

Ketogenic Diet Exacerbates L-Arginine-Induced Acute Pancreatitis and Reveals the Therapeutic Potential of Butyrate


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice and Models of Acute Pancreatitis (AP)
2.2. Treatments
2.3. β-Hydroxybutyric Acid (BHB) Assessment
2.4. Assessment in Serum and Tissue
2.5. Histology and Immunofluorescence Staining
2.6. Electron Microscopy (TEM)
2.7. Real-Time qPCR Analysis
2.8. 16s rRNA Gene Sequencing
2.9. 2bRad-M Gene Sequencing and Analysis
- S: the number of reads assigned to all 2bRAD markers belonging to species i within a sample.
- t: number of all 2bRAD markers of species i that have been sequenced within a sample.
2.10. Targeted (Short-Chain Fatty Acids) and Untargeted Metabolomics
2.11. Statistical Analysis
3. Results
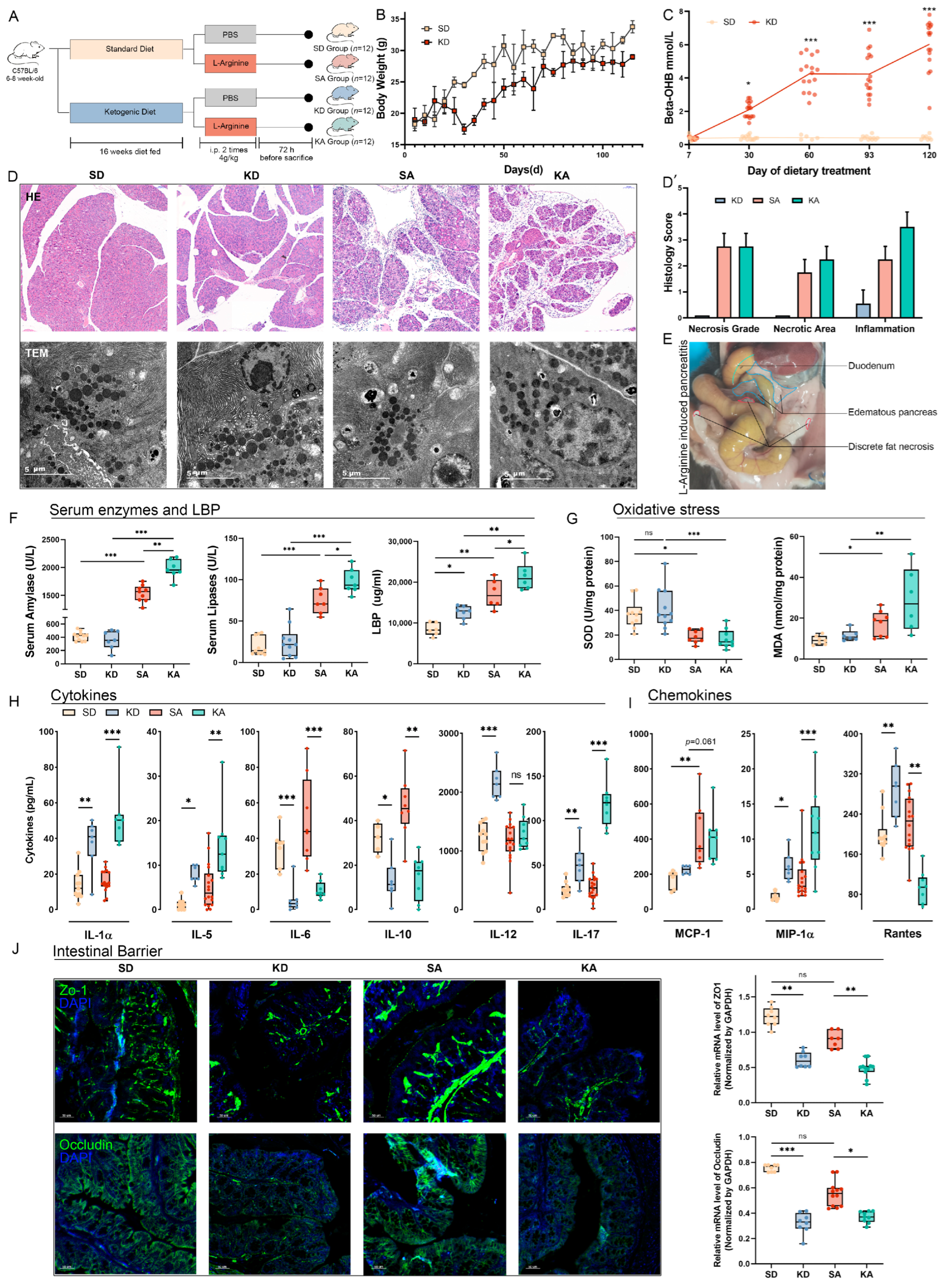
3.1. Ketogenic Diet Exacerbated L-Arginine-Induced Acute Pancreatitis and Induced Dysfunction of the Intestinal Barrier
