
Total Protein Intake in Patients with PKU: Adequacy Evaluation According to the European PKU Guidelines from 2017


 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
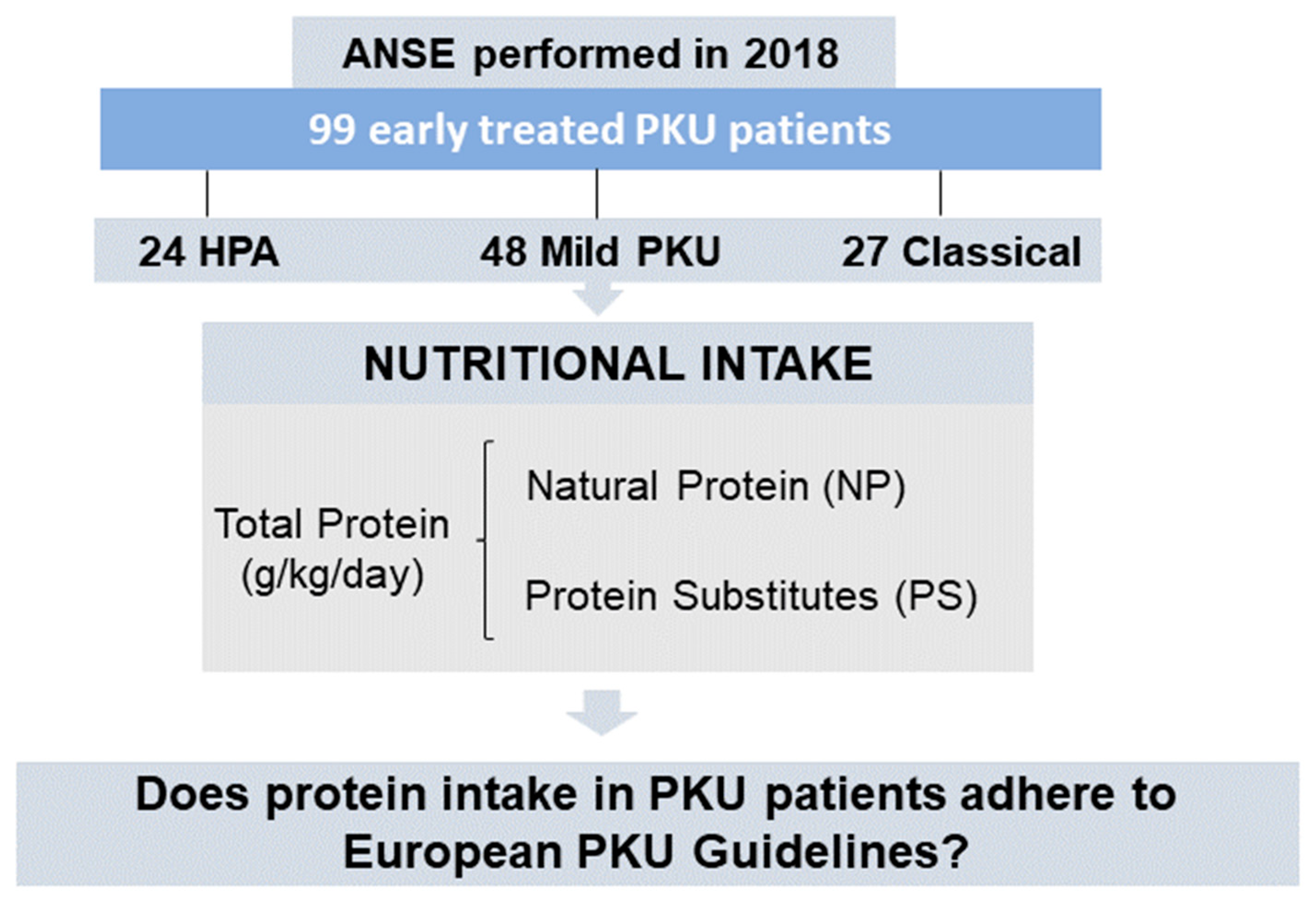
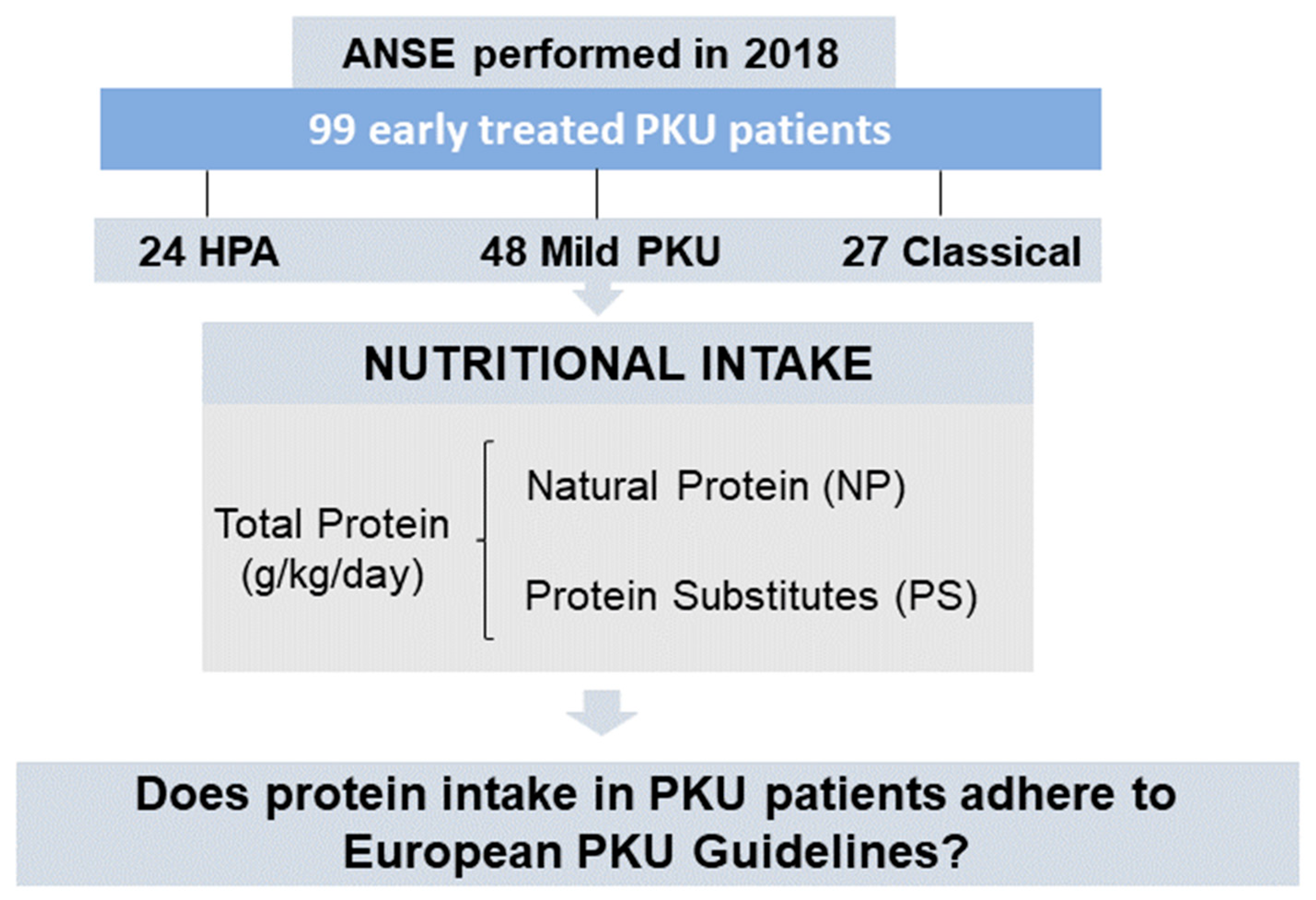
2.2. Participants
2.3. Eligibility
2.4. Data Collection
2.4.1. Anthropometry
2.4.2. Nutritional Intake
2.4.3. Metabolic Control
2.4.4. Ethical Statement
2.4.5. Statistical Analysis
3. Results
3.1. Subjects
3.2. Protein Intake
3.2.1. NP Intake Only
3.2.2. With PS Prescription
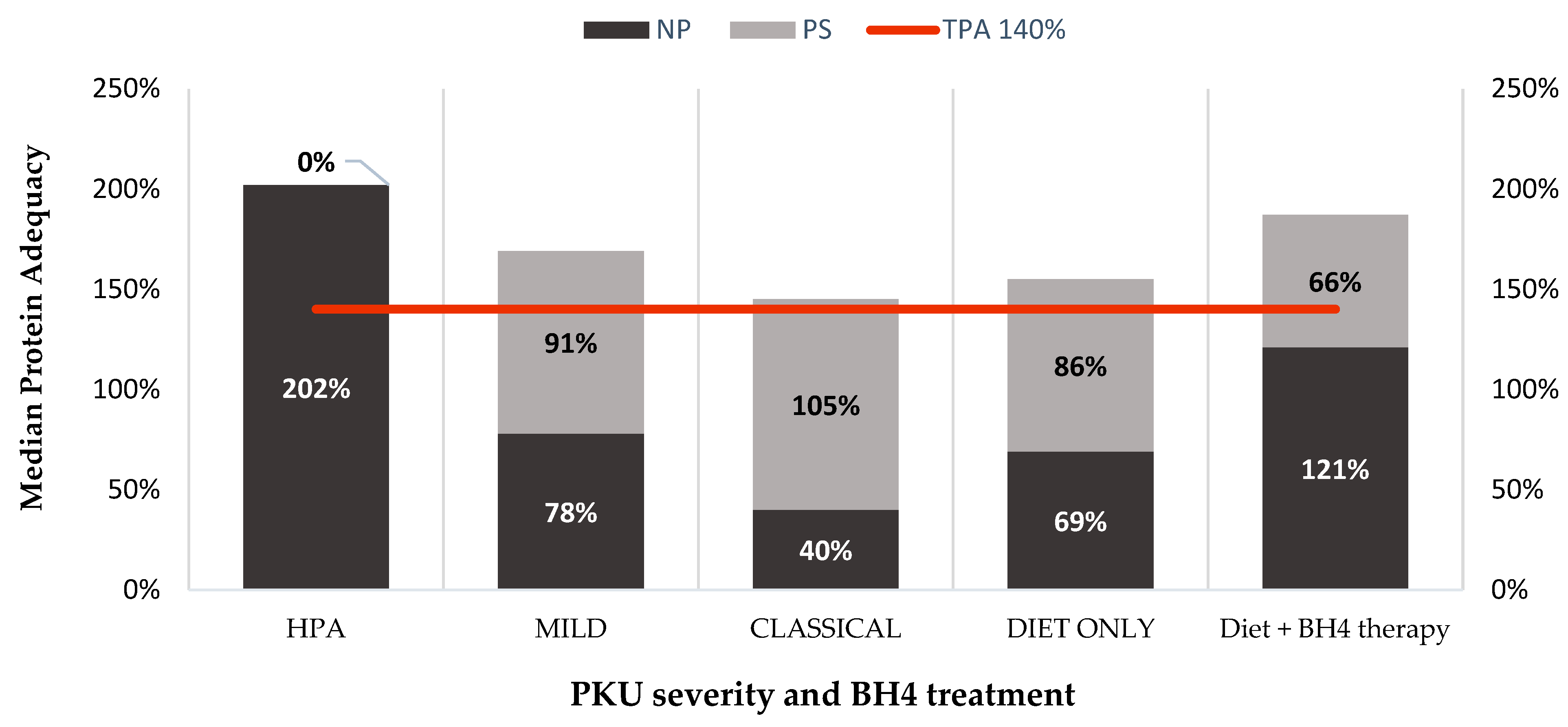
3.3. Protein Adequacy
3.4. Metabolic Control
3.4.1. Metabolic Control and Patients with TPA < 140%
3.4.2. Metabolic Control and Patients Taking PS
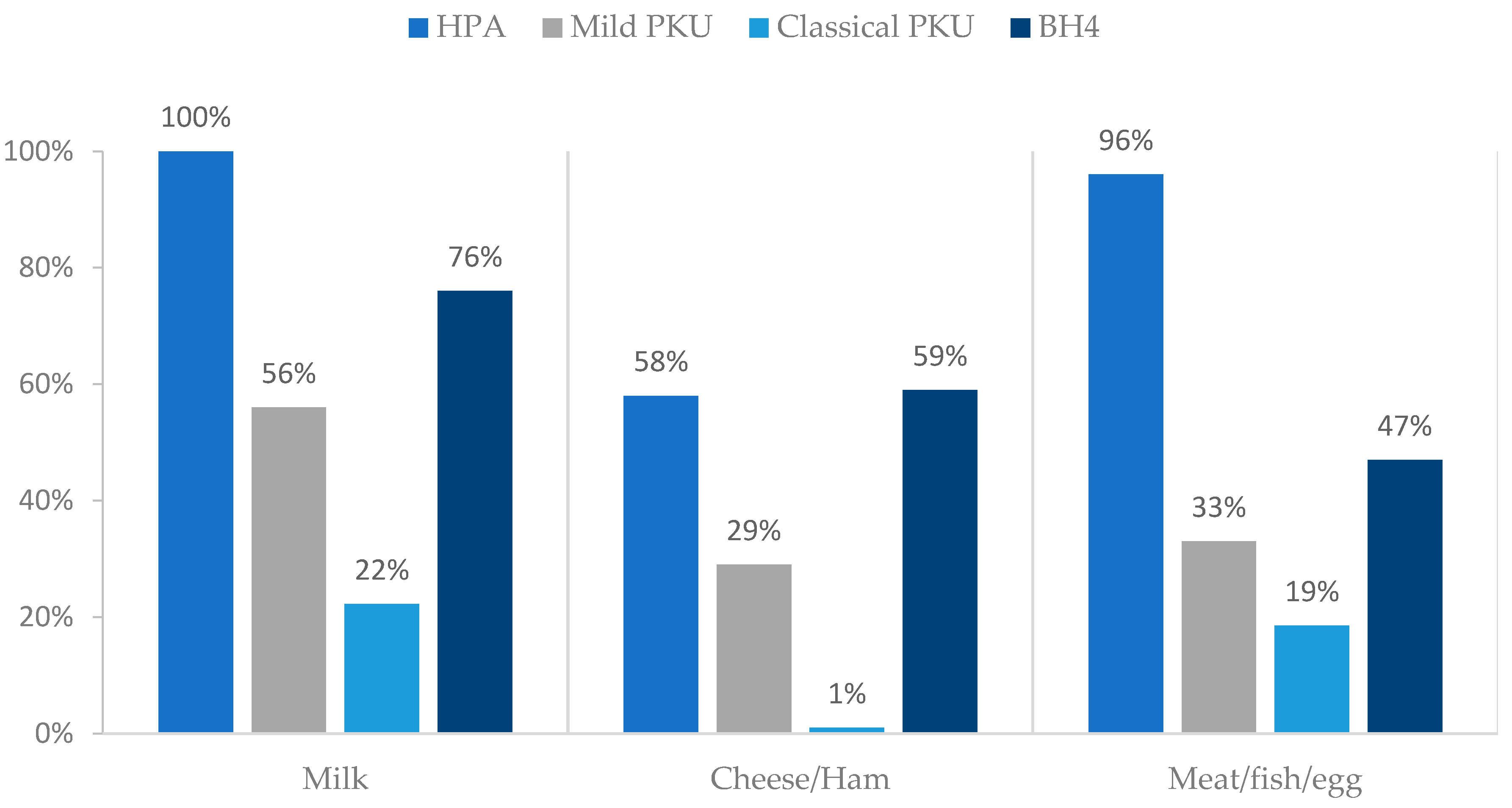
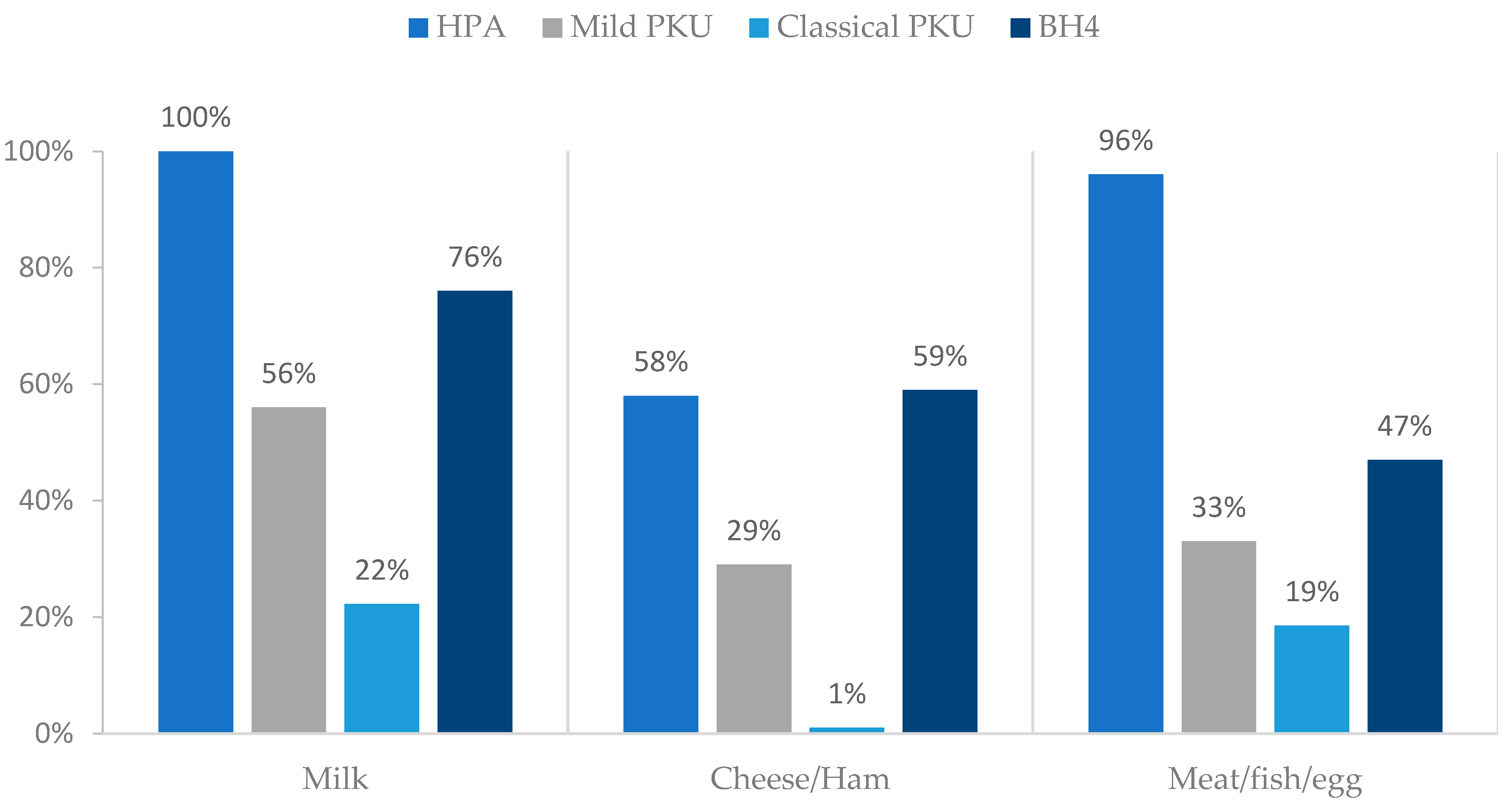
3.5. Natural Protein: Sources
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stone, W.L.; Basit, H.; Los, E. Phenylketonuria. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Qu, J.; Yang, T.; Wang, E.; Li, M.; Chen, C.; Ma, L.; Zhou, Y.; Cui, Y. Efficacy and safety of sapropterin dihydrochloride in patients with phenylketonuria: A meta-analysis of randomized controlled trials. Br. J. Clin. Pharmacol. 2019, 85, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Thiele, A.G.; Rohde, C.; Mütze, U.; Arelin, M.; Ceglarek, U.; Thiery, J.; Baerwald, C.; Kiess, W.; Beblo, S. The challenge of long-term tetrahydrobiopterin (BH4) therapy in phenylketonuria: Effects on metabolic control, nutritional habits and nutrient supply. Mol. Genet. Metab. Rep. 2015, 4, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Levy, H.; Amato, S.; Vockley, J.; Zori, R.; Dimmock, D.; Harding, C.O.; Bilder, D.A.; Weng, H.H.; Olbertz, J.; et al. Pegvaliase for the treatment of phenylketonuria: Results of a long-term phase 3 clinical trial program (PRISM). Mol. Genet. Metab. 