Esculeogenin A, a Glycan from Tomato, Alleviates Nonalcoholic Fatty Liver Disease in Rats through Hypolipidemic, Antioxidant, and Anti-Inflammatory Effects

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Diets
2.2. Preparation of the ESGA
2.3. Experimental Design
2.4. Analysis of Blood Glucose and Insulin Levels and Assessment of HOMA-IR
2.5. Liver Function Tests (LFTs)
2.6. Markers of Adipose Tissue Function and Inflammation
2.7. Tissue and Fat Pad Collection
2.8. Lipid Analysis in the Serum and Liver
2.9. Analysis of Markers of Oxidative Stress and Inflammation in Liver Homogenates
2.10. Assessment of Nuclear Activities of Nrf2 and NF-κB
2.11. Real-Time Polymerase Chain Reaction (q-pCR)
2.12. Liver Histology
2.13. Statistical Analysis
3. Results
3.1. ESGA Does Not Affect Body and Fat Weight, Hyperglycemia, Hyperinsulinemia, and IR, but Significantly Reduces Liver Weights in HFD-Fed Rats
3.2. ESGA Attenuates the Increase in Liver Function Test Enzymes in the Serum of HFD-Fed Rats
3.3. ESGA Represses Serum and Hepatic Lipids but Not Stool Lipids in Control and HFD-Fed Rats
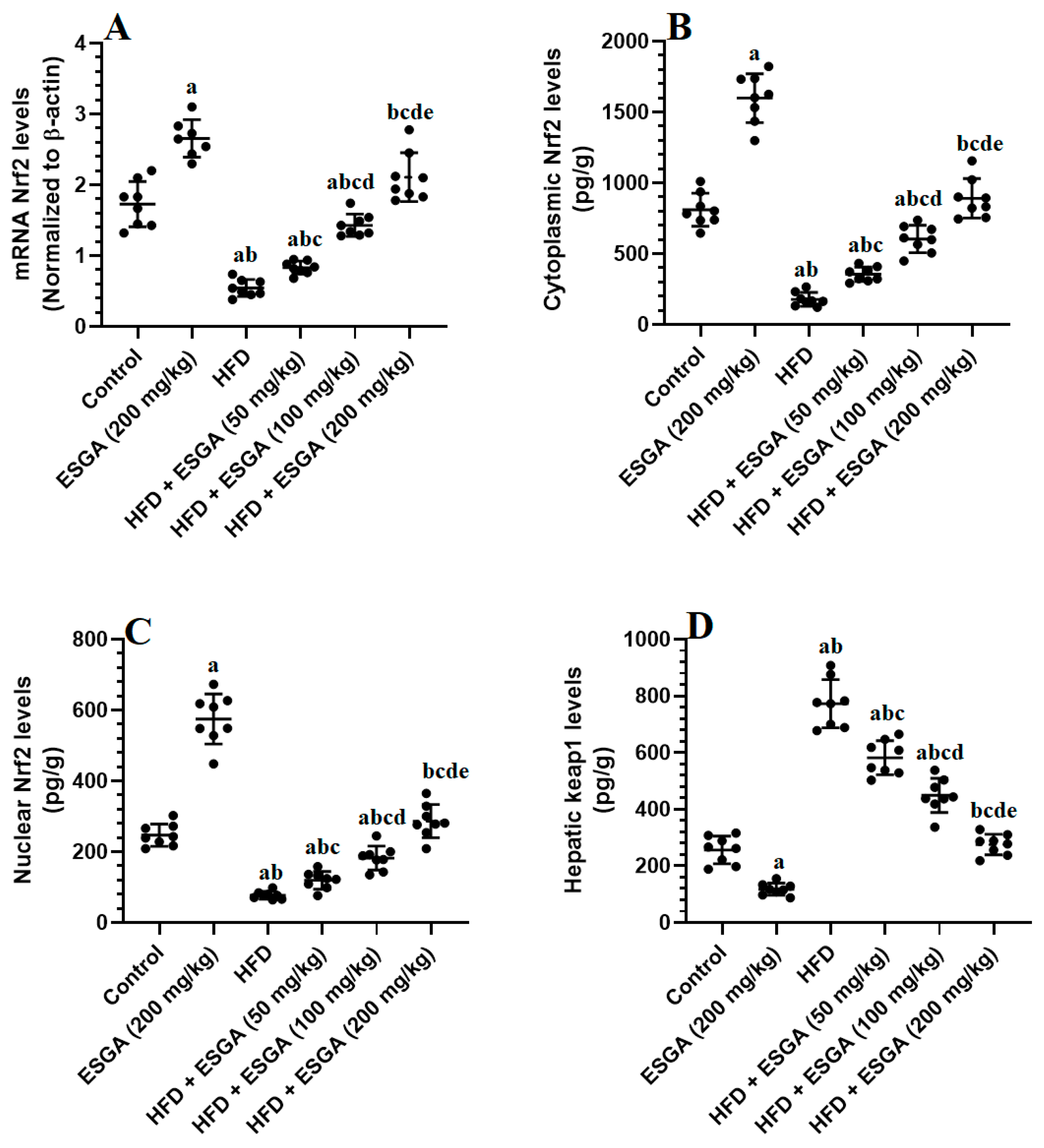
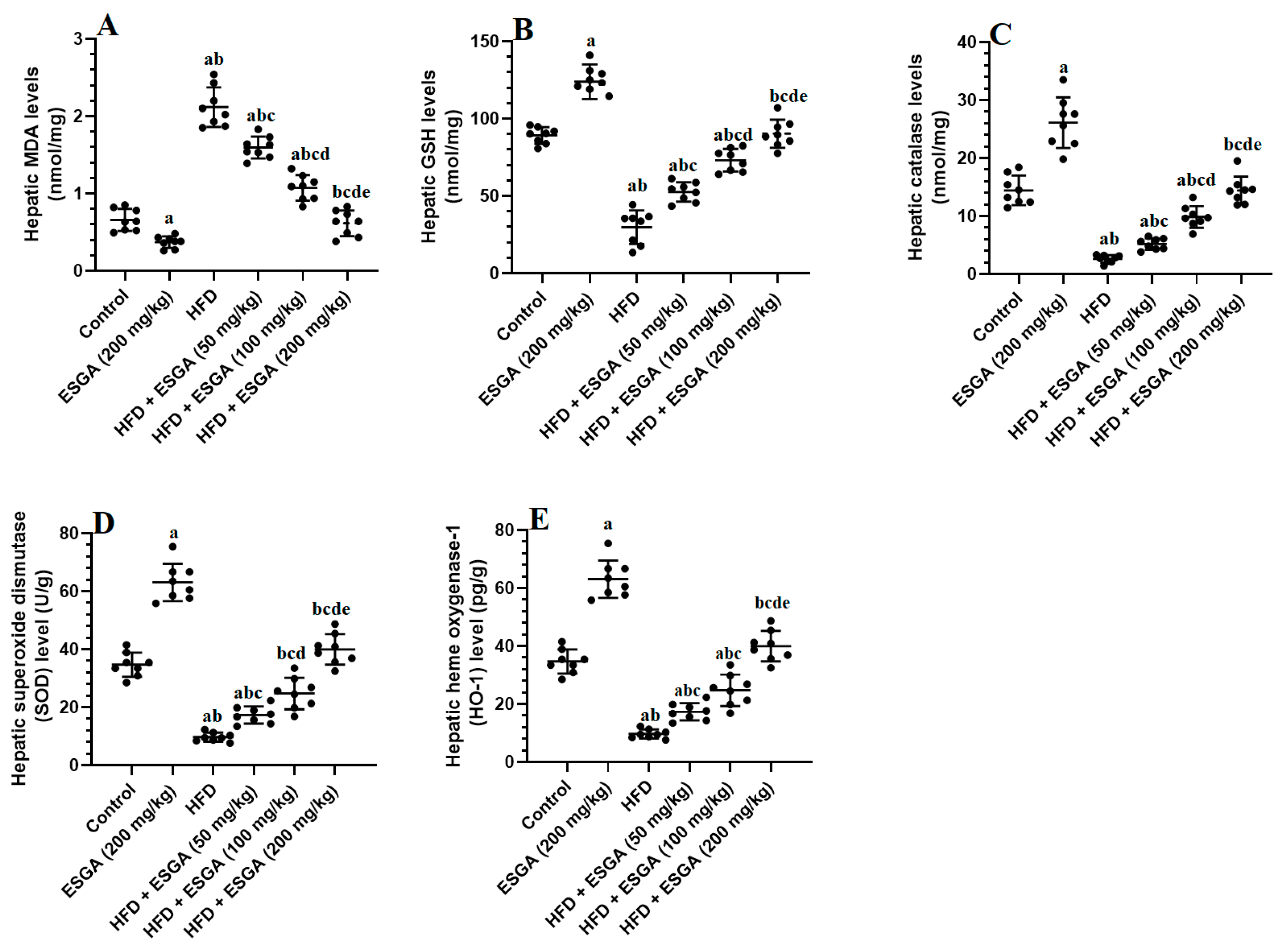
3.4. ESGA Stimulates the Nrf2/Antioxidant Axis in the Livers of Control and HFD-Fed Rats
3.5. ESGA Suppresses the IKK/NF-κB p65/Inflammatory Pathway of Nrf2/Antioxidant in the Livers of Control and HFD-Fed Rats
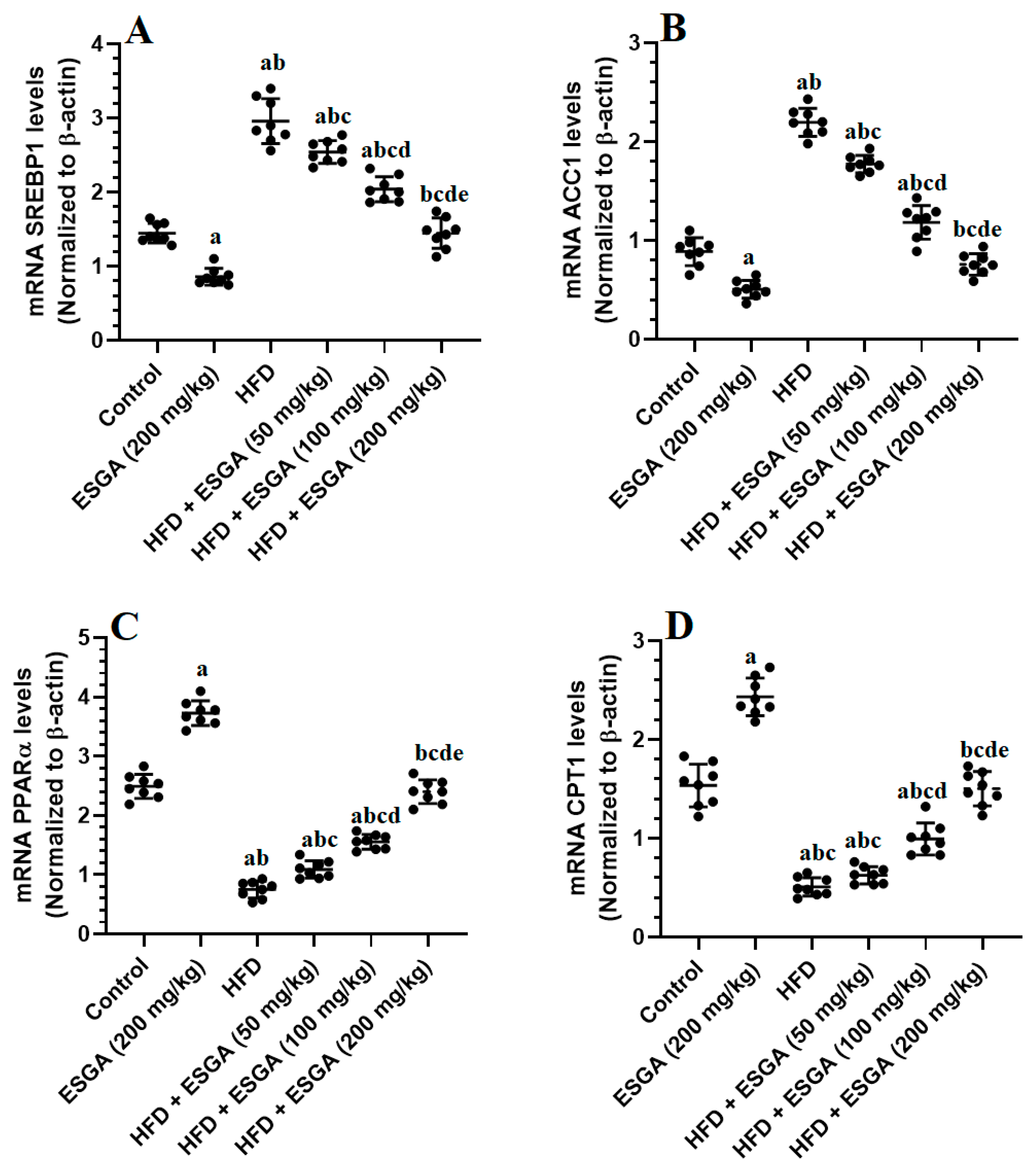
3.6. ESGA Suppresses the SREBP1/ACC Axis and Stimulates the PPARα/CPT-1 Axis in the Livers of the Control and HFD-Fed Rats
