptor agonist additively suppress T helper 17 cells and the related inflammatory response in the kidney. Am. J. Physiol. Physiol. 2014, 307, F1412–F1426. [Google Scholar] [CrossRef]
- Piemonti, L.; Monti, P.; Sironi, M.; Fraticelli, P.; Leone, B.E.; Dal Cin, E.; Allavena, P.; Di Carlo, V. Vitamin D3 affects differentiation, maturation, and function of human monocyte-derived dendritic cells. J. Immunol. 2000, 164, 4443–4451. [Google Scholar] [CrossRef] [PubMed]
- Rizka, A.; Setiati, S.; Sadikin, M.; Mansur, I.G. Immunomodulatory effect of in vitro calcitriol in fit and frail elderly. Int. Immunopharmacol. 2021, 96, 107737. [Google Scholar] [CrossRef] [PubMed]
- Kuro-O, M. Klotho. Pflug. Arch. Eur. J. Physiol. 2010, 459, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Forster, R.E.; Jurutka, P.W.; Hsieh, J.-C.; Haussler, C.A.; Lowmiller, C.L.; Kaneko, I.; Haussler, M.R.; Whitfield, G.K. Vitamin D receptor controls expression of the anti-aging klotho gene in mouse and human renal cells. Biochem. Biophys. Res. Commun. 2011, 414, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Mytych, J.; Romerowicz-Misielak, M.; Koziorowski, M. Klotho protects human monocytes from LPS-induced immune impairment associated with immunosenescent-like phenotype. Mol. Cell. Endocrinol. 2018, 470, 1–13. [Google Scholar] [CrossRef]
- Chen, J.; Fan, J.; Wang, S.; Sun, Z. Secreted Klotho Attenuates Inflammation-Associated Aortic Valve Fibrosis in Senescence-Accelerated Mice P1. Hypertension 2018, 71, 877–885. [Google Scholar] [CrossRef]
- Liu, F.; Wu, S.; Ren, H.; Gu, J. Klotho suppresses RIG-I-mediated senescence-associated inflammation. Nature 2011, 13, 254–262. [Google Scholar] [CrossRef]
- Cui, W.; Leng, B.; Wang, G. Klotho protein inhibits H2O2-induced oxidative injury in endothelial cells via regulation of PI3K/AKT/Nrf2/HO-1 pathways. Can. J. Physiol. Pharmacol. 2019, 97, 370–376. [Google Scholar] [CrossRef]
- Romero, A.; San Hipólito-Luengo, A.; Villalobos, L.A.; Vallejo, S.; Valencia, I.; Michalska, P.; Pajuelo-Lozano, N.; Sánchez-Pérez, I.; León, R.; Bartha, J.L.; et al. The angiotensin-(1-7)/Mas receptor axis protects from endothelial cell senescence via klotho and Nrf2 activation. Aging Cell 2019, 18, e12913. [Google Scholar] [CrossRef]
- He, D.; Wang, Y.; Liu, R.; He, A.; Li, S.; Fu, X.; Zhou, Z. 1,25(OH)2D3 Activates Autophagy to Protect against Oxidative Damage of INS-1 Pancreatic Beta Cells. Biol. Pharm. Bull. 2019, 42, 561–567. [Google Scholar] [CrossRef]
- Li, X.; Li, Z.; Li, B.; Zhu, X.; Lai, X. Klotho improves diabetic cardiomyopathy by suppressing the NLRP3 inflammasome pathway. Life Sci. 2019, 234, 116773. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Ma, J.; Ren, Y.; Xiang, S.; Jia, R. Secreted klotho from exosomes alleviates inflammation and apoptosis in acute pancreatitis. Am. J. Transl. Res. 2019, 11, 3375–3383. [Google Scholar] [PubMed]
- Izquierdo, M.C.; Perez-Gomez, M.V.; Sanchez-Niño, M.D.; Sanz, A.B.; Ruiz-Andres, O.; Poveda, J.; Moreno, J.A.; Egido, J.; Ortiz, A. Klotho, phosphate and inflammation/ageing in chronic kidney disease. Nephrol. Dial. Transplant. 2012, 27, iv6–iv10. [Google Scholar] [CrossRef] [PubMed]
- Araki, T.; Sasaki, Y.; Milbrandt, J. Increased Nuclear NAD Biosynthesis and SIRT1 Activation Prevent Axonal Degeneration. Science 2004, 305, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Watroba, M.; Szukiewicz, D. Sirtuins at the Service of Healthy Longevity. Front. Physiol. 2021, 12, 2006. [Google Scholar] [CrossRef]