2018, 124, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Hydery, T.; Coppenrath, V.A. A Comprehensive Review of Pegvaliase, an Enzyme Substitution Therapy for the Treatment of Phenylketonuria. Drug Target Insights 2019, 13, 1177392819857089. [Google Scholar] [CrossRef] [PubMed]
- Van Wegberg, A.M.J.; Macdonald, A.; Ahring, K.; BéLanger-Quintana, A.; Blau, N.; Bosch, A.M.; Burlina, A.; Campistol, J.; Feillet, F.; Giżewska, M.; et al. The complete European guidelines on phenylketonuria: Diagnosis and treatment. Orphanet J. Rare Dis. 2017, 12, 162. [Google Scholar] [CrossRef]
- Macleod, E.L.; Ney, D.M. Nutritional Management of Phenylketonuria. Ann. Nestle Eng. 2010, 68, 58–69. [Google Scholar] [CrossRef]
- Rocha, J.C.; MacDonald, A. Dietary intervention in the management of phenylketonuria: Current perspectives. Pediatric. Health Med. Ther. 2016, 7, 155–163. [Google Scholar] [CrossRef]
- Vockley, J.; Andersson, H.C.; Antshel, K.M.; Braverman, N.E.; Burton, B.K.; Frazier, D.M.; Mitchell, J.; Smith, W.E.; Thompson, B.H.; Berry, S.A.; et al. Phenylalanine hydroxylase deficiency: Diagnosis and management guideline. Genet. Med. 2014, 16, 188–200. [Google Scholar] [CrossRef]
- Onis, M.D.; Onyango, A.W.; Borghi, E.; Siyam, A.; Nishida, C.; Siekmann, J. Development of a WHO growth reference for school-aged children and adolescents. Bull World Health Organ 2007, 85, 660–667. [Google Scholar] [CrossRef]
- MacDonald, A.; Singh, R.H.; Rocha, J.C.; van Spronsen, F.J. Optimising amino acid absorption: Essential to improve nitrogen balance and metabolic control in phenylketonuria. Nutr. Res. Rev. 2019, 32, 70–78. [Google Scholar] [CrossRef]
- Firman, S.; Witard, O.C.; O’Keeffe, M.; Ramachandran, R. Dietary protein and protein substitute requirements in adults with phenylketonuria: A review of the clinical guidelines. Clin. Nutr. 2021, 40, 702–709. [Google Scholar] [CrossRef]