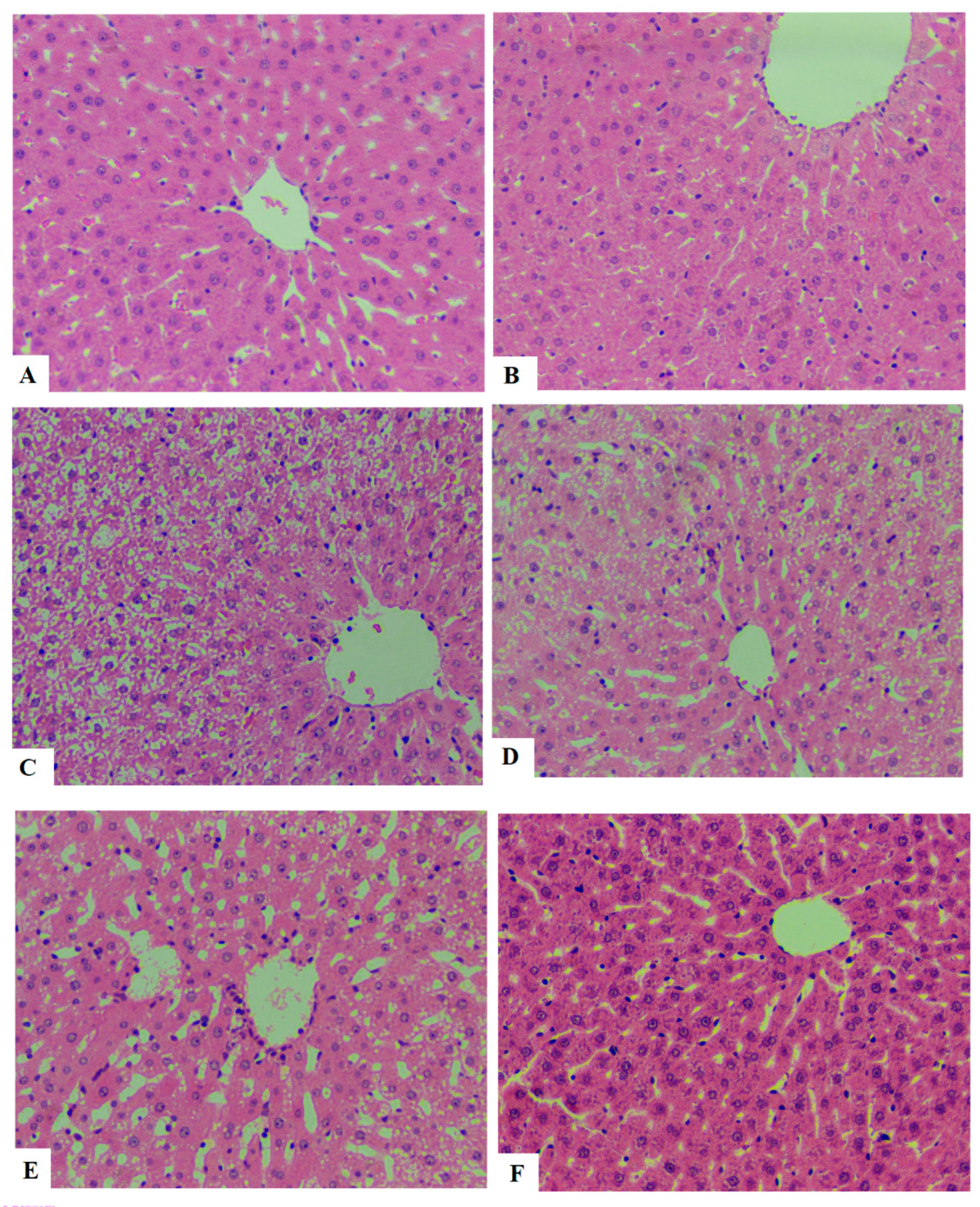
3.7. ESGA Improves Histological Features and Reduces Ballooning in the Hepatocytes of HFD-Fed Rats
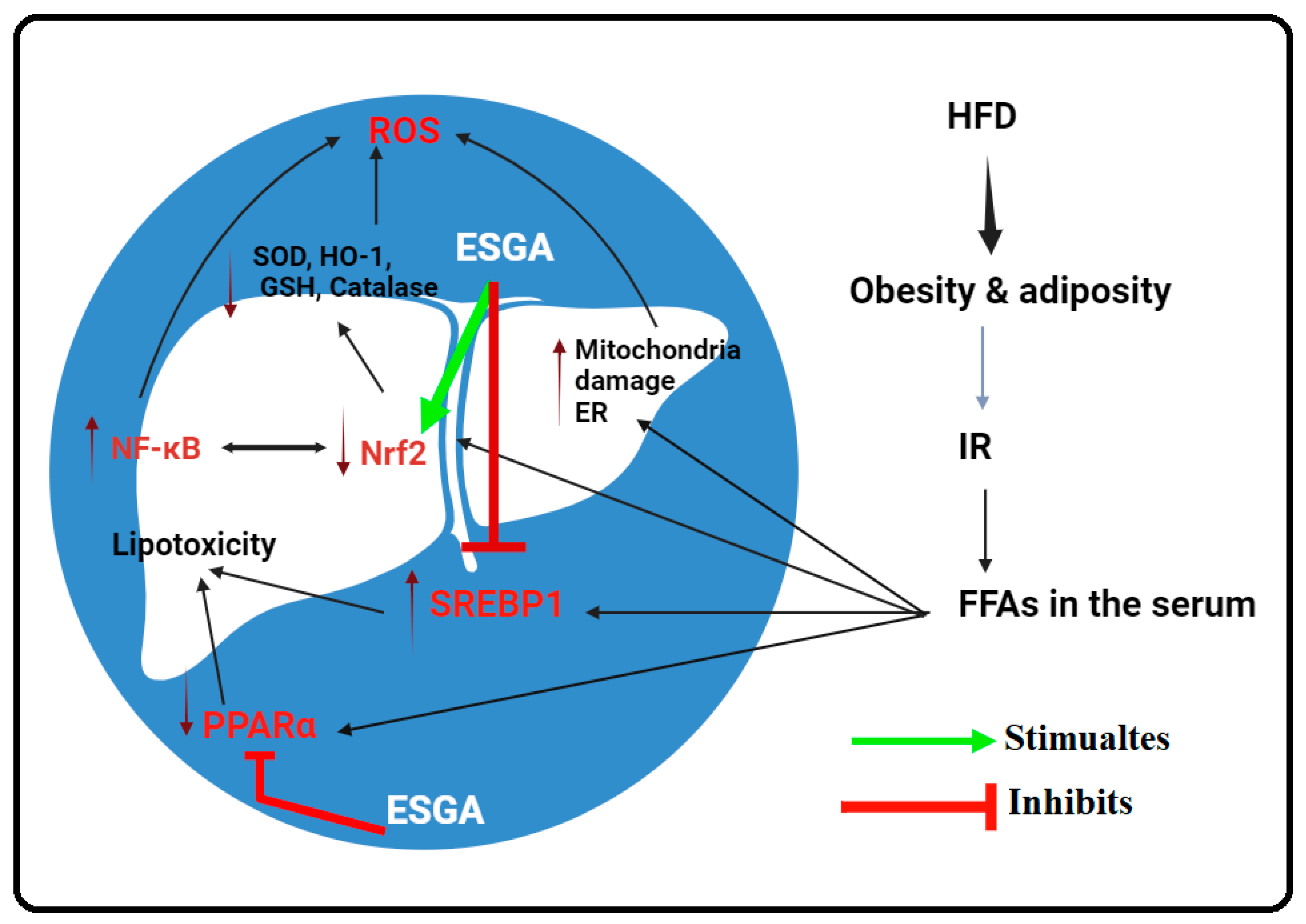
4. Discussion
5. Conclusions
6. Study Limitations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boutari, C.; Mantzoros, C.S. A 2022 update on the epidemiology of obesity and a call to action: As its twin COVID-19 pandemic appears to be receding, the obesity and dysmetabolism pandemic continues to rage on. Metabolism 2022, 133, 155217. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. World Health Organization Diabetes Country Profiles. Saudi Arabia. 2016. Available online: https://www.who.int/diabetes/country-profiles/sau_en.pdf (accessed on 31 May 2021).