- North, B.J.; Verdin, E. Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome Biol. 2004, 5, 224. [Google Scholar] [CrossRef]
- Chang, H.-C.; Guarente, L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol. Metab. 2014, 25, 138–145. [Google Scholar] [CrossRef]
- Chen, C.; Zhou, M.; Ge, Y.; Wang, X. SIRT1 and aging related signaling pathways. Mech. Ageing Dev. 2020, 187, 111215. [Google Scholar] [CrossRef]
- Edwards, J.R.; Perrien, D.; Fleming, N.; Nyman, J.S.; Ono, K.; Connelly, L.; Moore, M.M.; Lwin, S.T.; E Yull, F.; Mundy, G.R.; et al. Silent information regulator (Sir)T1 inhibits NF-κB signaling to maintain normal skeletal remodeling. J. Bone Miner. Res. 2013, 28, 960–969. [Google Scholar] [CrossRef]
- Zarzuelo, M.J.; López-Sepúlveda, R.; Sánchez, M.; Romero, M.; Gómez-Guzmán, M.; Ungvary, Z.; Pérez-Vizcaíno, F.; Jiménez, R.; Duarte, J. SIRT1 inhibits NADPH oxidase activation and protects endothelial function in the rat aorta: Implications for vascular aging. Biochem. Pharmacol. 2013, 85, 1288–1296. [Google Scholar] [CrossRef]
- Lu, C.-L.; Liao, M.-T.; Hou, Y.-C.; Fang, Y.-W.; Zheng, C.-M.; Liu, W.-C.; Chao, C.-T.; Lu, K.-C.; Ng, Y.-Y. Sirtuin-1 and Its Relevance in Vascular Calcification. Int. J. Mol. Sci. 2020, 21, 1593. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Kim, E.N.; Kim, M.Y.; Chung, S.; Shin, S.J.; Kim, H.W.; Yang, C.W.; Kim, Y.S.; Chang, Y.S.; Park, C.W.; et al. Age-Associated Molecular Changes in the Kidney in Aged Mice. Oxidative Med. Cell. Longev. 2012, 2012, 171383. [Google Scholar] [CrossRef] [PubMed]
- Ryu, D.R.; Yu, M.R.; Kong, K.H.; Kim, H.; Kwon, S.H.; Jeon, J.S.; Han, D.C.; Noh, H. Sirt1-hypoxia-inducible factor-1α interaction is a key mediator of tubulointerstitial damage in the aged kidney. Aging Cell 2019, 18, e12904. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; O’Keefe, J.H.; Bell, D.; Hensrud, D.D.; Holick, M.F. Vitamin D deficiency: An important, common, and easily treatable cardiovascular risk factor? J. Am. Coll. Cardiol. 2008, 52, 1949–1956. [Google Scholar] [CrossRef] [PubMed]
- Torregrosa, J.-V.; Bover, J.; Portillo, M.R.; Parra, E.G.; Arenas, M.D.; Caravaca, F.; Casaus, M.-L.G.; Martín-Malo, A.; Navarro-González, J.F.; Lorenzo, V.; et al. Recommendations of the Spanish Society of Nephrology for the management of mineral and bone metabolism disorders in patients with chronic kidney disease: 2021 (SEN-MM). Nefrología 2022, 42, 1–37. [Google Scholar] [CrossRef]
- Lucisano, S.; Buemi, M.; Passantino, A.; Aloisi, C.; Cernaro, V.; Santoro, D. New insights on the role of vitamin D in the progression of renal damage. Kidney Blood Press. Res. 2013, 37, 667–678. [Google Scholar] [CrossRef]
- Li, Y.C. Vitamin D in Chronic Kidney Disease; Karger: Basel, Switzerland, 2013; Volume 180, pp. 98–109. [Google Scholar] [CrossRef]
- Park, J.W.; Bae, E.H.; Kim, I.J.; Ma, S.K.; Choi, C.; Lee, J.; Kim, S.W. Paricalcitol attenuates cyclosporine-induced kidney injury in rats. Kidney Int. 2010, 77, 1076–1085. [Google Scholar] [CrossRef]
- Piao, S.; Song, J.; Lim, S.; Chung, B.; Choi, B.; Yang, C. Protective effect of paricalcitol on cyclosporine-induced renal injury in rats. Transplant. Proc. 2012, 44, 642–645. [Google Scholar] [CrossRef]