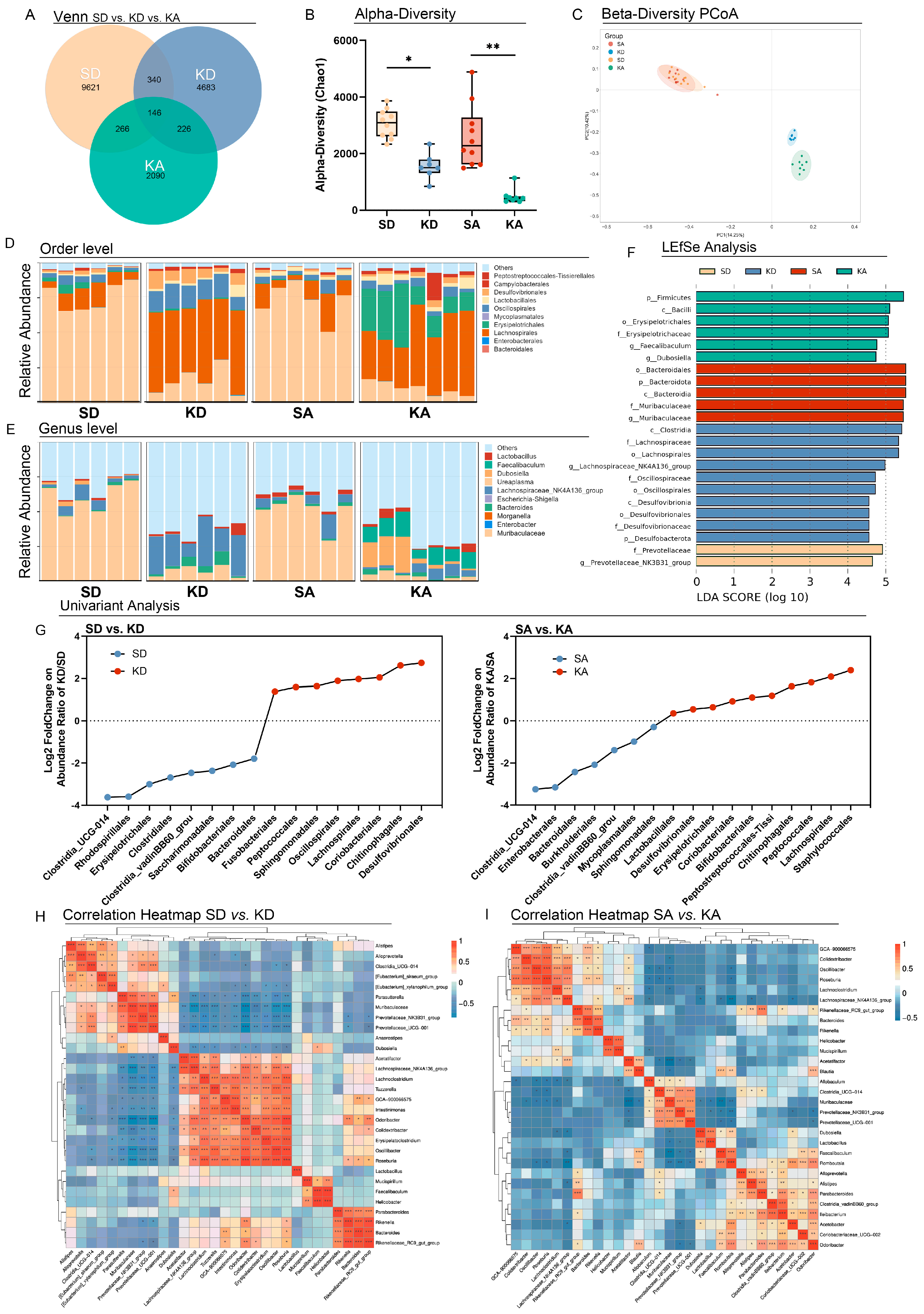
3.2. Ketogenic Diet Altered the Gut Microbiota in L-Arginine-Induced Acute Pancreatitis
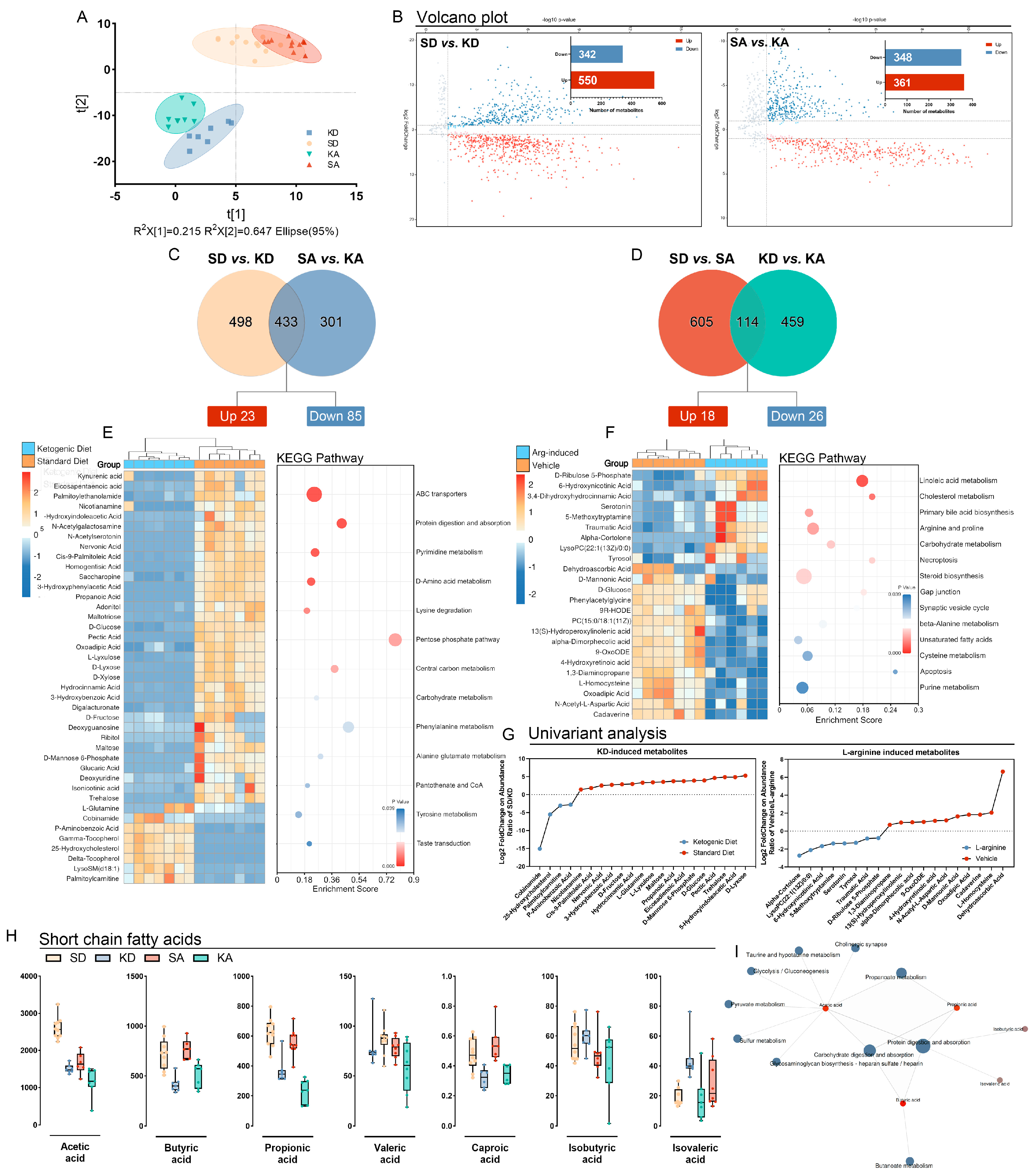
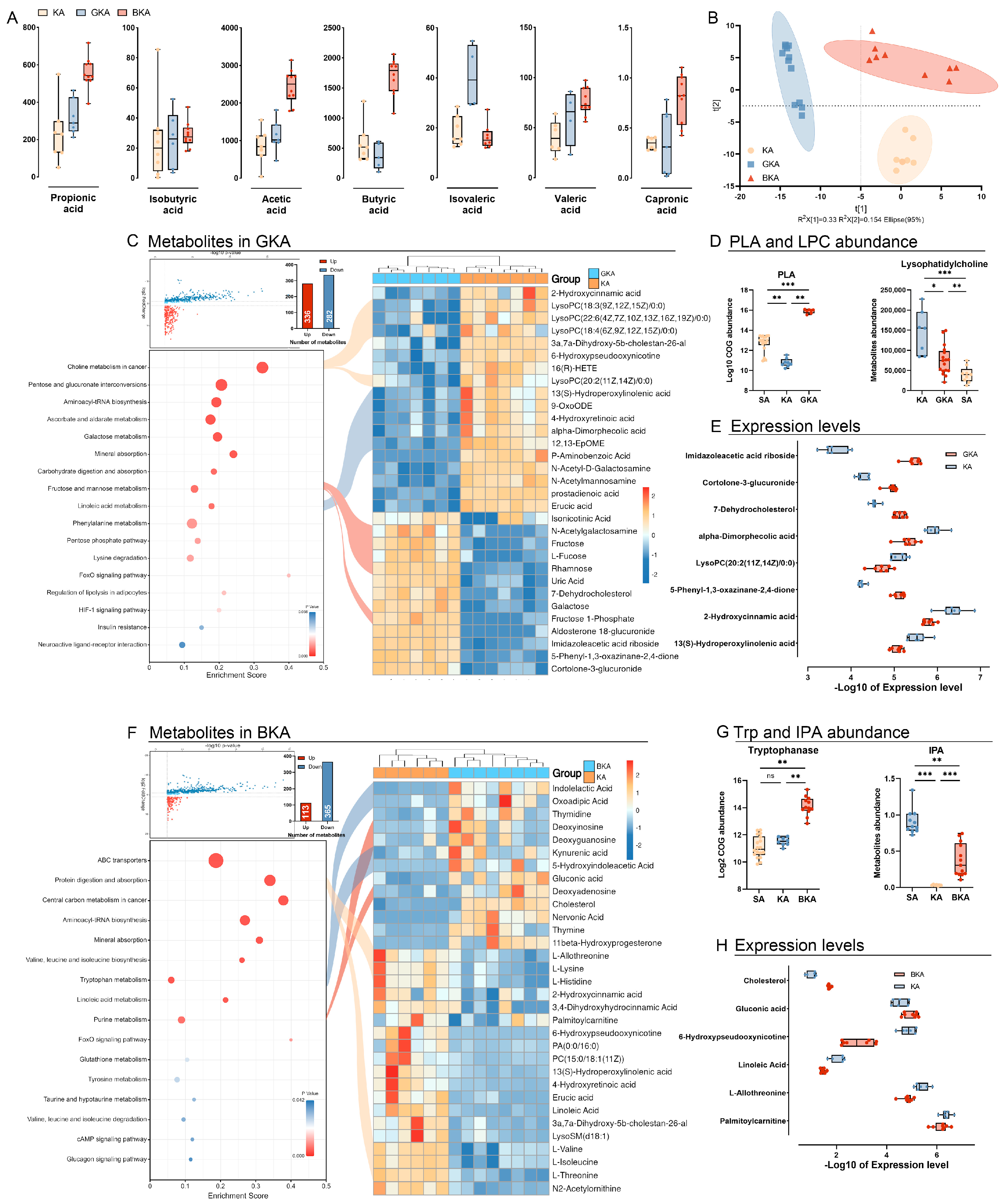
3.3. Ketogenic Diet Modulated Gut Metabolism and Resulted in the Depletion of Short-Chain Fatty Acids
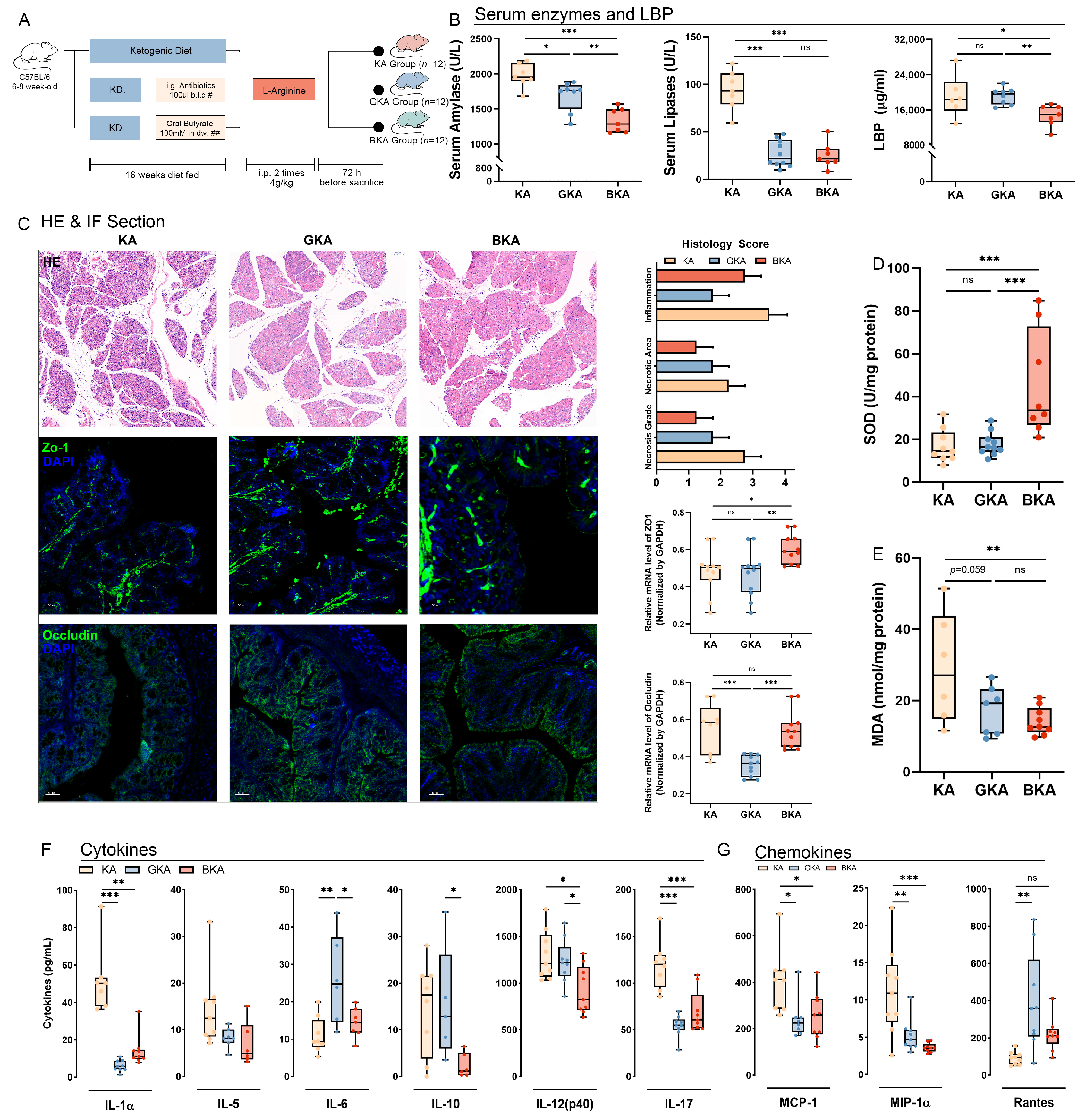
3.4. Germ-Depletion and Butyrate Supplementation Treatment Attenuate Necrosis of Pancreatic Tissue and Systemic Inflammation in L-Arginine-Induced Acute Pancreatitis
3.5. Germ-Depletion and Butyrate Supplement Treatment Altered Gut and Colonic Microbiota
3.6. Germ-Depletion and Butyric Acid Treatment Remodeling the Gut Metabolism
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cooder, H.R. Epilepsy in Children: With Particular Reference to the Ketogenic Diet. Calif. West. Med. 1933, 39, 169–173. [Google Scholar]