- Dangin, M.; Boirie, Y.; Garcia-Rodenas, C.; Gachon, P.; Fauquant, J.; Callier, P.; Ballèvre, O.; Beaufrère, B.; Dirks, M.L.; Wall, B.T.; et al. The digestion rate of protein is an independent regulating factor of postprandial protein retention. Am. J. Physiol.-Endocrinol. Metab. 2001, 280, E340–E348. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.; Truby, H.; Boneh, A. The relationship between dietary intake, growth and body composition in Phenylketonuria. Mol. Genet. Metab. 2017, 122, 36–42. [Google Scholar] [CrossRef]
- Pena, M.J.; de Almeida, M.F.; van Dam, E.; Ahring, K.; Bélanger-Quintana, A.; Dokoupil, K.; Gokmen-Ozel, H.; Lammardo, A.M.; MacDonald, A.; Robert, M.; et al. Protein substitutes for phenylketonuria in Europe: Access and nutritional composition. Eur. J. Clin. Nutr. 2016, 70, 785–789. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, A.; Ashmore, C.; Daly, A.; Pinto, A.; Evans, S. An Observational Study Evaluating the Introduction of a Prolonged-Release Protein Substitute to the Dietary Management of Children with Phenylketonuria. Nutrients 2020, 12, 2686. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.; Pinto, A.; Evans, S.; MacDonald, A. Glycomacropeptide in PKU-Does It Live Up to Its Potential? Nutrients 2022, 14, 807. [Google Scholar] [CrossRef] [PubMed]
- Van Calcar, S.C.; MacLeod, E.L.; Gleason, S.T.; Etzel, M.R.; Clayton, M.K.; Wolff, J.A.; Ney, D.M. Improved nutritional management of phenylketonuria by using a diet containing glycomacropeptide compared with amino acids. Am. J. Clin. Nutr. 2009, 89, 1068–1077. [Google Scholar] [CrossRef]
- WHO. Protein and amino acid requirements in human nutrition. World Health Organ. Tech. Rep. Ser. 2007, 935, 1–265. [Google Scholar]