- Hwang, H.S.; Yang, K.J.; Park, K.C.; Choi, H.S.; Kim, S.H.; Hong, S.Y.; Jeon, B.H.; Chang, Y.K.; Park, C.W.; Kim, S.Y.; et al. Pretreatment with paricalcitol attenuates inflammation in ischemia-reperfusion injury via the up-regulation of cyclooxygenase-2 and prostaglandin E2. Nephrol. Dial. Transplant. 2012, 28, 1156–1166. [Google Scholar] [CrossRef]
- Lee, J.-W.; Kim, S.C.; Ko, Y.S.; Lee, H.Y.; Cho, E.; Kim, M.-G.; Jo, S.-K.; Cho, W.Y.; Kim, H.K. Renoprotective effect of paricalcitol via a modulation of the TLR4-NF-κB pathway in ischemia/reperfusion-induced acute kidney injury. Biochem. Biophys. Res. Commun. 2014, 444, 121–127. [Google Scholar] [CrossRef]
- Tan, X.; Wen, X.; Liu, Y. Paricalcitol inhibits renal inflammation by promoting vitamin D receptor–mediated sequestration of NF-κB signaling. J. Am. Soc. Nephrol. 2008, 19, 1741–1752. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Niño, M.-D.; Bozic, M.; Córdoba-Lanús, E.; Valcheva, P.; Gracia, O.; Ibarz, M.; Fernández, E.; Navarro-González, J.F.; Ortiz, A.; Valdivielso, J.M.; et al. Beyond proteinuria: VDR activation reduces renal inflammation in experimental diabetic nephropathy. Am. J. Physiol. Physiol. 2012, 302, F647–F657. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Kang, Y.S.; Dai, C.; Liu, Y. Blockade of Wnt/β-catenin signaling by paricalcitol ameliorates proteinuria and kidney injury. J. Am. Soc. Nephrol. 2011, 22, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Lydia, A.; Asanuma, K.; Nonaka, K.; Takagi, M.; Jeong, K.-H.; Kodama, F.; Asao, R.; Asanuma, E.; Prodjosudjadi, W.; Tomino, Y. Effects of 22-oxa-calcitriol on podocyte injury in adriamycin-induced nephrosis. Am. J. Nephrol. 2011, 35, 58–68. [Google Scholar] [CrossRef]
- Sonneveld, R.; Ferrè, S.; Hoenderop, J.G.; Dijkman, H.B.; Berden, J.H.; Bindels, R.J.; Wetzels, J.F.; van der Vlag, J.; Nijenhuis, T. Vitamin D down-regulates TRPC6 expression in podocyte injury and proteinuric glomerular disease. Am. J. Pathol. 2013, 182, 1196–1204. [Google Scholar] [CrossRef]
- Jeong, K.H.; Asanuma, K.; Lydia, A.; Takagi, M.; Asao, R.; Kodama, F.; Asanuma, E.; Tomino, Y. Combination therapy with telmisartan and oxacalcitriol suppresses the progression of murine adriamycin nephropathy. Nephron 2015, 129, 143–154. [Google Scholar] [CrossRef]
- Zhang, Y.; Deb, D.K.; Kong, J.; Ning, G.; Wang, Y.; Li, G.; Chen, Y.; Zhang, Z.; Strugnell, S.; Sabbagh, Y.; et al. Long-term therapeutic effect of vitamin D analog doxercalciferol on diabetic nephropathy: Strong synergism with AT1 receptor antagonist. Am. J. Physiol. Physiol. 2009, 297, F791–F801. [Google Scholar] [CrossRef]
- Deb, D.K.; Sun, T.; Wong, K.E.; Zhang, Z.; Ning, G.; Zhang, Y.; Kong, J.; Shi, H.; Chang, A.; Li, Y.C. Combined vitamin D analog and AT1 receptor antagonist synergistically block the development of kidney disease in a model of type 2 diabetes. Kidney Int. 2010, 77, 1000–1009. [Google Scholar] [CrossRef]
- Sanz, A.B.; Sanchez-Niño, M.D.; Ramos, A.M.; Moreno, J.A.; Santamaria, B.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. NF-κB in renal inflammation. J. Am. Soc. Nephrol. 2010, 21, 1254–1262. [Google Scholar] [CrossRef]
- Cui, R.; Tieu, B.; Recinos, A.; Tilton, R.G.; Brasier, A.R. RhoA mediates angiotensin II–induced phospho-Ser536 nuclear factor κB/RelA subunit exchange on the interleukin-6 promoter in VSMCs. Circ. Res. 2006, 99, 723–730. [Google Scholar] [CrossRef]
