
Intermittent Fasting Attenuates Metabolic-Dysfunction-Associated Steatohepatitis by Enhancing the Hepatic Autophagy–Lysosome Pathway


Abstract
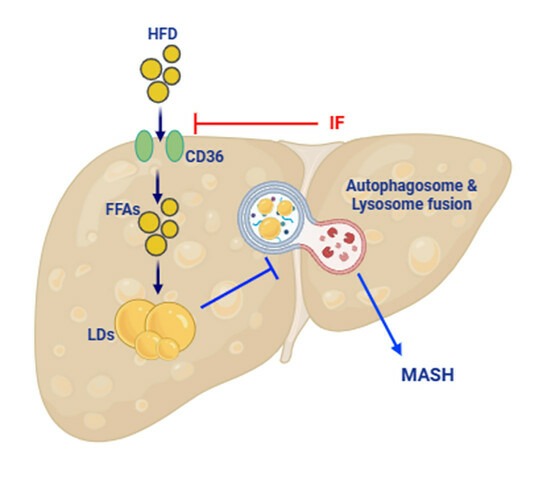
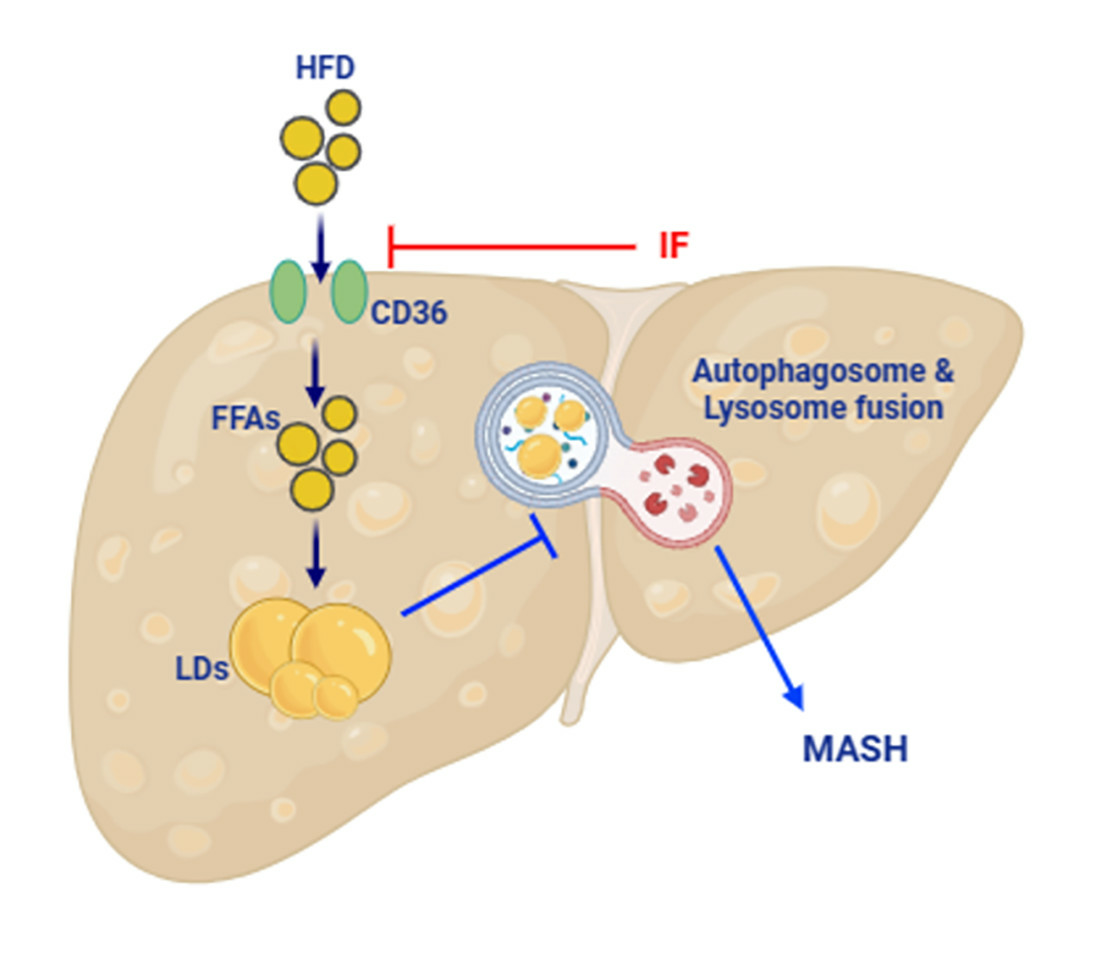
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals and IF Mouse Model
2.2. Measurement of Serum Metabolic Parameters
2.3. Tissue Collection and Histological Analysis
2.4. MASLD Activity Score Measurement
2.5. Hepatic TG Colorimetric Assay
2.6. Hepatic Hydroxyproline Assay
2.7. Lipid Profiling Analysis
2.8. RNA Isolation and Real-Time Polymerase Chain Reaction (RT-PCR) Analysis
2.9. Western Blot Analysis
2.10. Immunohistochemistry
2.11. Double Immunofluorescence
2.12. Transmission Electron Microscopy (TEM)
2.13. Statistical Analysis
3. Results
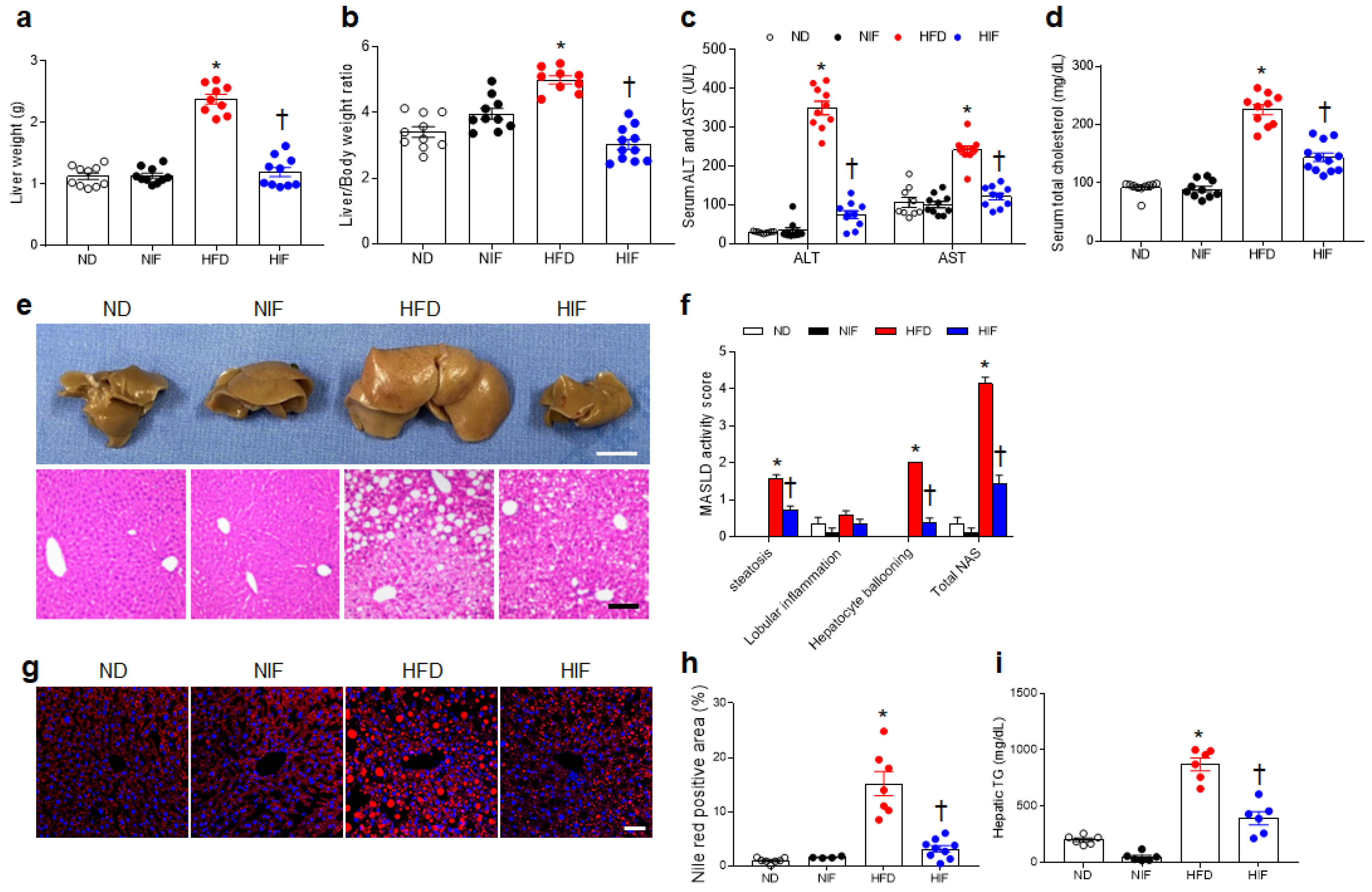
3.1. IF Reduces Body Weight and Insulin Resistance in HFD-Fed Mice
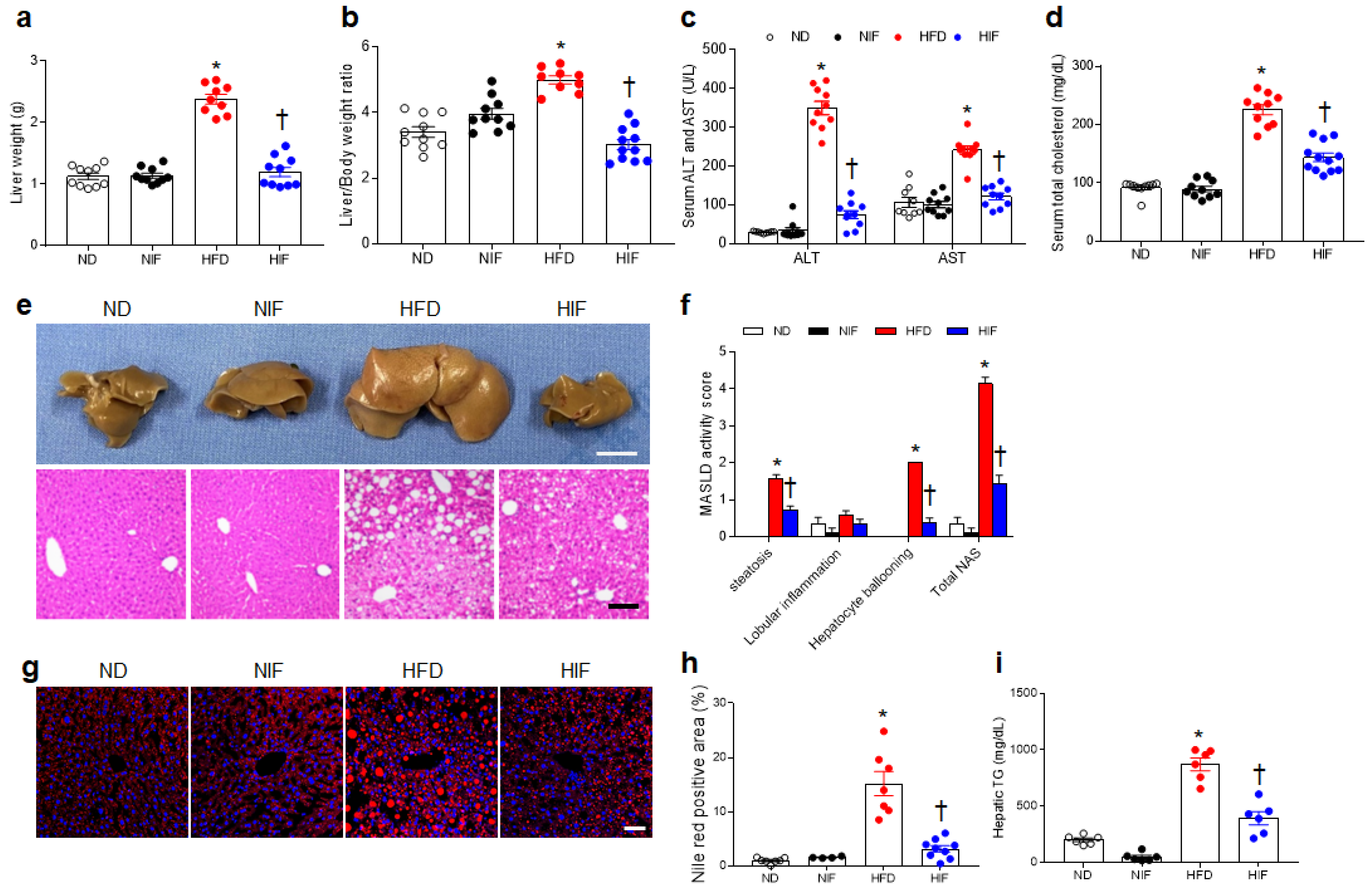
3.2. IF Attenuates MASLD Activity in HFD-Fed Mice
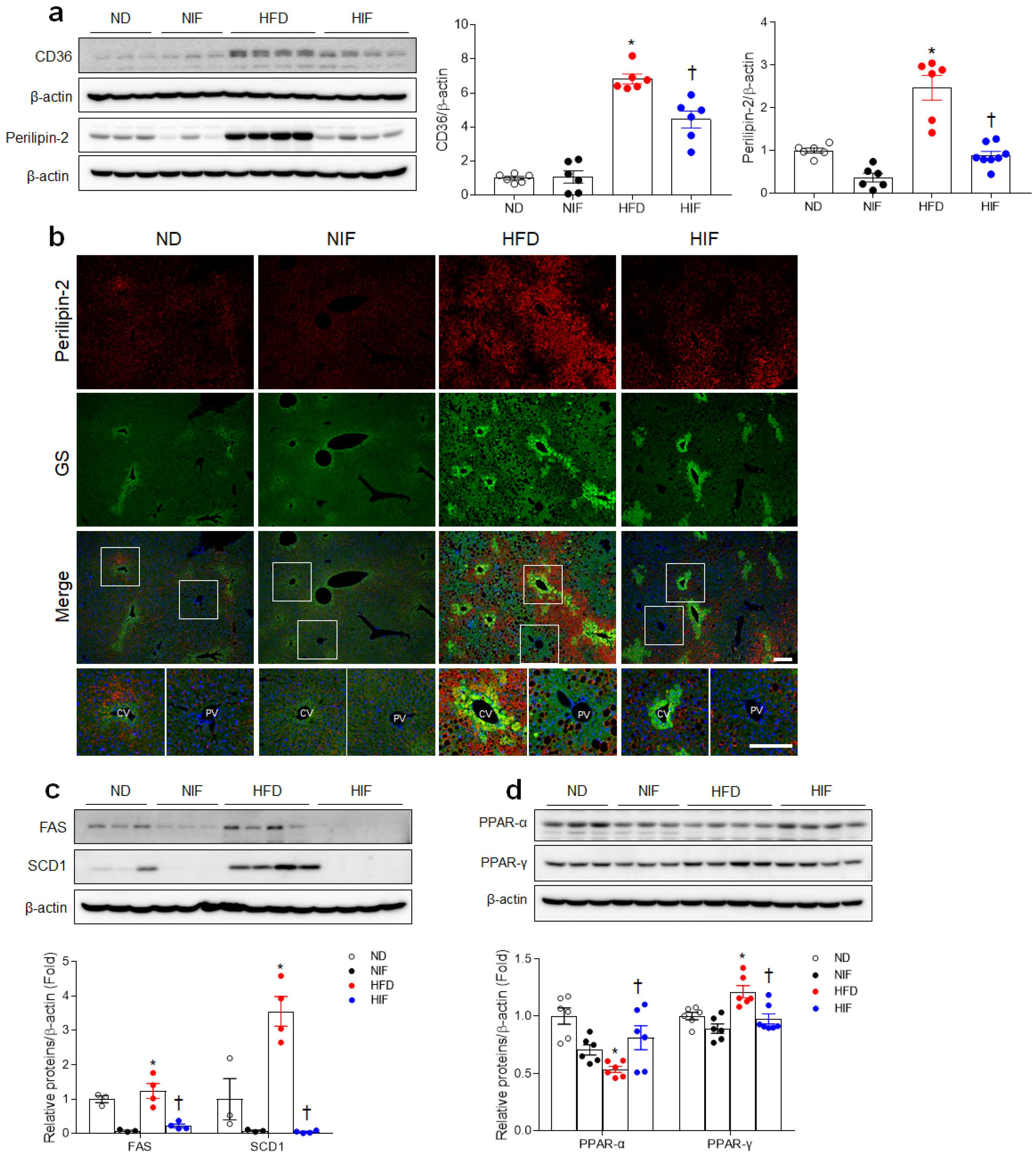
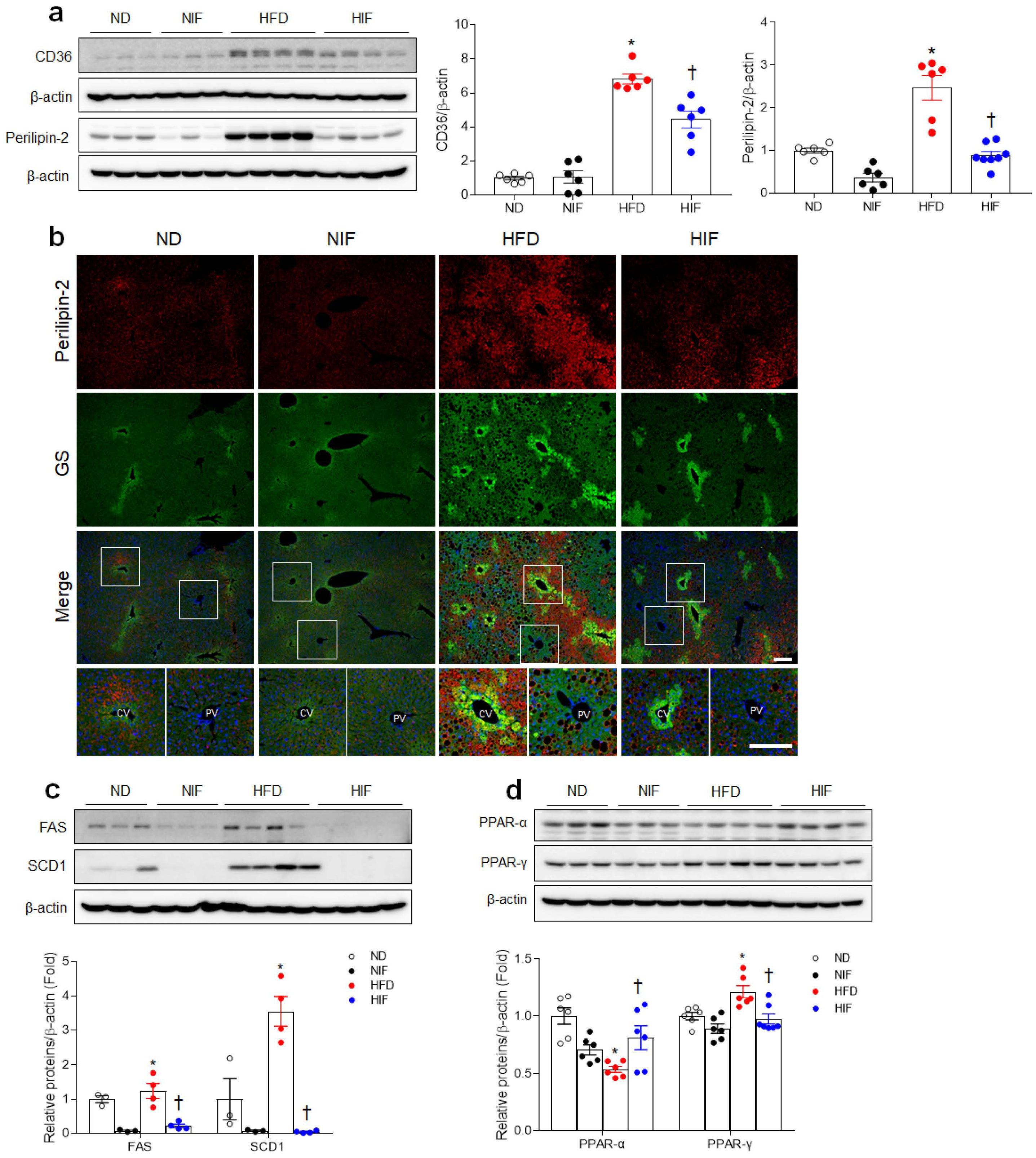
3.3. IF Reduces Hepatic Lipid Uptake and Improves Fatty Acid Oxidation in HFD-Fed Mice
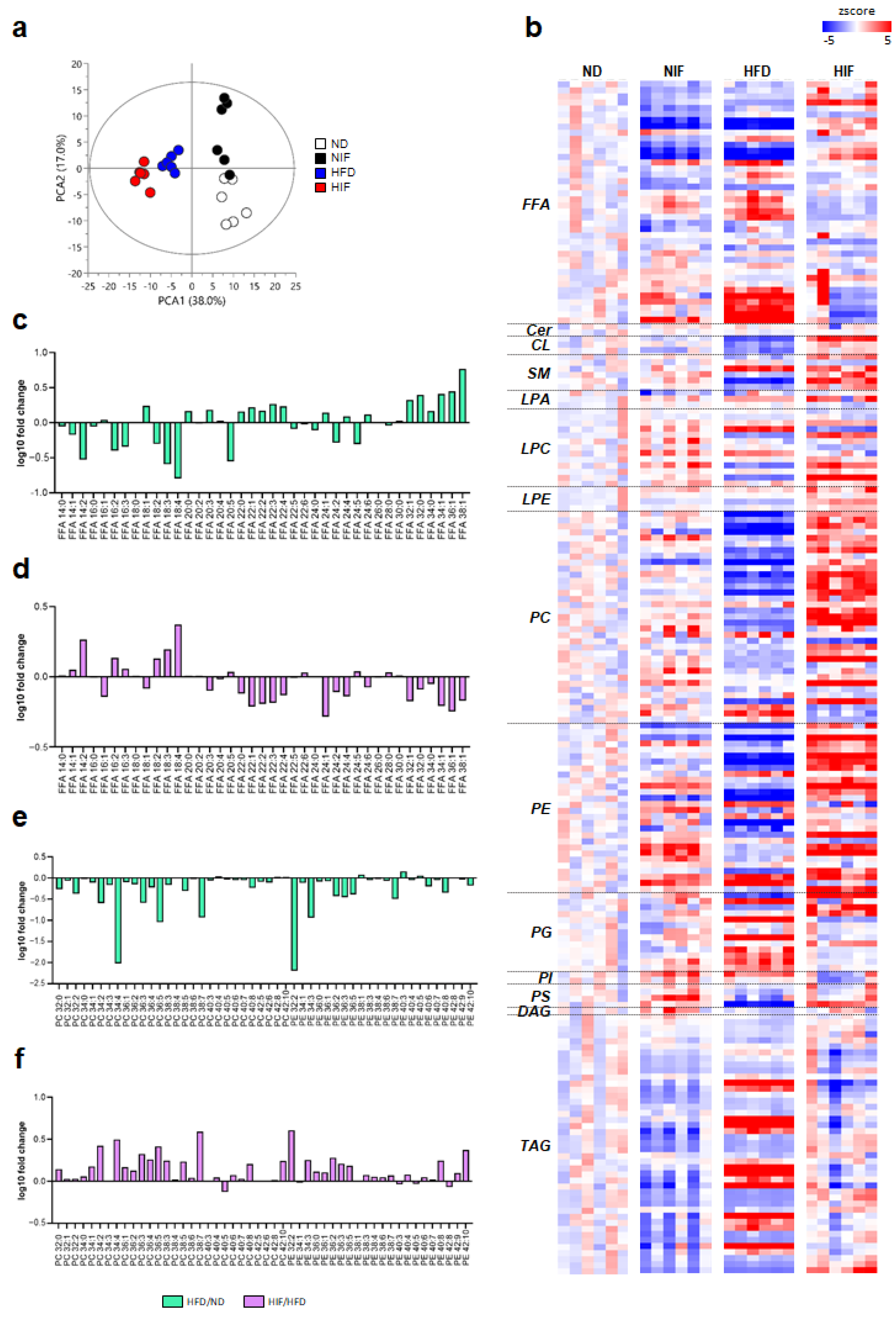
3.4. IF Alters Lipid Profiles in Livers with MASLD
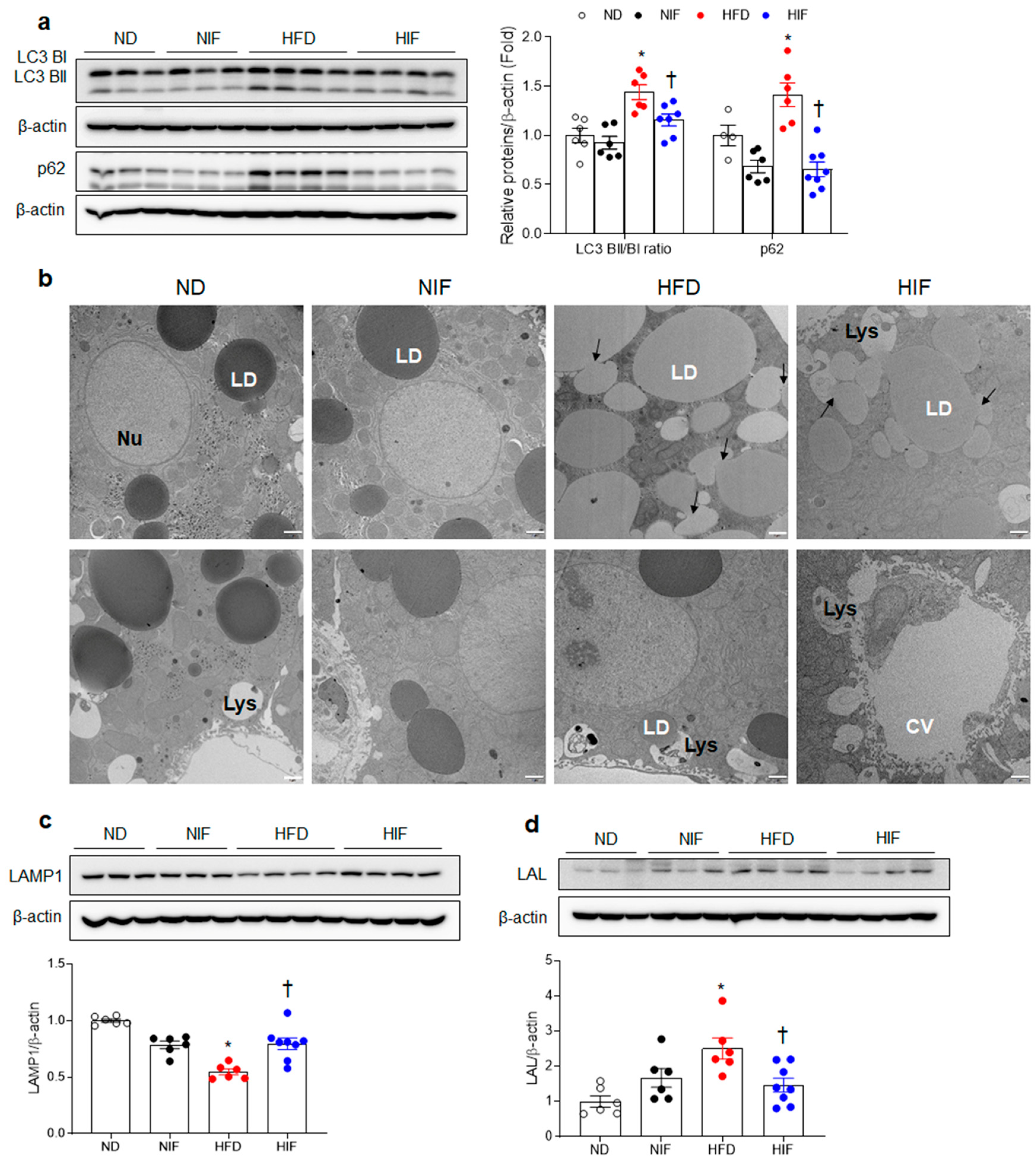
3.5. IF Improves Hepatic Autophagy Flux and Enhances Lysosomes in HFD-Fed Mice
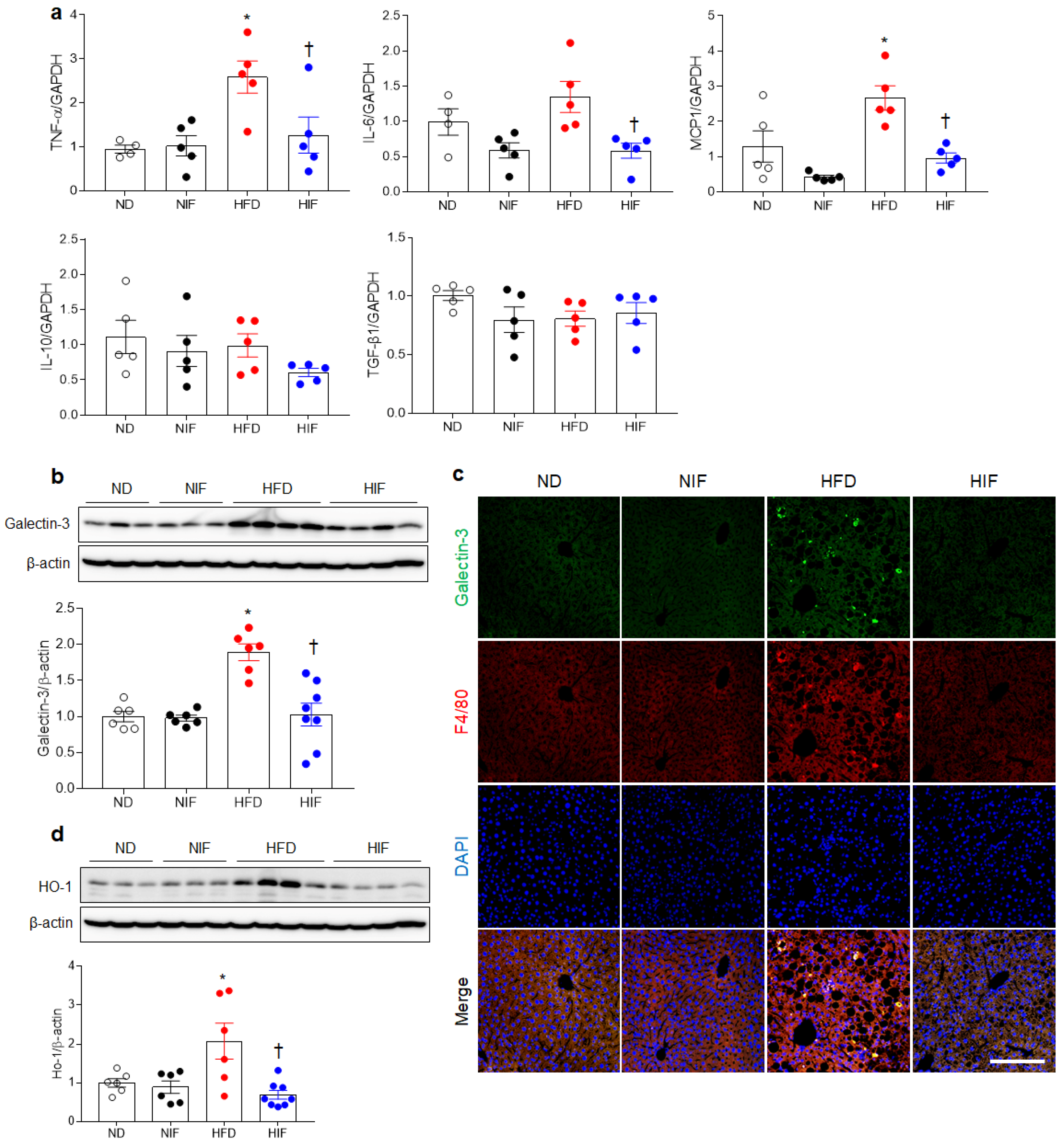
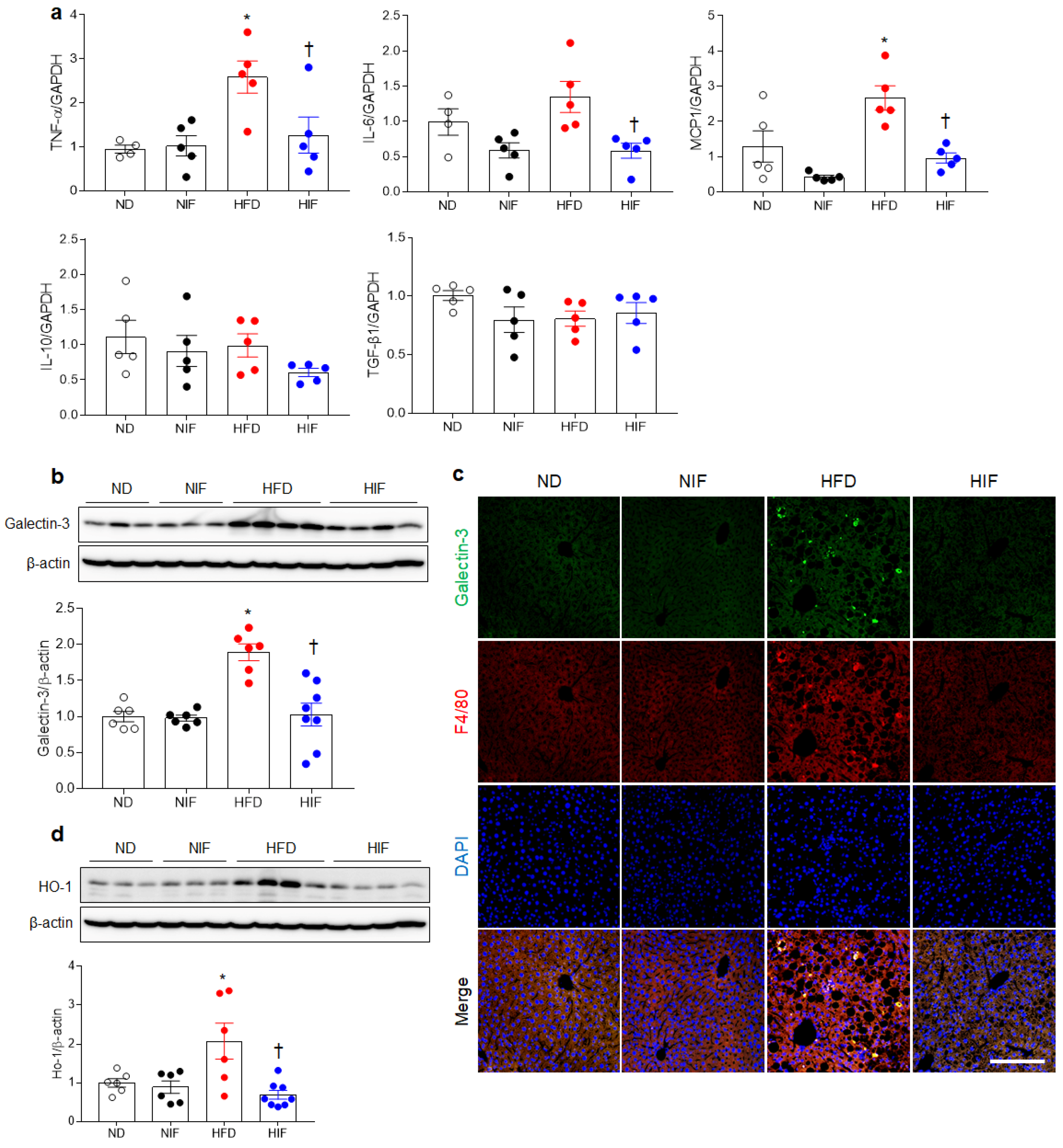
3.6. IF Reduces Hepatic Inflammation in HFD-Fed Mice
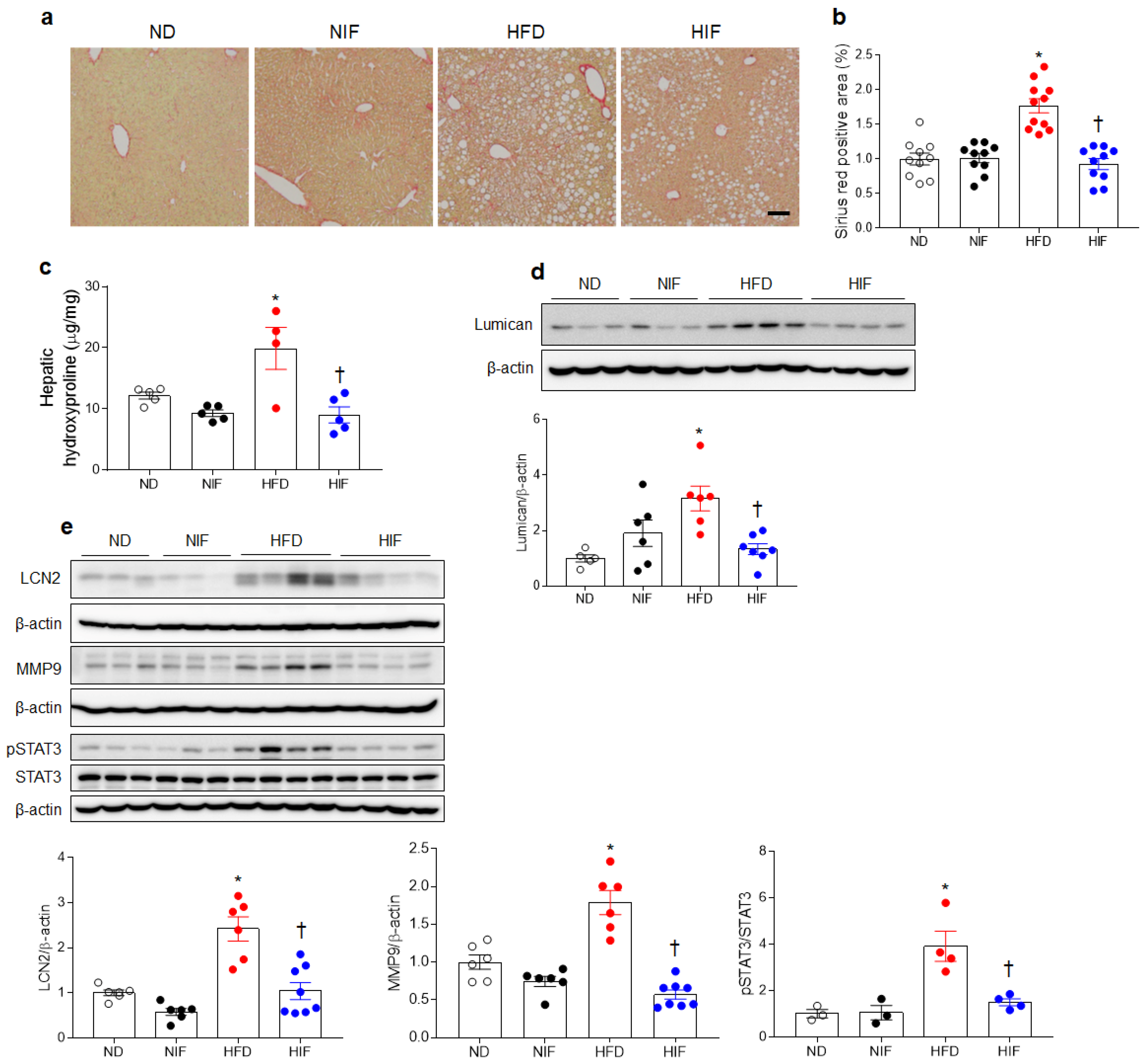
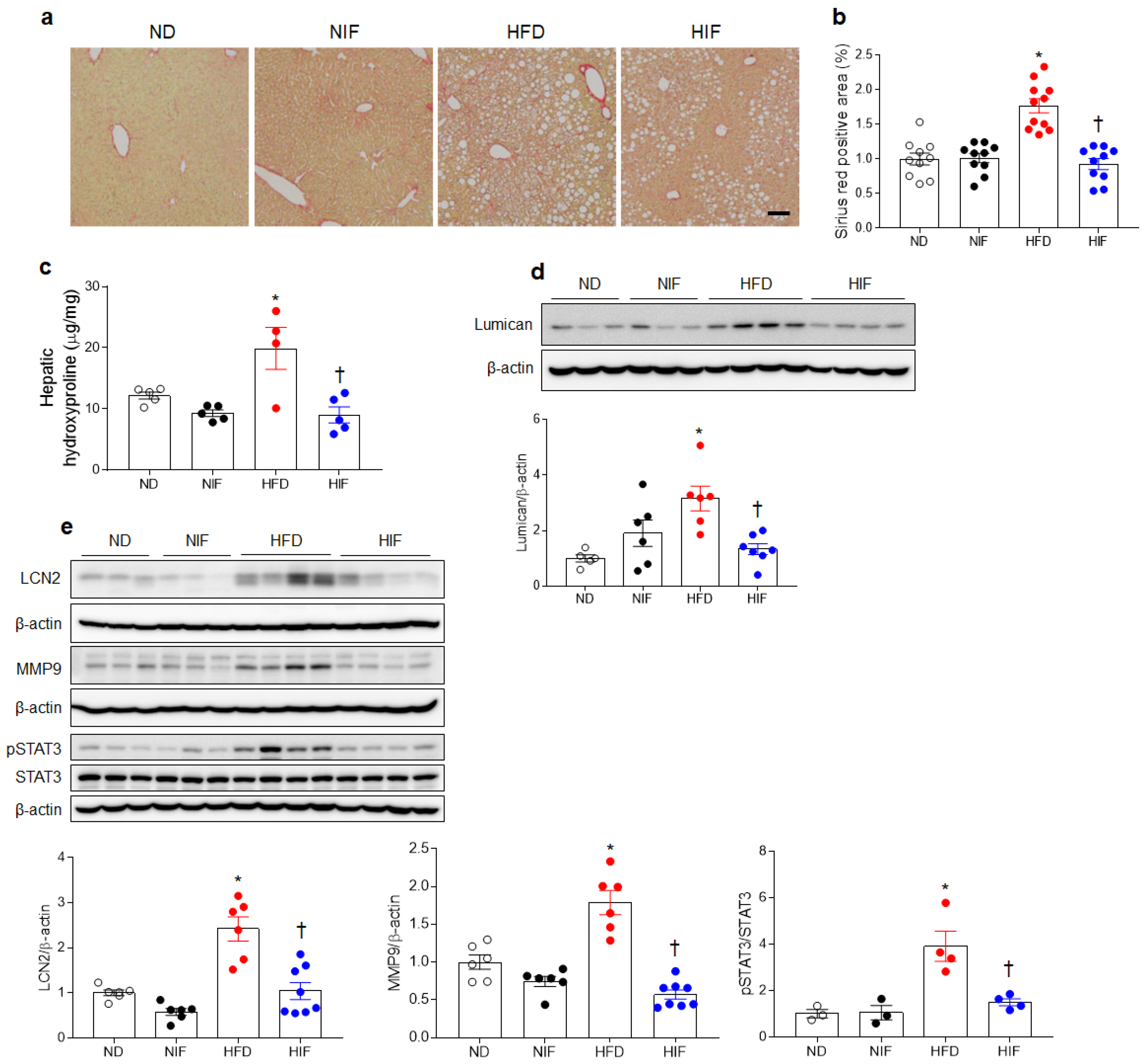
3.7. IF Reduces Hepatic Fibrosis in HFD-Fed Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wong, V.W.-S.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Mashek, D.G. Hepatic lipid droplets: A balancing act between energy storage and metabolic dysfunction in NAFLD. Mol. Metab. 2021, 50, 101115. [Google Scholar] [CrossRef]