- Dashti, H.M.; Mathew, T.C.; Al-Zaid, N.S. Efficacy of Low-Carbohydrate Ketogenic Diet in the Treatment of Type 2 Diabetes. Med. Princ. Pract. Int. J. Kuwait Univ. Health Sci. Cent. 2021, 30, 223–235. [Google Scholar] [CrossRef]
- Pérez-Guisado, J. [Ketogenic diets: Additional benefits to the weight loss and unfounded secondary effects]. Arch. Latinoam. Nutr. 2008, 58, 323–329. [Google Scholar]
- Bhanpuri, N.H.; Hallberg, S.J.; Williams, P.T.; McKenzie, A.L.; Ballard, K.D.; Campbell, W.W.; McCarter, J.P.; Phinney, S.D.; Volek, J.S. Cardiovascular disease risk factor responses to a type 2 diabetes care model including nutritional ketosis induced by sustained carbohydrate restriction at 1 year: An open label, non-randomized, controlled study. Cardiovasc. Diabetol. 2018, 17, 56. [Google Scholar] [CrossRef] [PubMed]
- Kong, C.; Yan, X.; Liu, Y.; Huang, L.; Zhu, Y.; He, J.; Gao, R.; Kalady, M.F.; Goel, A.; Qin, H.; et al. Ketogenic diet alleviates colitis by reduction