- Vila Cuenca, M.; Ferrantelli, E.; Meinster, E.; Pouw, S.M.; Kovačević, I.; de Menezes, R.X.; Niessen, H.W.; Beelen, R.H.; Hordijk, P.L.; Vervloet, M.G. Vitamin D Attenuates Endothelial Dysfunction in Uremic Rats and Maintains Human Endothelial Stability. J. Am. Heart Assoc. 2018, 7, e02709, Erratum for: J. Am. Heart Assoc. 2018, 7, e008776. [Google Scholar] [CrossRef]
- Zhang, Z.-H.; Luo, B.; Xu, S.; Xing, W.-Y.; Chen, Y.-H.; Zhang, C.; Wang, H.; Xie, D.-D.; Xu, D.-X. Long-term vitamin D deficiency promotes renal fibrosis and functional impairment in middle-aged male mice. Br. J. Nutr. 2020, 125, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Kong, J.; Wei, M.; Chen, Z.-F.; Liu, S.Q.; Cao, L.-P. 1,25-Dihydroxyvitamin D3 is a negative endocrine regulator of the renin-angiotensin system. J. Clin. Investig. 2002, 110, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Takeyama, K.-I.; Kitanaka, S.; Murayama, A.; Sekine, K.; Yoshizawa, T. In vivo function of VDR in gene expression-VDR knock-out mice. J. Steroid Biochem. Mol. Biol. 1999, 69, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Xiang, W.; Kong, J.; Chen, S.; Cao, L.-P.; Qiao, G.; Zheng, W.; Liu, W.; Li, X.; Gardner, D.G.; Li, Y.C. Cardiac hypertrophy in vitamin D receptor knockout mice: Role of the systemic and cardiac renin-angiotensin systems. Am. J. Physiol. Metab. 2005, 288, E125–E132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kong, J.; Deb, D.K.; Chang, A.; Li, Y.C. Vitamin D receptor attenuates renal fibrosis by suppressing the renin-angiotensin system. J. Am. Soc. Nephrol. 2010, 21, 966–973. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Li, Y.; Liu, Y. Paricalcitol attenuates renal interstitial fibrosis in obstructive nephropathy. J. Am. Soc. Nephrol. 2006, 17, 3382–3393. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Watanabe, H.; Fujimura, R.; Nishida, K.; Nakamura, R.; Oshiro, S.; Imafuku, T.; Komori, H.; Miyahisa, M.; Tanaka, M.; et al. A downstream molecule of 1,25-dihydroxyvitamin D3, alpha-1-acid glycoprotein, protects against mouse model of renal fibrosis. Sci. Rep. 2018, 8, 17329. [Google Scholar] [CrossRef]
- Wang, S.; Huang, S.; Liu, X.; He, Y.; Liu, Y. Paricalcitol Ameliorates Acute Kidney Injury in Mice by Suppressing Oxidative Stress and Inflammation via Nrf2/HO-1 Signaling. Int. J. Mol. Sci. 2023, 24, 969. [Google Scholar] [CrossRef]
- Shi, S.; Lei, S.; Tang, C.; Wang, K.; Xia, Z. Melatonin attenuates acute kidney ischemia/reperfusion injury in diabetic rats by activation of the SIRT1/Nrf2/HO-1 signaling pathway. Biosci. Rep. 2019, 39, BSR20181614. [Google Scholar] [CrossRef]
- Chuang, P.Y.; Cai, W.; Li, X.; Fang, L.; Xu, J.; Yacoub, R.; He, J.C.; Lee, K. Reduction in podocyte SIRT1 accelerates kidney injury in aging mice. Am. J. Physiol. Ren. Physiol. 2017, 313, F621–F628. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.; Kim, J.H.; Jung, M.H.; Jang, S.J.; Lee, T.W.; Jung, S.; Lee, S.; Jang, H.N.; Chang, S.-H.; Park, D.J. Paricalcitol Attenuates Contrast-Induced Acute Kidney Injury by Regulating Mitophagy and Senescence. Oxidative Med. Cell. Longev. 2020, 2020, 7627934. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.; Ayasolla, K.; Wen, H.; Lan, X.; Haque, S.; Saleem, M.A.; Malhotra, A.; Singhal, P.C. Vitamin D receptor deficit induces activation of renin angiotensin system via SIRT1 modulation in podocytes. Exp. Mol. Pathol. 2017, 102, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Duranton, F.; Rodriguez-Ortiz, M.E.; Duny, Y.; Rodriguez, M.; Daurès, J.-P.; Argilés, A. Vitamin D treatment and mortality in chronic kidney disease: A systematic review and meta-analysis. Am. J. Nephrol. 2013, 37, 239–248. [Google Scholar] [CrossRef]