- Brunt, E.M.; Tiniakos, D.G. Histopathology of nonalcoholic fatty liver disease. World J. Gastroenterol. 2010, 16, 5286–5296. [Google Scholar] [CrossRef] [PubMed]
- Pierantonelli, I.; Svegliati-Baroni, G. Nonalcoholic Fatty Liver Disease: Basic Pathogenetic Mechanisms in the Progression From NAFLD to NASH. Transplantation 2019, 103, e1–e13. [Google Scholar] [CrossRef] [PubMed]
- Grefhorst, A.; van de Peppel, I.P.; Larsen, L.E.; Jonker, J.W.; Holleboom, A.G. The Role of Lipophagy in the Development and Treatment of Non-Alcoholic Fatty Liver Disease. Front. Endocrinol. 2020, 11, 601627. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Luedde, T.; Trautwein, C. Inflammatory pathways in liver homeostasis and liver injury. Clin. Rev. Allergy Immunol. 2009, 36, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Gross, D.A.; Silver, D.L. Cytosolic lipid droplets: From mechanisms of fat storage to disease. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 304–326. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef]
- Jonas, W.; Schwerbel, K.; Zellner, L.; Jahnert, M.; Gottmann, P.; Schurmann, A. Alterations of Lipid Profile in Livers with Impaired Lipophagy. Int. J. Mol. Sci. 2022, 23, 11863. [Google Scholar] [CrossRef]
- Chiappini, F.; Coilly, A.; Kadar, H.; Gual, P.; Tran, A.; Desterke, C.; Samuel, D.; Duclos-Vallee, J.C.; Touboul, D.; Bertrand-Michel, J.; et al. Metabolism dysregulation induces a specific lipid signature of nonalcoholic steatohepatitis in patients. Sci. Rep. 2017, 7, 46658. [Google Scholar] [CrossRef]