- Althumiri, N.A.; Basyouni, M.H.; AlMousa, N.; AlJuwaysim, M.F.; Almubark, R.A.; BinDhim, N.F.; Alkhamaali, Z.; Alqahtani, S.A. Obesity in Saudi Arabia in 2020: Prevalence, Distribution, and Its Current Association with Various Health Conditions. Healthcare 2021, 9, 311. [Google Scholar] [CrossRef] [PubMed]
- Almubark, R.A.; Alqahtani, S.; Isnani, A.C.; Alqarni, A.; Shams, M.; Yahia, M.; Alfadda, A.A. Gender Differences in the Attitudes and Management of People with Obesity in Saudi Arabia: Data from the ACTION-IO Study. Risk Manag. Healthc Policy 2022, 15, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.G.; Kim, S.U.; Wong, V.W. New trends on obesity and NAFLD in Asia. J. Hepatol. 2017, 67, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Pouwels, S.; Sakran, N.; Graham, Y.; Leal, A.; Pintar, T.; Yang, W.; Kassir, R.; Singhal, R.; Mahawar, K.; Ramnarain, D. Non-alcoholic fatty liver disease (NAFLD): A review of pathophysiology, clinical management and effects of weight loss. BMC Endocr. Disord. 2022, 22, 63. [Google Scholar] [CrossRef] [PubMed]
- Cai, G.S.; Su, H.; Zhang, J. Protective effect of probiotics in patients with non-alcoholic fatty liver disease. Medicine 2020, 99, e21464. [Google Scholar] [CrossRef] [PubMed]
- Godoy-Matos, A.F.; Silva Júnior, W.S.; Valerio, C.M. NAFLD as a continuum: From obesity to metabolic syndrome and diabetes. Diabetol. Metab. Syndr. 2020, 12, 60. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Jinjuvadia, R.; Antaki, F.; Lohia, P.; Liangpunsakul, S. The Association between Nonalcoholic Fatty Liver Disease and Metabolic Abnormalities in The United States Population. J. Clin. Gastroenterol. 2017, 51, 160–166. [Google Scholar] [CrossRef]
- Virtue, S.; Vidal-Puig, A. It’s not how fat you are, it’s what you do with it that counts. PLoS Biol. 2008, 6, e237. [Google Scholar] [CrossRef]
- Rinaldi, L.; Pafundi, P.C.; Galiero, R.; Caturano, A.; Morone, M.V.; Silvestri, C.; Giordano, M.; Salvatore, T.; Sasso, F.C. Mechanisms of Non-Alcoholic Fatty Liver Disease in the Metabolic Syndrome. A Narrative Review. Antioxidants 2021, 10, 270. [Google Scholar] [CrossRef]
- Linden, A.G.; Li, S.; Choi, H.Y.; Fang, F.; Fukasawa, M.; Uyeda, K.; Hammer, R.E.; Horton, J.D.; Engelking, L.J.; Liang, G. Interplay between ChREBP and SREBP-1c coordinates postprandial glycolysis and lipogenesis in livers of mice. J. Lipid. Res. 2018, 59, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Palma, R.; Pronio, A.; Romeo, M.; Scognamiglio, F.; Ventriglia, L.; Ormando, V.M.; Lamazza, A.; Pontone, S.; Federico, A.; Dallio, M. The Role of Insulin Resistance in Fueling NAFLD Pathogenesis: From Molecular Mechanisms to Clinical Implications. J. Clin. Med. 2022, 11, 3649. [Google Scholar] [CrossRef] [PubMed]
- Solano-Urrusquieta, A.; Morales-González, J.A.; Castro-Narro, G.E.; Cerda-Reyes, E.; Flores-Rangel, P.D.; Fierros-Oceguera, R. NRF-2 and nonalcoholic fatty liver disease. Ann Hepatol. 2020, 19, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Ghareghomi, S.; Habibi-Rezaei, M.; Arese, M.; Saso, L.; Moosavi-Movahedi, A.A. Nrf2 Modulation in Breast Cancer. Biomedicines 2022, 10, 2668. [Google Scholar] [CrossRef] [PubMed]
- Tossetta, G.; Fantone, S.; Piani, F.; Crescimanno, C.; Ciavattini, A.; Giannubilo, S.R.; Marzioni, D. Modulation of NRF2/KEAP1 Signaling in Preeclampsia. Cells 2023, 12, 1545. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [PubMed]
- Ferramosca, A.; Di Giacomo, M.; Zara, V. Antioxidant dietary approach in treatment of fatty liver: New insights and updates. World J. Gastroenterol. 2017, 23, 4146–4157. [Google Scholar] [CrossRef] [PubMed]
- Doumas, M.; Imprialos, K.; Dimakopoulou, A.; Stavropoulos, K.; Binas, A.; Athyros, V.G. The Role of Statins in the Management of Nonalcoholic Fatty Liver Disease. Curr. Pharm. Des. 2018, 24, 4587–4592. [Google Scholar] [CrossRef]
- Fock, K.M.; Khoo, J. Diet and exercise in management of obesity and overweight. J. Gastroenterol. Hepatol. 2013, 28 (Suppl. S4), 59–63. [Google Scholar] [CrossRef]
- Tyrovolas, S.; Panagiotakos, D.B.; Georgousopoulou, E.N.; Chrysohoou, C.; Skoumas, J.; Pan, W.; Tousoulis, D.; Pitsavos, C. The anti-inflammatory potential of diet and nonalcoholic fatty liver disease: The ATTICA study. Therap. Adv. Gastroenterol. 2019, 12, 1756284819858039. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.; Jin, L.; Qin, X.; He, B. Natural flavonoids: Potential therapeutic strategies for non-alcoholic fatty liver disease. Front. Pharmacol. 2022, 13, 1005312. [Google Scholar] [CrossRef] [PubMed]
- Collins, E.J.; Bowyer, C.; Tsouza, A.; Chopra, M. Tomatoes: An Extensive Review of the Associated Health Impacts of Tomatoes and Factors That Can Affect Their Cultivation. Biology 2022, 11, 239. [Google Scholar] [CrossRef] [PubMed]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants 2021, 10, 174. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhang, L.; Liu, J.; Lu, F.; Wang, L.; Chen, Y.; Li, D. Hypoglycemic effects of esculeoside A are mediated via activation of AMPK and upregulation of IRS-1. BMC Complement. Altern. Med. 2019, 19, 136. [Google Scholar] [CrossRef] [PubMed]
- Nohara, T.; Ono, M.; Ikeda, T.; Fujiwara, Y.; El-Aasr, M. The tomato saponin, esculeoside A. J. Nat. Prod. 2010, 73, 1734–1741. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Kiyota, N.; Hori, M.; Matsushita, S.; Iijima, Y.; Aoki, K.; Shibata, D.; Takeya, M.; Ikeda, T.; Nohara, T.; et al. Esculeogenin A, a new tomato sapogenol, ameliorates hyperlipidemia and atherosclerosis in ApoE-deficient mice by inhibiting ACAT. Arter. Thromb. Vasc. Biol. 2007, 27, 2400–2406. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Kiyota, N.; Tsurushima, K.; Yoshitomi, M.; Horlad, H.; Ikeda, T.; Nohara, T.; Takeya, M.; Nagai, R. Tomatidine, a tomato sapogenol, ameliorates hyperlipidemia and atherosclerosis in apoE-deficient mice by inhibiting acyl-CoA:cholesterol acyl-transferase (ACAT). J. Agric. Food Chem. 2012, 60, 2472–2479. [Google Scholar] [CrossRef]