- Rocha, J.C.; Cabral, A.; Osório, R.V.; Almeida, M.F. Consensus for the nutritional treatment of phenylketonuria. Acta Pediátrica Port. 2007, 38, 44–54. [Google Scholar]
- WHO. Child Growth Standards based on length/height, weight and age. Acta Paediatr. Suppl. 2006, 450, 76–85. [Google Scholar]
- WHO. Physical status: The Use of and Interpretation of Anthropometry, Report of a WHO Expert Committee; World Health Organization: Geneva, Switzerland, 1995. [Google Scholar]
- Gatel, F. Protein quality of legume seeds for non-ruminant animals: A literature review. Anim. Feed. Sci. Technol. 1994, 45, 317–348. [Google Scholar] [CrossRef]
- Ilgaz, F.; Marsaux, C.; Pinto, A.; Singh, R.; Rohde, C.; Karabulut, E.; Gökmen-Özel, H.; Kuhn, M.; MacDonald, A. Protein Substitute Requirements of Patients with Phenylketonuria on BH4 Treatment: A Systematic Review and Meta-Analysis. Nutrients 2021, 13, 1040. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, A.; Rocha, J.C.; van Rijn, M.; Feillet, F. Nutrition in phenylketonuria. Mol. Genet. Metab. 2011, 104, S10–S18. [Google Scholar] [CrossRef] [PubMed]
- Turki, A.; Ueda, K.; Cheng, B.; Giezen, A.; Salvarinova, R.; Stockler-Ipsiroglu, S.; Elango, R. The Indicator Amino Acid Oxidation Method with the Use of l-[1-13C]Leucine Suggests a Higher than Currently Recommended Protein Requirement in Children with Phenylketonuria. J. Nutr. 2017, 147, 211–217. [Google Scholar] [CrossRef]
- Daly, A.; Evans, S.; Pinto, A.; Ashmore, C.; MacDonald, A. Protein substitutes in PKU; their historical evolution. Nutrients 2021, 13, 484. [Google Scholar] [CrossRef]
- MacDonald, A.; Gokmen-Ozel, H.; van Rijn, M.; Burgard, P. The reality of dietary compliance in the management of phenylketonuria. J. Inherit. Metab. Dis. 2010, 33, 665–670. [Google Scholar] [CrossRef]