- Puri, P.; Baillie, R.A.; Wiest, M.M.; Mirshahi, F.; Choudhury, J.; Cheung, O.; Sargeant, C.; Contos, M.J.; Sanyal, A.J. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007, 46, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Agellon, L.B.; Allen, T.M.; Umeda, M.; Jewell, L.; Mason, A.; Vance, D.E. The ratio of phosphatidylcholine to phosphatidylethanolamine influences membrane integrity and steatohepatitis. Cell Metab. 2006, 3, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kang, H.; Heo, R.W.; Jeon, B.T.; Yi, C.O.; Shin, H.J.; Kim, J.; Jeong, S.Y.; Kwak, W.; Kim, W.H.; et al. Caloric restriction improves diabetes-induced cognitive deficits by attenuating neurogranin-associated calcium signaling in high-fat diet-fed mice. J. Cereb. Blood Flow Metab. 2016, 36, 1098–1110. [Google Scholar] [CrossRef] [PubMed]
- Jeon, B.T.; Heo, R.W.; Jeong, E.A.; Yi, C.O.; Lee, J.Y.; Kim, K.E.; Kim, H.; Roh, G.S. Effects of caloric restriction on O-GlcNAcylation, Ca(2+) signaling, and learning impairment in the hippocampus of ob/ob mice. Neurobiol. Aging 2016, 44, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.E.; Jung, Y.; Min, S.; Nam, M.; Heo, R.W.; Jeon, B.T.; Song, D.H.; Yi, C.O.; Jeong, E.A.; Kim, H.; et al. Caloric restriction of db/db mice reverts hepatic steatosis and body weight with divergent hepatic metabolism. Sci. Rep. 2016, 6, 30111. [Google Scholar] [CrossRef] [PubMed]
- Krasnoff, J.B.; Painter, P.L.; Wallace, J.P.; Bass, N.M.; Merriman, R.B. Health-related fitness and physical activity in patients with nonalcoholic fatty liver disease. Hepatology 2008, 47, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Anson, R.M.; Guo, Z.; de Cabo, R.; Iyun, T.; Rios, M.; Hagepanos, A.; Ingram, D.K.; Lane, M.A.; Mattson, M.P. Intermittent fasting dissociates beneficial effects of dietary restriction on glucose metabolism and neuronal resistance to injury from calorie intake. Proc. Natl. Acad. Sci. USA 2003, 100, 6216–6220. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Jung, Y.; Cho, Y.; Kim, N.; Oh, I.Y.; Kang, S.W.; Choi, E.K.; Hwang, G.S. Lipidomic profiling reveals free fatty acid alterations in plasma from patients with atrial fibrillation. PLoS ONE 2018, 13, e0196709. [Google Scholar] [CrossRef]