- Zhou, J.R.; Kanda, Y.; Tanaka, A.; Manabe, H.; Nohara, T.; Yokomizo, K. Anti-hyaluronidase Activity in Vitro and Amelioration of Mouse Experimental Dermatitis by Tomato Saponin, Esculeoside A. J. Agric. Food Chem. 2016, 64, 403–408. [Google Scholar] [CrossRef]
- Fan, S.; Zhang, Y.; Sun, Q.; Yu, L.; Li, M.; Zheng, B.; Wu, X.; Yang, B.; Li, Y.; Huang, C. Extract of okra lowers blood glucose and serum lipids in high-fat diet-induced obese C57BL/6 mice. J. Nutr. Biochem. 2014, 25, 702–709. [Google Scholar] [CrossRef]
- Li, Y.; Xu, J.; Lu, Y.; Bian, H.; Yang, L.; Wu, H.; Zhang, X.; Zhang, B.; Xiong, M.; Chang, Y.; et al. DRAK2 aggravates nonalcoholic fatty liver disease progression through SRSF6-associated RNA alternative splicing. Cell Metab. 2021, 33, 2004–2020.e9. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Y.; Ming, Y.; Song, Y.; Zhang, J.; Chen, X.; Zeng, M.; Mao, Y. S100A9: A Potential Biomarker for the Progression of Non-Alcoholic Fatty Liver Disease and the Diagnosis of Non-Alcoholic Steatohepatitis. PLoS ONE 2015, 10, e0127352. [Google Scholar] [CrossRef] [PubMed]
- Akbari, G.; Mard, S.A.; Savari, F.; Barati, B.; Sameri, M.J. Characterization of diet based nonalcoholic fatty liver disease/nonalcoholic steatohepatitis in rodent models: Histological and biochemical outcomes. Histol. Histopathol. 2022, 37, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Alwadani, A.H.; Almasri, S.A.; Aloud, A.A.; Albadr, N.A.; Alshammari, G.M.; Yahya, M.A. The Synergistic Protective Effect of γ-Oryzanol (OZ) and N-Acetylcysteine (NAC) against Experimentally Induced NAFLD in Rats Entails Hypoglycemic, Antioxidant, and PPARα Stimulatory Effects. Nutrients 2022, 15, 106. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Felemban, A.H.; Alshammari, G.M.; Yagoub, A.E.A.; Al-Harbi, L.N.; Alhussain, M.H.; Yahya, M.A. Activation of AMPK Entails the Protective Effect of Royal Jelly against High-Fat-Diet-Induced Hyperglycemia, Hyperlipidemia, and Non-Alcoholic Fatty Liver Disease in Rats. Nutrients 2023, 15, 1471. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.; Zhou, X.; Xu, J.; Gao, L. Rodent Models of Nonalcoholic Fatty Liver Disease. Digestion 2020, 101, 522–535. [Google Scholar] [CrossRef]
- Jacobs, A.; Warda, A.S.; Verbeek, J.; Cassiman, D.; Spincemaille, P. An Overview of Mouse Models of Nonalcoholic Steatohepatitis: From Past to Present. Curr. Protoc. Mouse Biol. 2016, 6, 185–200. [Google Scholar] [CrossRef]
- Radhakrishnan, S.; Ke, J.Y.; Pellizzon, M.A. Targeted Nutrient Modifications in Purified Diets Differentially Affect Nonalcoholic Fatty Liver Disease and Metabolic Disease Development in Rodent Models. Curr. Dev. Nutr. 2020, 4, nzaa078. [Google Scholar] [CrossRef]
- Neuman, M.G.; Cohen, L.B.; Nanau, R.M. Biomarkers in nonalcoholic fatty liver disease. Can. J. Gastroenterol. Hepatol. 2014, 28, 607–618. [Google Scholar] [CrossRef]
- Soret, P.A.; Magusto, J.; Housset, C.; Gautheron, J. In Vitro and In Vivo Models of Non-Alcoholic Fatty Liver Disease: A Critical Appraisal. J. Clin. Med. 2020, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Kanuri, G.; Bergheim, I. In vitro and in vivo models of non-alcoholic fatty liver disease (NAFLD). Int. J. Mol. Sci. 2013, 14, 11963–11980. [Google Scholar] [CrossRef] [PubMed]
- Kořínková, L.; Pražienková, V.; Černá, L.; Karnošová, A.; Železná, B.; Kuneš, J.; Maletínská, L. Pathophysiology of NAFLD and NASH in Experimental Models: The Role of Food Intake Regulating Peptides. Front. Endocrinol. 2020, 11, 597583. [Google Scholar] [CrossRef] [PubMed]
- Santhekadur, P.K.; Kumar, D.P.; Sanyal, A.J. Preclinical models of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Li, J.; Lu, C.; Wang, J.; Ge, J.; Huang, Y.; Zhang, L.; Wang, Y. High-fat emulsion-induced rat model of nonalcoholic steatohepatitis. Life Sci. 2006, 79, 1100–1107. [Google Scholar] [CrossRef] [PubMed]
- Larter, C.Z.; Yeh, M.M. Animal models of NASH: Getting both pathology and metabolic context right. J. Gastroenterol. Hepatol. 2008, 23, 1635–1648. [Google Scholar] [CrossRef] [PubMed]
- Eng, J.M.; Estall, J.L. Diet-Induced Models of Non-Alcoholic Fatty Liver Disease: Food for Thought on Sugar, Fat, and Cholesterol. Cells 2021, 10, 1805. [Google Scholar] [CrossRef]
- Koch, C.E.; Lowe, C.; Pretz, D.; Steger, J.; Williams, L.M.; Tups, A. High-fat diet induces leptin resistance in leptin-deficient mice. J. Neuroendocrinol. 2014, 26, 58–67. [Google Scholar] [CrossRef]
- Kitade, H.; Chen, G.; Ni, Y.; Ota, T. Nonalcoholic Fatty Liver Disease and Insulin Resistance: New Insights and Potential New Treatments. Nutrients 2017, 9, 387. [Google Scholar] [CrossRef]
- Guerrero-Romero, F.; Simental-Mendía, L.E.; Guerra Rosas, M.I.; Sayago-Monreal, V.I.; Morales Castro, J.; Gamboa-Gómez, C.I. Hypoglycemic and antioxidant effects of green tomato (Physalis ixocarpa Brot.) calyxes’ extracts. J. Food Biochem. 2021, 45, e13678. [Google Scholar] [CrossRef]
- Zhang, Q.Q.; Lu, L.G. Nonalcoholic Fatty Liver Disease: Dyslipidemia, Risk for Cardiovascular Complications, and Treatment Strategy. J. Clin. Transl. Hepatol. 2015, 3, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Lang, S.; Goeser, T.; Demir, M.; Steffen, H.M.; Kasper, P. Management of Dyslipidemia in Patients with Non-Alcoholic Fatty Liver Disease. Curr. Atheroscler. Rep. 2022, 24, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Chatrath, H.; Vuppalanchi, R.; Chalasani, N. Dyslipidemia in patients with nonalcoholic fatty liver disease. Semin. Liver Dis. 2012, 32, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Viscarra, J.; Kim, S.J.; Sul, H.S. Transcriptional regulation of hepatic lipogenesis. Nat. Rev. Mol. Cell Biol. 2015, 16, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Ramatchandirin, B.; Pearah, A.; He, L. Regulation of Liver Glucose and Lipid Metabolism by Transcriptional Factors and Coactivators. Life 2023, 13, 515. [Google Scholar] [CrossRef] [PubMed]