- Pena, M.J.; Pinto, A.; de Almeida, M.F.; Barbosa, C.d.S.; Ramos, P.C.; Rocha, S.; Guimas, A.; Ribeiro, R.; Martins, E.; Bandeira, A.; et al. Continuous use of glycomacropeptide in the nutritional management of patients with phenylketonuria: A clinical perspective. Orphanet J. Rare Dis. 2021, 16, 84. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.; Almeida, M.F.; Ramos, P.C.; Rocha, S.; Guimas, A.; Ribeiro, R.; Martins, E.; Bandeira, A.; MacDonald, A.; Rocha, J.C. Nutritional status in patients with phenylketonuria using glycomacropeptide as their major protein source. Eur. J. Clin. Nutr. 2017, 71, 1230–1234. [Google Scholar] [CrossRef]
- van Rijn, M.; Hoeksma, M.; Sauer, P.J.; Modderman, P.; Reijngoud, D.-J.; van Spronsen, F.J. Adult patients with well-controlled phenylketonuria tolerate incidental additional intake of phenylalanine. Ann. Nutr. Metab. 2011, 58, 94–100. [Google Scholar] [CrossRef]
- Pinto, A.; Almeida, M.F.; Donald, M.; Ramos, P.C.; Rocha, S.; Guimas, A.; Ribeiro, R.; Martins, E.; Bandeira, A.; Jackson, R.; et al. Over restriction of dietary protein allowance: The importance of ongoing reassessment of natural protein tolerance in phenylketonuria. Nutrients 2019, 11, 995. [Google Scholar] [CrossRef]
- MacLeod, E.L.; Gleason, S.T.; van Calcar, S.C.; Ney, D.M. Reassessment of phenylalanine tolerance in adults with phenylketonuria is needed as body mass changes. Mol. Genet. Metab. 2009, 98, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Gama, M.I.; Adam, S.; Adams, S.; Allen, H.; Ashmore, C.; Bailey, S.; Cochrane, B.; Dale, C.; Daly, A.; De Sousa, G.; et al. Suitability and Allocation of Protein-Containing Foods According to Protein Tolerance in PKU: A 2022 UK National Consensus. Nutrients 2022, 14, 4987. [Google Scholar] [CrossRef] [PubMed]
- Rocha, J.C.; MacDonald, A. Treatment options and dietary supplements for patients with phenylketonuria. Expert Opin. Orphan Drugs 2018, 6, 667–681. [Google Scholar] [CrossRef]