- Miquilena-Colina, M.E.; Lima-Cabello, E.; Sanchez-Campos, S.; Garcia-Mediavilla, M.V.; Fernandez-Bermejo, M.; Lozano-Rodriguez, T.; Vargas-Castrillon, J.; Buque, X.; Ochoa, B.; Aspichueta, P.; et al. Hepatic fatty acid translocase CD36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non-alcoholic steatohepatitis and chronic hepatitis C. Gut 2011, 60, 1394–1402. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Leopold, C.; Duta-Mare, M.; Sachdev, V.; Goeritzer, M.; Maresch, L.K.; Kolb, D.; Reicher, H.; Wagner, B.; Stojakovic, T.; Ruelicke, T.; et al. Hepatocyte-specific lysosomal acid lipase deficiency protects mice from diet-induced obesity but promotes hepatic inflammation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Jeftic, I.; Jovicic, N.; Pantic, J.; Arsenijevic, N.; Lukic, M.L.; Pejnovic, N. Galectin-3 Ablation Enhances Liver Steatosis, but Attenuates Inflammation and IL-33-Dependent Fibrosis in Obesogenic Mouse Model of Nonalcoholic Steatohepatitis. Mol. Med. 2015, 21, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Iacobini, C.; Menini, S.; Ricci, C.; Blasetti Fantauzzi, C.; Scipioni, A.; Salvi, L.; Cordone, S.; Delucchi, F.; Serino, M.; Federici, M.; et al. Galectin-3 ablation protects mice from diet-induced NASH: A major scavenging role for galectin-3 in liver. J. Hepatol. 2011, 54, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.H.; Zheng, K.I.; Wang, X.D.; Qiao, J.; Li, Y.Y.; Zhang, L.; Zheng, M.H.; Wu, J. LC-MS-based lipidomic analysis in distinguishing patients with nonalcoholic steatohepatitis from nonalcoholic fatty liver. Hepatobiliary Pancreat. Dis. Int. 2021, 20, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.E.; Lee, J.; Shin, H.J.; Jeong, E.A.; Jang, H.M.; Ahn, Y.J.; An, H.S.; Lee, J.Y.; Shin, M.C.; Kim, S.K.; et al. Lipocalin-2 activates hepatic stellate cells and promotes nonalcoholic steatohepatitis in high-fat diet-fed Ob/Ob mice. Hepatology 2023, 77, 888–901. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.A.; Kang, P.B. PLIN2 inhibits insulin-induced glucose uptake in myoblasts through the activation of the NLRP3 inflammasome. Int. J. Mol. Med. 2015, 36, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Ikura, Y.; Arimoto, J.; Sugioka, K.; Iezzoni, J.C.; Park, S.H.; Naruko, T.; Itabe, H.; Kawada, N.; Caldwell, S.H.; et al. Expression of perilipin and adipophilin in nonalcoholic fatty liver disease; relevance to oxidative injury and hepatocyte ballooning. J. Atheroscler. Thromb. 2009, 16, 893–901. [Google Scholar] [CrossRef]
- Najt, C.P.; Senthivinayagam, S.; Aljazi, M.B.; Fader, K.A.; Olenic, S.D.; Brock, J.R.; Lydic, T.A.; Jones, A.D.; Atshaves, B.P. Liver-specific loss of Perilipin 2 alleviates diet-induced hepatic steatosis, inflammation, and fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G726–G738. [Google Scholar] [CrossRef]
- Jin, Y.; Tan, Y.; Chen, L.; Liu, Y.; Ren, Z. Reactive Oxygen Species Induces Lipid Droplet Accumulation in HepG2 Cells by Increasing Perilipin 2 Expression. Int. J. Mol. Sci. 2018, 19, 3445. [Google Scholar] [CrossRef]
- Greco, D.; Kotronen, A.; Westerbacka, J.; Puig, O.; Arkkila, P.; Kiviluoto, T.; Laitinen, S.; Kolak, M.; Fisher, R.M.; Hamsten, A.; et al. Gene expression in human NAFLD. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G1281–G1287. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, C.; Luo, X.; Wang, P.; Zhou, W.; Zhong, S.; Xie, Y.; Jiang, Y.; Yang, P.; Tang, R.; et al. CD36 palmitoylation disrupts free fatty acid metabolism and promotes tissue inflammation in non-alcoholic steatohepatitis. J. Hepatol. 