- Shimano, H.; Sato, R. SREBP-regulated lipid metabolism: Convergent physiology—Divergent pathophysiology. Nat. Rev. Endocrinol. 2017, 13, 710–730. [Google Scholar] [CrossRef] [PubMed]
- Bitter, A.; Nüssler, A.K.; Thasler, W.E.; Klein, K.; Zanger, U.M.; Schwab, M.; Burk, O. Human sterol regulatory element-binding protein 1a contributes significantly to hepatic lipogenic gene expression. Cell Physiol. Biochem. 2015, 35, 803–815. [Google Scholar] [CrossRef]
- Horton, J.D.; Shimomura, I.; Ikemoto, S.; Bashmakov, Y.; Hammer, R.E. Overexpression of sterol regulatory element-binding protein-1a in mouse adipose tissue produces adipocyte hypertrophy, increased fatty acid secretion, and fatty liver. J. Biol. Chem. 2003, 278, 36652–36660. [Google Scholar] [CrossRef]
- Yang, B.; Sun, J.; Liang, S.; Wu, P.; Lv, R.; He, Y.; Li, D.; Sun, W.; Song, X. Prediction of Srebp-1 as a Key Target of Qing Gan San Against MAFLD in Rats via RNA-Sequencing Profile Analysis. Front. Pharmacol. 2021, 12, 680081. [Google Scholar] [CrossRef]
- Kwak, J.H.; Lee, H.J.; Jeong, S.T.; Lee, J.Y.; Lee, M.; Paik, J.K. Effect of fermented Rhus verniciflua stokes extract on liver function parameters in healthy Korean adults: A double-blind randomized controlled trial. Trials 2021, 22, 830. [Google Scholar] [CrossRef]
- Brocker, C.N.; Patel, D.P.; Velenosi, T.J.; Kim, D.; Yan, T.; Yue, J.; Li, G.; Krausz, K.W.; Gonzalez, F.J. Extrahepatic PPARα modulates fatty acid oxidation and attenuates fasting-induced hepatosteatosis in mice. J. Lipid. Res. 2018, 59, 2140–2152. [Google Scholar] [CrossRef] [PubMed]
- Delerive, P.; De Bosscher, K.; Besnard, S.; Vanden Berghe, W.; Peters, J.M.; Gonzalez, F.J.; Fruchart, J.C.; Tedgui, A.; Haegeman, G.; Staels, B. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J. Biol. Chem. 1999, 274, 32048–32054. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Chaudhary, A.; Sethi, S. Oxidized omega-3 fatty acids inhibit NF-kappaB activation via a PPARalpha-dependent pathway. Arter. Thromb. Vasc. Biol. 2004, 24, 1621–1627. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Gao, Y.; Lu, B.; Zhang, Q.; Hu, Y.; Chen, Y. Negatively regulating TLR4/NF-κB signaling via PPARα in endotoxin-induced uveitis. Biochim. Biophys. Acta. 2014, 1842, 1109–1120. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Yoo, S.H.; Henderson, L.E.; Gonzalez, F.J.; Woodcroft, K.J.; Song, B.J. PPARalpha expression protects male mice from high fat-induced nonalcoholic fatty liver. J. Nutr. 2011, 141, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, Y.; Zhuo, Q.; Tseng, Y.; Wang, J.; Ma, Y.; Zhang, J.; Liu, J. TET1 promotes fatty acid oxidation and inhibits NAFLD progression by hydroxymethylation of PPARα promoter. Nutr. Metab. 2020, 17, 46. [Google Scholar] [CrossRef] [PubMed]
- Régnier, M.; Polizzi, A.; Smati, S.; Lukowicz, C.; Fougerat, A.; Lippi, Y.; Fouché, E.; Lasserre, F.; Naylies, C.; Bétoulières, C.; et al. Hepatocyte-specific deletion of Pparα promotes NAFLD in the context of obesity. Sci. Rep. 2020, 10, 6489. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, A.; Moallem, S.A.; Johnston, T.P.; Sahebkar, A. Liver Protective Effect of Fenofibrate in NASH/NAFLD Animal Models. PPAR Res. 2022, 2022, 5805398. [Google Scholar] [CrossRef]
- Sekiya, M.; Hiraishi, A.; Touyama, M.; Sakamoto, K. Oxidative stress induced lipid accumulation via SREBP1c activation in HepG2 cells. Biochem. Biophys. Res. Commun. 2008, 375, 602–607. [Google Scholar] [CrossRef]
- Su, W.; Cao, R.; He, Y.C.; Guan, Y.F.; Ruan, X.Z. Crosstalk of Hyperglycemia and Dyslipidemia in Diabetic Kidney Disease. Kidney Dis. 2017, 3, 171–180. [Google Scholar] [CrossRef]
- Kusnadi, A.; Park, S.H.; Yuan, R.; Pannellini, T.; Giannopoulou, E.; Oliver, D.; Lu, T.; Park-Min, K.H.; Ivashkiv, L.B. The Cytokine TNF Promotes Transcription Factor SREBP Activity and Binding to Inflammatory Genes to Activate Macrophages and Limit Tissue Repair. Immunity 2019, 51, 241–257.e9. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.C.; Waxman, D.J. Cross-talk between Janus kinase-signal transducer and activator of transcription (JAK-STAT) and peroxisome proliferator-activated receptor-α (PPARα) signaling pathways: Growth hormone inhibition of PPARα transcriptional activity mediated by STAT5b. J. Biol. Chem. 1999, 274, 2672–2681. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Considine, R.V.; Leone, T.C.; Kelly, D.P.; Crabb, D.W. Role of adiponectin in the protective action of dietary saturated fat against alcoholic fatty liver in mice. Hepatology 2005, 42, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Steineger, H.H.; Sørensen, H.N.; Tugwood, J.D.; Skrede, S.; Spydevold, Ø.; Gautvik, K.M. Dexamethasone and Insulin Demonstrate Marked and Opposite Regulation of the Steady-State mRNA Level of the Peroxisomal Proliferator-Activated Receptor (PPAR) in Hepatic Cells: Hormonal Modulation of Fatty-Acid-Induced Transcription. Eur. J. Biochem. 1994, 225, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Delli Bovi, A.P.; Marciano, F.; Mandato, C.; Siano, M.A.; Savoia, M.; Vajro, P. Oxidative Stress in Non-alcoholic Fatty Liver Disease. An Updated Mini Review. Front. Med. 2021, 8, 595371. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [PubMed]