- Leuzzi, V.; Chiarotti, F.; Nardecchia, F.; Van Vliet, D.; Van Spronsen, F.J. Predictability and inconsistencies of cognitive outcome in patients with phenylketonuria and personalised therapy: The challenge for the future guidelines. J. Med. Genet. 2020, 57, 145–150. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Sample Size | Age | Gender | Disease Classification |
|---|---|---|---|
| N = 99 | 19.3 ± 8.2 years | Male: n = 53 | HPA PKU: n = 24 |
| <12 years: 7.2 ± 2.6 years | Female: n = 46 | Mild PKU: n = 48 | |
| ≥12 years: 22 ± 6.3 years | Classical PKU: n = 27 |
| Anthropometry Data | HPA | Mild PKU | Classical PKU | ||||
|---|---|---|---|---|---|---|---|
| <12 years (n = 6) | ≥12 years (n = 18) | <12 years (n = 12) | ≥12 years (n = 36) | <12 years | ≥12 years (n = 27) | ||
| Weight (kg) | Mean ± SD | 27.9 ± 11.1 | 60.6 ± 13.9 | 28.5 ± 11.5 | 62.9 ± 14.2 | -- | 62.0 ± 13.4 |
| z-score (<10 y) | 0.4 ± 0.64 | -- | 0.49 ± 1.27 | -- | -- | -- | |
| Height (cm) | Mean ± SD | 126 ± 17.1 | 165.9 ± 7.78 | 127.8 ± 18.5 | 164.9 ± 11.0 | -- | 163.3 ± 8.06 |
| z-score (<19 y) | 0.28 ± 0.88 | −0.01 ± 1.16 | 0.42 ± 0.95 | −0.02 ± 1.18 | -- | −1.01 ± 0.29 | |
| BMI (kg/m2) | Mean ± SD | 16.9 ± 2.41 | 21.9 ± 4.12 | 16.8 ± 2.17 | 22.9 ± 3.78 | -- | 22.3 ± 6.05 |
| z-score (<19 y) | 0.34 ± 0.8 | −0.1 ± 1.41 | 0.33 ± 1.11 | −0.18 ± 1.0 | -- | −0.18 ± 1.71 | |
| TP Intake (g/kg) Median [P25–P75] | PS Intake (g/kg) Median [P25–P75] | NP Intake (g/kg) Median [P25–P75] | ||
|---|---|---|---|---|
| Age | <12 years (n = 18) | 1.68 [1.42–2.03] | 0.9 [0.05–1.06] | 0.6 [0.49–1.65] |
| ≥12 years (n = 81) | 1.42 [1.17–1.71] | 0.67 [0–0.87] | 0.73 [0.39–1.4] | |
| PKU severity | HPA (n = 24) | 1.8 [1.42–2.08] | 0 [0–0] | 1.8 [1.42–2.08] |
| Mild (n = 48) | 1.53 [1.29–1.73] | 0.78 [0.26–1.02] | 0.68 [0.48–1.15] | |