2018, 69, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Sheedfar, F.; Sung, M.M.; Aparicio-Vergara, M.; Kloosterhuis, N.J.; Miquilena-Colina, M.E.; Vargas-Castrillon, J.; Febbraio, M.; Jacobs, R.L.; de Bruin, A.; Vinciguerra, M.; et al. Increased hepatic CD36 expression with age is associated with enhanced susceptibility to nonalcoholic fatty liver disease. Aging 2014, 6, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.G.; Tran, J.L.; Erion, D.M.; Vera, N.B.; Febbraio, M.; Weiss, E.J. Hepatocyte-Specific Disruption of CD36 Attenuates Fatty Liver and Improves Insulin Sensitivity in HFD-Fed Mice. Endocrinology 2016, 157, 570–585. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Febbraio, M.; Wada, T.; Zhai, Y.; Kuruba, R.; He, J.; Lee, J.H.; Khadem, S.; Ren, S.; Li, S.; et al. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis. Gastroenterology 2008, 134, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Ohtake, T.; Motomura, W.; Takahashi, N.; Hosoki, Y.; Miyoshi, S.; Suzuki, Y.; Saito, H.; Kohgo, Y.; Okumura, T. Increased expression of PPARgamma in high fat diet-induced liver steatosis in mice. Biochem. Biophys. Res. Commun. 2005, 336, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Yoo, S.H.; Henderson, L.E.; Gonzalez, F.J.; Woodcroft, K.J.; Song, B.J. PPARalpha expression protects male mice from high fat-induced nonalcoholic fatty liver. J. Nutr. 2011, 141, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Devaux, P.F. Static and dynamic lipid asymmetry in cell membranes. Biochemistry 1991, 30, 1163–1173. [Google Scholar] [CrossRef]