- Daniel, P.V.; Dogra, S.; Rawat, P.; Choubey, A.; Khan, A.S.; Rajak, S.; Kamthan, M.; Mondal, P. NF-κB p65 regulates hepatic lipogenesis by promoting nuclear entry of ChREBP in response to a high carbohydrate diet. J. Biol. Chem. 2021, 296, 100714. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Hung, M.C. Beyond NF-κB activation: Nuclear functions of IκB kinase α. J. Biomed. Sci. 2013, 20, 3. [Google Scholar] [CrossRef]
- Ngo, V.; Duennwald, M.L. Nrf2 and Oxidative Stress: A General Overview of Mechanisms and Implications in Human Disease. Antioxidants 2022, 11, 2345. [Google Scholar] [CrossRef]
- Mendonca, P.; Soliman, K.F.A. Flavonoids Activation of the Transcription Factor Nrf2 as a Hypothesis Approach for the Prevention and Modulation of SARS-CoV-2 Infection Severity. Antioxidants 2020, 9, 659. [Google Scholar] [CrossRef]
- Suraweera, T.L.; Rupasinghe, H.P.V.; Dellaire, G.; Xu, Z. Regulation of Nrf2/ARE Pathway by Dietary Flavonoids: A Friend or Foe for Cancer Management? Antioxidants 2020, 9, 973. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Jeon, J.H. Recent Advances in Understanding Nrf2 Agonism and Its Potential Clinical Application to Metabolic and Inflammatory Diseases. Int. J. Mol. Sci. 2022, 23, 2846. [Google Scholar] [CrossRef] [PubMed]
- Chambel, S.S.; Santos-Gonçalves, A.; Duarte, T.L. The Dual Role of Nrf2 in Nonalcoholic Fatty Liver Disease: Regulation of Antioxidant Defenses and Hepatic Lipid Metabolism. Biomed. Res. Int. 2015, 2015, 597134. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Liu, P.; Yang, B.; Wang, M.; Wang, T.; Sun, W.; Wang, X.; Zheng, W.; Song, X.; Li, J. A network pharmacology approach: Inhibition of the NF-κB signaling pathway contributes to the NASH preventative effect of an Oroxylum indicum seed extract in oleic acid-stimulated HepG2 cells and high-fat diet-fed rats. Phytomedicine 2021, 88, 153498. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Tang, Y.; He, J.; Yang, Y.; Cao, X.; Kou, S.; Wang, L.; Sheng, L.; Xue, J. Alleviation of non-alcoholic fatty liver disease by Huazhi Fugan Granules is associated with suppression of TLR4/NF-κB signaling pathway. Clin. Investig. Arterioscler. 2021, 33, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Xing, J.; Pan, D.; Gao, P. Effect of Chinese Herbal Medicine Mixture 919 Syrup on Regulation of the Ghrelin Pathway and Intestinal Microbiota in Rats With Non-alcoholic Fatty Liver Disease. Front. Microbiol. 2021, 12, 793854. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zheng, Q.; Chen, Z. The Nrf2 Pathway in Liver Diseases. Front. Cell Dev. Biol. 2022, 10, 826204. [Google Scholar] [CrossRef] [PubMed]
- Bukke, V.N.; Moola, A.; Serviddio, G.; Vendemiale, G.; Bellanti, F. Nuclear factor erythroid 2-related factor 2-mediated signaling and metabolic associated fatty liver disease. World J. Gastroenterol. 2022, 28, 6909–6921. [Google Scholar] [CrossRef]
- Sahin, K.; Orhan, C.; Tuzcu, M.; Tastan, H.; Bilir, B.; Sahin, N.; Oner, D.A.; Kucuk, O. Tomato Powder Modulates NF-κB, mTOR, and Nrf2 Pathways during Aging in Healthy Rats. J. Aging Res. 2019, 2019, 1643243. [Google Scholar] [CrossRef]
- Carranza, A.D.V.; Bonacci, G.; Moran, Y.; Asprelli, P.; Carrari, F.; Asis, R. Assessment and characterization of tomato lipophilic electrophiles and their potential contribution to nutraceutical properties via SKN-1/Nrf2 signaling activation. Food Chem. 2022, 366, 130531. [Google Scholar] [CrossRef]
- Kolberg, M.; Pedersen, S.; Bastani, N.E.; Carlsen, H.; Blomhoff, R.; Paur, I. Tomato paste alters NF-κB and cancer-related mRNA expression in prostate cancer cells, xenografts, and xenograft microenvironment. Nutr. Cancer 2015, 67, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Guo, L.; Yang, Y.; Wang, Y.; Xia, S.; Gong, H.; Zhang, B.K.; Yan, M. Dissecting the Crosstalk Between Nrf2 and NF-κB Response Pathways in Drug-Induced Toxicity. Front. Cell Dev. Biol. 2022, 9, 809952. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Martín-Moldes, Z.; Ye, J.; Lastres-Becker, I. Transcription factors NRF2 and NF-κB are coordinated effectors of the Rho family, GTP-binding protein RAC1 during inflammation. J. Biol. Chem. 2014, 289, 15244–15258. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.F.; Kuo, H.P.; Liu, M.; Chou, C.K.; Xia, W.; Du, Y.; Shen, J.; Chen, C.T.; Huo, L.; Hsu, M.C.; et al. KEAP1 E3 ligase-mediated downregulation of NF-kappaB signaling by targeting IKKbeta. Mol. Cell. 2009, 36, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, S.A.; MacEwan, D.J.; O’Connell, M.A. Lipopolysaccharide-induced expression of NAD(P)H:quinone oxidoreductase 1 and heme oxygenase-1 protects against excessive inflammatory responses in human monocytes. J. Immunol. 2008, 181, 6730–6737. [Google Scholar] [CrossRef] [PubMed]
- Bellezza, I.; Tucci, A.; Galli, F.; Grottelli, S.; Mierla, A.L.; Pilolli, F.; Minelli, A. Inhibition of NF-κB nuclear translocation via HO-1 activation underlies α-tocopheryl succinate toxicity. J. Nutr. Biochem. 2012, 23, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Wu, X.; Yan, D.; Peng, C.; Rao, C.; Yan, H. Acrylamide-induced oxidative stress and inflammatory response are alleviated by N-acetylcysteine in PC12 cells: Involvement of the crosstalk between Nrf2 and NF-κB pathways regulated by MAPKs. Toxicol. Lett. 2018, 288, 55–64. [Google Scholar] [CrossRef]
- Jin, W.; Wang, H.; Yan, W.; Xu, L.; Wang, X.; Zhao, X.; Yang, X.; Chen, G.; Ji, Y. Disruption of Nrf2 enhances upregulation of nuclear factor-kappaB activity, proinflammatory cytokines, and intercellular adhesion molecule-1 in the brain after traumatic brain injury. Mediat. Inflamm. 2008, 2008, 725174. [Google Scholar] [CrossRef]
- Minelli, A.; Grottelli, S.; Mierla, A.; Pinnen, F.; Cacciatore, I.; Bellezza, I. Cyclo(His-Pro) exerts anti-inflammatory effects by modulating NF-κB and Nrf2 signalling. Int. J. Biochem. Cell Biol. 2012, 44, 525–535. [Google Scholar] [CrossRef]