| Classical (n = 27) | 1.28 [1.12–1.41] | 0.87 [0.72–0.98] | 0.34 [0.25–0.49] | |
| Type of treatment | Diet only (n = 82) | 1.43 [1.24–1.79] | 0.72 [0–0.98] | 0.6 [0.35–1.45] |
| Diet + BH4 therapy (n = 17) | 1.57 [1.29–1.71] | 0.5 [0.25–0.78] | 1 [0.79–1.2] |
| PKU Severity | TPA | Pediatrics (<12 Years) n = 18 | Adolescents (12–18 Years) n = 32 | Adults (>19 Years) (n = 49) | Total Percentage of Patients |
|---|---|---|---|---|---|
| HPA (N = 24) | TPA < 100% | 1/24 (4%) | 1/24 (4%) | 1/24 (4%) | 12% |
| TPA ≥ 100–120% | - | 1/24 (4%) | --- | 4% | |
| TPA ≥ 120–140% | - | --- | --- | ||
| TPA ≥ 140% | 5/24 (21%) | 11/24 (46%) | 4/24 (17%) | 84% | |
| MILD PKU (N = 48) | TPA < 100% | -- | --- | 1/48 (2%) | 2% |
| TPA ≥ 100–120% | -- | --- | 3/48 (6%) | 6% | |
| TPA ≥ 120–140% | --- | 2/48 (4%) | 2/48 (4%) | 8% | |
| TPA ≥ 140% | 12/48 (25%) | 11/48 (23%) | 17/48 (36%) | 74% | |
| CLASSICAL PKU (N = 27) | TPA < 100% | -- | --- | 2/27(7%) | 7% |
| TPA ≥ 100–120% | -- | --- | 1/27 (4%) | 4% | |
| TPA ≥ 120–140% | -- | 2/27(7%) | 5/27 (19%) | 26% | |
| TPA ≥ 140% | -- | 4/27 (15%) | 13/27 (48%) | 67% |
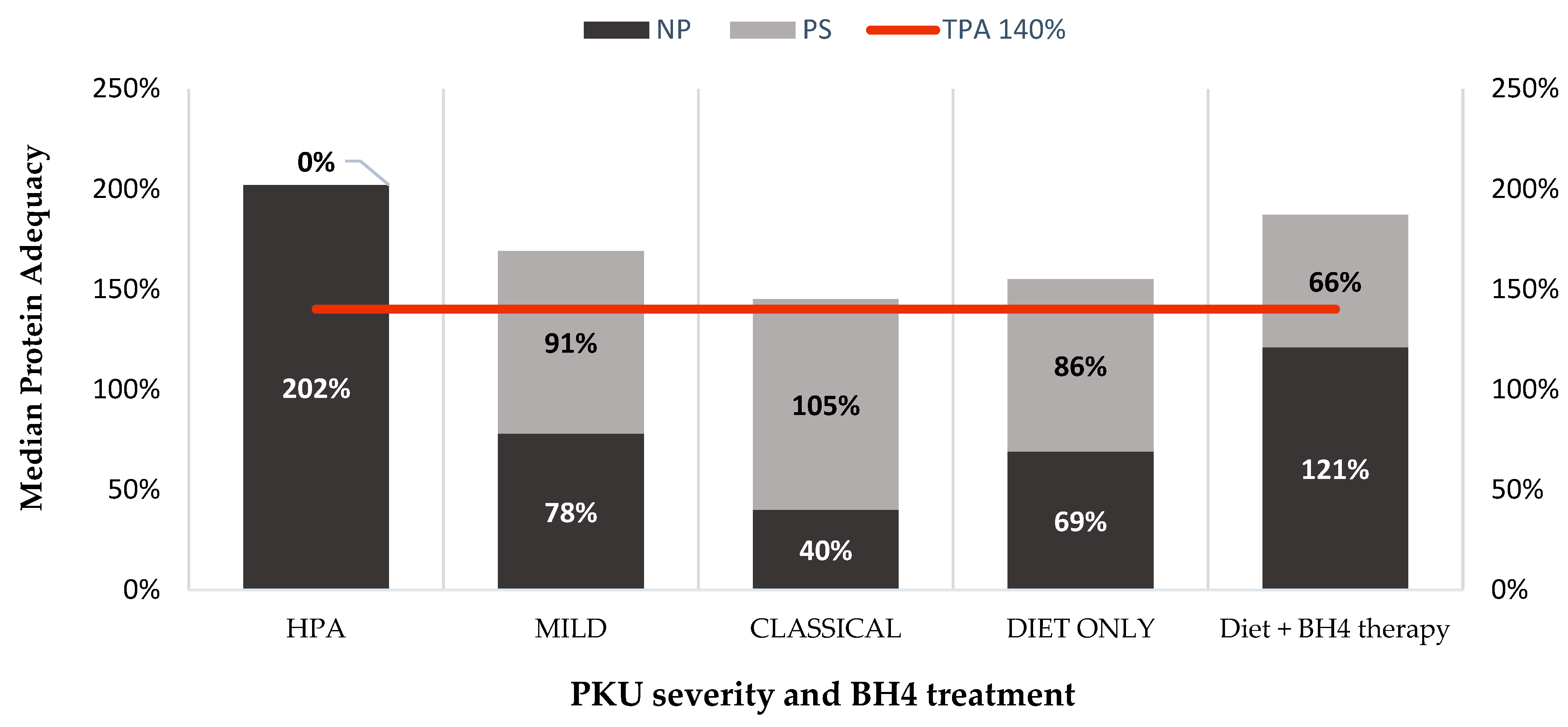
| TPA (%) Median [P25–P75] | PS Contribution (%) Median [P25–P75] | NP Contribution (%) Median [P25–P75] | ||
|---|---|---|---|---|
| Age | <12 years (n = 18) | 184 [158–223] | 96 [6–117] | 73 [55–183] |
| ≥12 years (n = 81) | 166 [140–201] | 80 [0–105] | 88 [17–164] | |
| PKU severity | HPA (n = 24) | 202 [165–230] | 0 [0–0] | 202 [165–234] |
| Mild (n = 48) | 178 [151–204] | 91 [31–117] | 78 [57–133] | |
| Classical (n = 27) | 154 [135–170] | 105 [86–117] | 40 [30–59] | |
| Type of treatment | Diet only (n = 82) | 168 [144–202] | 86 [0–116] | 69 [41–169] |
| Diet + BH4 (n = 17) | 182 [155–206] | 66 [30–90] | 121 [89–143] |