- Ogawa, Y.; Kobayashi, T.; Honda, Y.; Kessoku, T.; Tomeno, W.; Imajo, K.; Nakahara, T.; Oeda, S.; Nagaoki, Y.; Amano, Y.; et al. Metabolomic/lipidomic-based analysis of plasma to diagnose hepatocellular ballooning in patients with non-alcoholic fatty liver disease: A multicenter study. Hepatol. Res. 2020, 50, 955–965. [Google Scholar] [CrossRef]
- Lee, H.S.; Nam, Y.; Chung, Y.H.; Kim, H.R.; Park, E.S.; Chung, S.J.; Kim, J.H.; Sohn, U.D.; Kim, H.C.; Oh, K.W.; et al. Beneficial effects of phosphatidylcholine on high-fat diet-induced obesity, hyperlipidemia and fatty liver in mice. Life Sci. 2014, 118, 7–14. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, C.; Yan, T.; Yu, C.; Li, Y. Omega-3 Fatty acids reverse lipotoxity through induction of autophagy in nonalcoholic fatty liver disease. Nutrition 2015, 31, 1423–1429.e2. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rodriguez, A.; Mayoral, R.; Agra, N.; Valdecantos, M.P.; Pardo, V.; Miquilena-Colina, M.E.; Vargas-Castrillon, J.; Lo Iacono, O.; Corazzari, M.; Fimia, G.M.; et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014, 5, e1179. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.X.; Chen, X.; Hsu, D.K.; Baghy, K.; Serizawa, N.; Scott, F.; Takada, Y.; Takada, Y.; Fukada, H.; Chen, J.; et al. Galectin-3 modulates phagocytosis-induced stellate cell activation and liver fibrosis in vivo. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G439–G446. [Google Scholar] [CrossRef] [PubMed]
- Pejnovic, N.N.; Pantic, J.M.; Jovanovic, I.P.; Radosavljevic, G.D.; Milovanovic, M.Z.; Nikolic, I.G.; Zdravkovic, N.S.; Djukic, A.L.; Arsenijevic, N.N.; Lukic, M.L. Galectin-3 deficiency accelerates high-fat diet-induced obesity and amplifies inflammation in adipose tissue and pancreatic islets. Diabetes 2013, 62, 1932–1944. [Google Scholar] [CrossRef]
- Pejnovic, N.; Jeftic, I.; Jovicic, N.; Arsenijevic, N.; Lukic, M.L. Galectin-3 and IL-33/ST2 axis roles and interplay in diet-induced steatohepatitis. World J. Gastroenterol. 2016, 22, 9706–9717. [Google Scholar] [CrossRef] [PubMed]