- Kim, S.W.; Lee, H.K.; Shin, J.H.; Lee, J.K. Up-down regulation of HO-1 and iNOS gene expressions by ethyl pyruvate via recruiting p300 to Nrf2 and depriving It from p65. Free. Radic. Biol. Med. 2013, 65, 468–476. [Google Scholar] [CrossRef]
- Bellezza, I.; Mierla, A.L.; Minelli, A. Nrf2 and NF-κB and Their Concerted Modulation in Cancer Pathogenesis and Progression. Cancers 2010, 2, 483–497. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Primer Sequence 5′→3′ | Accession No. | Base Pair Length |
|---|---|---|---|
| PPARα | F:GAAGTCAAAGCCGACCCAAT R: AGGGTTCTTCCTTCGCACAC | NM_019142 | 116 |
| CPT1 | F:TCCGAGGCAGGAGCCCCATC R: TCTCGGTCCAGTTTGCGGCG | NM_013200.1 | 124 |
| SREBP1c | F: GCTCACAAAAGCAAATCACT R: GCGTTTCTACCACTTCAGG | NM_001276707.1 | 191 |
| ACC-1 | F:TGAGGAGGACCGCATTTATC R: AAGCTTCCTTCGTGACCAGA | NM_022193.1 | 221 |
| Nrf2 | F:AAAATCATTAACCTCCCTGTTGAT R: CGGCGACTTTATTCTTACCTCTC | NM_031789 | 118 |
| NF-κB | F:GTGCAGAAAGAAGACATTGAGGTG R:AGGCTAGGGTCAGCGTATGG | XM_342346.4 | 176 |
| β-actin | F: CGAGTACAACCTTCTTGCAGC R: CCTTCTGACCCATACCCACC | NM_031144.3 | 209 |
| Parameter | Control | ESGA (200 mg/kg) | HFD | HFD + ESGA (50 mg/kg) | HFD + ESGA (100 mg/kg) | HFD + ESGA (200 mg/kg) |
|---|---|---|---|---|---|---|
| Final body weight (g) | 506.2 ± 43.2 | 519.4 ± 55.9 | 634.5 ± 58.2 ab | 653.4 ± 61.9 ab | 638.7 ± 52.3 ab | 659.2 ± 54.7 ab |
| Liver weight (g) | 15.8 ± 1.3 | 16.4 ± 1.6 | 24.7 ± 1.9 ab | 19.8 ± 1.8 abc | 17.0 ± 1.4 abcd | 15.8 ± 1.1 bcde |
| Liver/body weight % | 3.04 ± 0.32 | 3.16 ± 1.6 | 3.93 ± 0.57 ab | 2.95 ± 0.11 bc | 2.68 ± 0.7 abcd | 2.34 ± 0.31 abcde |
| Subcutaneous fat (g) | 5.6 ± 0.46 | 5.8 ± 0.45 | 9.3 ± 0.89 ab | 8.7 ± 0.93 ab | 8.9 ± 0.85 ab | 9.2 ± 1.3 ab |
| Epididymal fat (g) | 7.2 ± 1.3 | 7.5 ± 1.1 | 13.4 ± 1.3 ab | 12.9 ± 1.4 ab | 14.2 ± 1.8 ab | 12.6 ± 1.2 ab |
| Peritoneal fat (g) | 4.7 ± 0.64 | 4.3 ± 0.48 | 8.45 ± 0.74 ab | 9.2 ± 1.1 ab | 8.7 ± 0.95 ab | 9.0 ± 1.2 ab |
| Serum adiponectin (µg/mL) | 45.3 ± 6.7 | 41.7 ± 8.1 | 20.1 ± 2.1 ab | 25.5 ± 3.5 ab | 19.6 ± 3.7 ab | 23.7 ± 3.1 ab |
| Serum leptin (ng/mL) | 22.7 ± 2.8 | 25.6 ± 4.8 | 64.5 ± 5.3 ab | 61.2 ± 7.8 ab | 58.9 ± 6.9 ab | 63.9 ± 6.5 ab |
| Serum IL-6 (pg/mL) | 2.4 ± 0.54 | 2.7 ± 0.18 | 28.6 ± 3.1 ab | 33.1 ± 4.3 ab | 30.8 ± 2.7 ab | 31.2 ± 4.1 ab |
| Serum TNF-α (pg/mL) | 0.82 ± 0.13 | 0.73 ± 0.09 | 4.9 ± 0.53 ab | 5.1 ± 0.84 ab | 4.4 ± 0.72 ab | 5.3 ± 0.69 ab |
| Serum FFAs (mmol/L) | 3.8 ± 0.57 | 4.3 ± 0.52 | 16.8 ± 2.1 ab | 18.2 ± 1.9 ab | 15.8 ± 2.6 ab | 16.9 ± 1.6 ab |
| Parameter | Control | ESGA (200 mg/kg) | HFD | HFD + ESGA (50 mg/kg) | HFD + ESGA (100 mg/kg) | HFD + ESGA (200 mg/kg) |
|---|---|---|---|---|---|---|
| FPG (mg/dL) | 114.2 ± 9.4 | 110.8 ± 12.8 | 183.4 ± 22.3 ab | 176.7 ± 16.5 ab | 171.3 ± 18.6 ab | 180.1 ± 18.3 ab |
| FPI (mg/dL) | 3.8 ± 0.53 | 4.2 ± 0.49 | 7.3 ± 0.82 ab | 8.1 ± 0.94 ab | 7.1 ± 0.82 ab | 7.5 ± 0.88 ab |
| HOMA-IR | 1.03 ± 0.1 | 1.16 ± 0.23 | 3.31 ± 0.43 ab | 3.52 ± 0.31 ab | 3.11 ± 0.52 ab | 3.44 ± 4.72 ab |
| Serum ALT (U/L) | 29.9 ± 1.7 | 22.4 ± 2.8 | 127.3 ± 11.6 ab | 98.3 ± 13.2 abc | 57.3 ± 6.4 abcd | 26.3 ± 2.3 bcde |
| Serum AST (U/L) | 41.3 ± 5.2 | 47.6 ± 5.8 | 153.2 ± 14.1 ab | 103.5 ± 15.7 ab | 72.5 ± 8.4 abcd | 45.8 ± 18.4 bcd |
| Serum GTT (U/L) | 25.8 ±2.3 | 23.7 ± 3.6 | 88.3 ± 8.3 ab | 68.4 ± 5.4 ab | 46.9 ± 6.3 abcd | 27.3 ± 3.4 bcde |
| Parameter | Control | ESGA (200 mg/kg) | HFD | HFD + ESGA (50 mg/kg) | HFD + ESGA (100 mg/kg) | HFD + ESGA (200 mg/kg) | |
|---|---|---|---|---|---|---|---|
| Serum | TGs (mg/dL) | 76.4 ± 5.8 | 59.8 ± 5.4 | 225 ± 19.4 ab | 176.4 ± 15.6 abc | 127.2 ± 11.9 abcd | 85.6 ± 7.4 abcde |
| CHOL (mg/dL) | 109.5 ± 8.6 | 88.2 ± 6.8 ab | 296.7 ± 18.9 ab | 206.3 ± 17.8 abc | 156.4 ± 13.8 abcd | 114.2 ± 12.3 abcde | |
| LDL-c (mg/dL) | 56.4 ± 5.7 | 33.7 ± 4.3 ab | 164.5 ± 13.8 abc | 128.4 ± 10.8 abc | 93.2 ± 8.4 abcd | 67.8 ± 6.2 abcde | |
| Liver | TGs (ng/g tissue) | 403.1 ± 41.5 | 311.3 ± 28.5 ab | 1242.1 ± 134.5 abc | 892.1 ± 93.5 abcd | 638.1 ± 54.3 abcd | 459 ± 51.4 abcde |
| CHOL (ng/g tissue) | 119.2 ± 12.5 | 89.2 ± 58.7 ab | 434.2 ± 56.1 abc | 309.2 ± 33.1 abc | 212.2 ± 22.4 abcd | 153.4 ± 15.2 abcde | |
| Stool | TGs (ng/g) | 3.18 ± 0.46 | 3.3 ± 3.71 | 6.83 ± 0.73 ab | 7.2 ± 0.92 ab | 6.54 ± 0.83 ab | 6.99 ± 0.73 ab |
| CHOL (ng/g) | 2.95 ± 0.36 | 2.72 ± 0.41 | 4.83 ± 0.39 ab | 4.66 ± 0.53 ab | 4.51 ± 0.68 ab | 4.77 ± 0.41 ab |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Jadani, J.M.; Albadr, N.A.; Alshammari, G.M.; Almasri, S.A.; Alfayez, F.F.; Yahya, M.A. Esculeogenin A, a Glycan from Tomato, Alleviates Nonalcoholic Fatty Liver Disease in Rats through Hypolipidemic, Antioxidant, and Anti-Inflammatory Effects. Nutrients 2023, 15, 4755. https://doi.org/10.3390/nu15224755
Al Jadani JM, Albadr NA, Alshammari GM, Almasri SA, Alfayez FF, Yahya MA. Esculeogenin A, a Glycan from Tomato, Alleviates Nonalcoholic Fatty Liver Disease in Rats through Hypolipidemic, Antioxidant, and Anti-Inflammatory Effects. Nutrients. 2023; 15(22):4755. https://doi.org/10.3390/nu15224755
Chicago/Turabian StyleAl Jadani, Jwharah M., Nawal A. Albadr, Ghedeir M. Alshammari, Soheir A. Almasri, Farah Fayez Alfayez, and Mohammed Abdo Yahya. 2023. "Esculeogenin A, a Glycan from Tomato, Alleviates Nonalcoholic Fatty Liver Disease in Rats through Hypolipidemic, Antioxidant, and Anti-Inflammatory Effects" Nutrients 15, no. 22: 4755. https://doi.org/10.3390/nu15224755
APA StyleAl Jadani, J. M., Albadr, N. A., Alshammari, G. M., Almasri, S. A., Alfayez, F. F., & Yahya, M. A. (2023). Esculeogenin A, a Glycan from Tomato, Alleviates Nonalcoholic Fatty Liver Disease in Rats through Hypolipidemic, Antioxidant, and Anti-Inflammatory Effects. Nutrients, 15(22), 4755. https://doi.org/10.3390/nu15224755