| Median µmol/L [P25–P75] <12 Years | Median µmol/L [P25–P75] ≥12 Years | Median % of Blood Phe Levels within Target Range | ||
|---|---|---|---|---|
| Age | <12 years (n = 18) | 294 [234–354] | -- | 65 [50–83.0] |
| ≥12 years (n = 81) | -- | 450 [270–600] | 61 [15.4–100] | |
| PKU severity | HPA (n = 24) | 247 [234–279] | 217 [186–249] | 100 [98.3–100] |
| Mild (n = 48) | 331 [257–360] | 462 [313–553] | 57 [22.7–92.8] | |
| Classical (n = 27) | -- | 610 [462–806] | 17 [0–55.9] | |
| Type of treatment | Diet only (n = 82) | 295 [236–344] | 462 [240–636] | 59 [15.5–100] |
| Diet + BH4 (n = 17) | 296 [266–325] | 421 [347–503] | 77 [50.9–94.1] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, M.; Almeida, M.F.; Barbosa, C.S.; Gama, M.I.; Peres, M.; Pinto, É.; MacDonald, A.; Rocha, J.C. Total Protein Intake in Patients with PKU: Adequacy Evaluation According to the European PKU Guidelines from 2017. Nutrients 2023, 15, 4883. https://doi.org/10.3390/nu15234883
Gomes M, Almeida MF, Barbosa CS, Gama MI, Peres M, Pinto É, MacDonald A, Rocha JC. Total Protein Intake in Patients with PKU: Adequacy Evaluation According to the European PKU Guidelines from 2017. Nutrients. 2023; 15(23):4883. https://doi.org/10.3390/nu15234883
Chicago/Turabian StyleGomes, Melanie, Manuela Ferreira Almeida, Catarina Sousa Barbosa, Maria Inês Gama, Maria Peres, Élia Pinto, Anita MacDonald, and Júlio César Rocha. 2023. "Total Protein Intake in Patients with PKU: Adequacy Evaluation According to the European PKU Guidelines from 2017" Nutrients 15, no. 23: 4883. https://doi.org/10.3390/nu15234883
APA StyleGomes, M., Almeida, M. F., Barbosa, C. S., Gama, M. I., Peres, M., Pinto, É., MacDonald, A., & Rocha, J. C. (2023). Total Protein Intake in Patients with PKU: Adequacy Evaluation According to the European PKU Guidelines from 2017. Nutrients, 15(23), 4883. https://doi.org/10.3390/nu15234883