- Al Attar, A.; Antaramian, A.; Noureddin, M. Review of galectin-3 inhibitors in the treatment of nonalcoholic steatohepatitis. Expert Rev. Clin. Pharmacol. 2021, 14, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.; Li, X.; Kao, W.Y.; Viker, K.; Butters, K.; Masuoka, H.; Knudsen, B.; Gores, G.; Charlton, M. Lumican, an extracellular matrix proteoglycan, is a novel requisite for hepatic fibrosis. Lab. Investig. 2012, 92, 1712–1725. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, K.E.; Shin, H.J.; Ju, Y.; Jung, Y.; An, H.S.; Lee, S.J.; Jeong, E.A.; Lee, J.; Hwang, G.-S.; Roh, G.S. Intermittent Fasting Attenuates Metabolic-Dysfunction-Associated Steatohepatitis by Enhancing the Hepatic Autophagy–Lysosome Pathway. Nutrients 2023, 15, 4574. https://doi.org/10.3390/nu15214574
Kim KE, Shin HJ, Ju Y, Jung Y, An HS, Lee SJ, Jeong EA, Lee J, Hwang G-S, Roh GS. Intermittent Fasting Attenuates Metabolic-Dysfunction-Associated Steatohepatitis by Enhancing the Hepatic Autophagy–Lysosome Pathway. Nutrients. 2023; 15(21):4574. https://doi.org/10.3390/nu15214574
Chicago/Turabian StyleKim, Kyung Eun, Hyun Joo Shin, Yeajin Ju, Youngae Jung, Hyeong Seok An, So Jeong Lee, Eun Ae Jeong, Jaewoong Lee, Geum-Sook Hwang, and Gu Seob Roh. 2023. "Intermittent Fasting Attenuates Metabolic-Dysfunction-Associated Steatohepatitis by Enhancing the Hepatic Autophagy–Lysosome Pathway" Nutrients 15, no. 21: 4574. https://doi.org/10.3390/nu15214574
APA StyleKim, K. E., Shin, H. J., Ju, Y., Jung, Y., An, H. S., Lee, S. J., Jeong, E. A., Lee, J., Hwang, G.-S., & Roh, G. S. (2023). Intermittent Fasting Attenuates Metabolic-Dysfunction-Associated Steatohepatitis by Enhancing the Hepatic Autophagy–Lysosome Pathway. Nutrients, 15(21), 4574. https://doi.org/10.3390/nu15214574