- Cubbon, R.M.; Lowry, J.E.; Drozd, M.; Hall, M.; Gierula, J.; Paton, M.F.; Byrom, R.; Kearney, L.C.; Barth, J.H.; Kearney, M.T.; et al. Vitamin D deficiency is an independent predictor of mortality in patients with chronic heart failure. Eur. J. Nutr. 2018, 58, 2535–2543. [Google Scholar] [CrossRef]
- Thiele, K.; Cornelissen, A.; Florescu, R.; Kneizeh, K.; Brandenburg, V.M.; Witte, K.; Marx, N.; Schuh, A.; Stöhr, R. The Role of Vitamin D3 as an Independent Predicting Marker for One-Year Mortality in Patients with Acute Heart Failure. J. Clin. Med. 2022, 11, 2733. [Google Scholar] [CrossRef]
- Kuo, Y.-T.; Kuo, L.-K.; Chen, C.-W.; Yuan, K.-C.; Fu, C.-H.; Chiu, C.-T.; Yeh, Y.-C.; Liu, J.-H.; Shih, M.-C. Score-based prediction model for severe vitamin D deficiency in patients with critical illness: Development and validation. Crit. Care 2022, 26, 394. [Google Scholar] [CrossRef]
- Michael, W.; Couture, A.D.; Swedlund, M.; Hampton, A.; Eglash, A.; Schrager, S. An Evidence-Based Review of Vitamin D for Common and High-Mortality Conditions. J. Am. Board Fam. Med. 2022, 35, 1217–1229. [Google Scholar] [CrossRef]
- Wimalawansa, S.J. Vitamin D and cardiovascular diseases: Causality. J. Steroid Biochem. Mol. Biol. 2018, 175, 29–43. [Google Scholar] [CrossRef]
- Zheng, Z.; Shi, H.; Jia, J.; Li, D.; Lin, S. Vitamin D supplementation and mortality risk in chronic kidney disease: A meta-analysis of 20 observational studies. BMC Nephrol. 2013, 14, 199. [Google Scholar] [CrossRef] [PubMed]
- Kalantar-Zadeh, K.; Kuwae, N.; Regidor, D.; Kovesdy, C.; Kilpatrick, R.; Shinaberger, C.; McAllister, C.; Budoff, M.; Salusky, I.; Kopple, J. Survival predictability of time-varying indicators of bone disease in maintenance hemodialysis patients. Kidney Int. 2006, 70, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Wu, X. Effect of Vitamin D Supplementation on Vascular Function and Inflammation in Patients with Chronic Kidney Disease: A Controversial Issue. Ther. Apher. Dial. 2020, 24, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Dou, D.; Yang, B.; Gan, H.; Xie, D.; Lei, H.; Ye, N. Vitamin D supplementation for the improvement of vascular function in patients with chronic kidney disease: A meta-analysis of randomized controlled trials. Int. Urol. Nephrol. 2019, 51, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Lundwall, K.; Jacobson, S.H.; Jörneskog, G.; Spaak, J. Treating endothelial dysfunction with vitamin D in chronic kidney disease: A meta-analysis. BMC Nephrol. 2018, 19, 247. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, D.; Chitalia, N.; Ster, I.C.; Appelbaum, E.; Thadhani, R.; Kaski, J.C.; Goldsmith, D. Impact of vitamin D on cardiac structure and function in chronic kidney disease patients with hypovitaminosis D: A randomized controlled trial and meta-analysis. Eur. Heart J. Cardiovasc. Pharmacother. 2021, 7, 302–311. [Google Scholar] [CrossRef]
- Hu, X.; Shang, J.; Yuan, W.; Zhang, S.; Jiang, Y.; Zhao, B.; Duan, Y.; Xiao, J.; Zhao, Z. Effects of paricalcitol on cardiovascular outcomes and renal function in patients with chronic kidney disease: A meta-analysis. Herz 2017, 43, 518–528. [Google Scholar] [CrossRef]
- Christodoulou, M.; Aspray, T.J.; Schoenmakers, I. Vitamin D Supplementation for Patients with Chronic Kidney Disease: A Systematic Review and Meta-analyses of Trials Investigating the Response to Supplementation and an Overview of Guidelines. Calcif. Tissue Int. 2021, 109, 157–178. [Google Scholar] [CrossRef]
- Fernandez-Fernandez, B.; Ortiz, A. Paricalcitol and albuminuria: Tread carefully. Lancet Diabetes Endocrinol. 2018, 6, 3–5. [Google Scholar] [CrossRef]
- Ortiz, A.; Niño, M.D.S.; Rojas, J.; Egido, J. Paricalcitol for reduction of albuminuria in diabetes. Lancet 2011, 377, 635–636. [Google Scholar] [CrossRef]
- Su, G.; Liu, Z.; Qin, X.; Hong, X.; Liu, X.; Wen, Z.; Lindholm, B.; Carrero, J.-J.; Johnson, D.W.; Brusselaers, N.; et al. Vitamin D deficiency and treatment versus risk of infection in end-stage renal disease patients under dialysis: A systematic review and meta-analysis. Nephrol. Dial. Transplant. 2018, 34, 146–156. [Google Scholar] [CrossRef]
- Milajerdi, A.; Ostadmohammadi, V.; Amirjani, S.; Kolahdooz, F.; Asemi, Z. The effects of vitamin D treatment on glycemic control, serum lipid profiles, and C-reactive protein in patients with chronic kidney disease: A systematic review and meta-analysis of randomized controlled trials. Int. Urol. Nephrol. 2019, 51, 1567–1580. [Google Scholar] [CrossRef] [PubMed]
- Jayedi, A.; Soltani, S.; Shab-Bidar, S. Vitamin D status and all-cause mortality in patients with chronic kidney disease: A systematic review and dose-response meta-analysis. J. Clin. Endocrinol. Metab. 2017, 102, 2136–2145. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.J.; Zhu, S.M.; Tang, F.L.; Zhu, X.S.; Fan, Z.D.; Wang, G.L.; Jiang, Y.F.; Zhang, Y. Effects of vitamin D or its analogues on the mortality of patients with chronic kidney disease: An updated systematic review and meta-analysis. Eur. J. Clin. Nutr. 2017, 71, 683–693. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinelli, R.P.; Rayego-Mateos, S.; Alique, M.; Márquez-Expósito, L.; Tejedor-Santamaria, L.; Ortiz, A.; González-Parra, E.; Ruiz-Ortega, M. Vitamin D, Cellular Senescence and Chronic Kidney Diseases: What Is Missing in the Equation? Nutrients 2023, 15, 1349. https://doi.org/10.3390/nu15061349
Martinelli RP, Rayego-Mateos S, Alique M, Márquez-Expósito L, Tejedor-Santamaria L, Ortiz A, González-Parra E, Ruiz-Ortega M. Vitamin D, Cellular Senescence and Chronic Kidney Diseases: What Is Missing in the Equation? Nutrients. 2023; 15(6):1349. https://doi.org/10.3390/nu15061349
Chicago/Turabian StyleMartinelli, Romina P., Sandra Rayego-Mateos, Matilde Alique, Laura Márquez-Expósito, Lucia Tejedor-Santamaria, Alberto Ortiz, Emilio González-Parra, and Marta Ruiz-Ortega. 2023. "Vitamin D, Cellular Senescence and Chronic Kidney Diseases: What Is Missing in the Equation?" Nutrients 15, no. 6: 1349. https://doi.org/10.3390/nu15061349
APA StyleMartinelli, R. P., Rayego-Mateos, S., Alique, M., Márquez-Expósito, L., Tejedor-Santamaria, L., Ortiz, A., González-Parra, E., & Ruiz-Ortega, M. (2023). Vitamin D, Cellular Senescence and Chronic Kidney Diseases: What Is Missing in the Equation? Nutrients, 15(6), 1349. https://doi.org/10.3390/nu15061349