of colonic group 3 innate lymphoid cells through altering gut microbiome. Signal Transduct. Target. Ther. 2021, 6, 154. [Google Scholar] [CrossRef]
- Li, S.; Zhuge, A.; Wang, K.; Lv, L.; Bian, X.; Yang, L.; Xia, J.; Jiang, X.; Wu, W.; Wang, S.; et al. Ketogenic diet aggravates colitis, impairs intestinal barrier and alters gut microbiota and metabolism in DSS-induced mice. Food Funct. 2021, 12, 10210–10225. [Google Scholar] [CrossRef] [PubMed]
- Tagliabue, A.; Ferraris, C.; Uggeri, F.; Trentani, C.; Bertoli, S.; de Giorgis, V.; Veggiotti, P.; Elli, M. Short-term impact of a classical ketogenic diet on gut microbiota in GLUT1 Deficiency Syndrome: A 3-month prospective observational study. Clin. Nutr. ESPEN 2017, 17, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Paoli, A. Ketogenic diet for obesity: Friend or foe? Int. J. Environ. Res. Public Health 2014, 11, 2092–2107. [Google Scholar] [CrossRef] [PubMed]
- Romano, L.; Marchetti, M.; Gualtieri, P.; Di Renzo, L.; Belcastro, M.; De Santis, G.L.; Perrone, M.A.; De Lorenzo, A. Effects of a Personalized VLCKD on Body Composition and Resting Energy Expenditure in the Reversal of Diabetes to Prevent Complications. Nutrients 2019, 11, 1526. [Google Scholar] [CrossRef]
- Mavropoulos, J.C.; Yancy, W.S.; Hepburn, J.; Westman, E.C. The effects of a low-carbohydrate, ketogenic diet on the polycystic ovary syndrome: A pilot study. Nutr. Metab. 2005, 2, 35. [Google Scholar] [CrossRef] [PubMed]
- Lankisch, P.G.; Apte, M.; Banks, P.A. Acute pancreatitis. Lancet 2015, 386, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.J.; Papachristou, G.I. New insights into acute pancreatitis. Nature reviews. Gastroenterol. Hepatol. 2019, 16, 479–496. [Google Scholar]
- Gupta, M.; Liti, B.; Barrett, C.; Thompson, P.D.; Fernandez, A.B. Prevention and Management of Hypertriglyceridemia-Induced Acute Pancreatitis During Pregnancy: A Systematic Review. Am. J. Med. 2022, 135, 709–714. [Google Scholar] [CrossRef]
- Guo, Y.Y.; Li, H.X.; Zhang, Y.; He, W.H. Hypertriglyceridemia-induced acute pancreatitis: Progress on disease mechanisms and treatment modalities. Discov. Med. 2019, 27, 101–109. [Google Scholar]
- Biczo, G.; Vegh, E.T.; Shalbueva, N.; Mareninova, O.A.; Elperin, J.; Lotshaw, E.; Gretler, S.; Lugea, A.; Malla, S.R.; Dawson, D.; et al. Mitochondrial Dysfunction, Through Impaired Autophagy, Leads to Endoplasmic Reticulum Stress, Deregulated Lipid Metabolism, and Pancreatitis in Animal Models. Gastroenterology 2018, 154, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Long, W.B.; Grider, J.R., Jr. Amylase isoenzyme clearances in normal subjects and in patients with acute pancreatitis. Gastroenterology 1976, 71, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Meyer, E.; McKay, J.K.; Matz, M.V. 2b-RAD: A simple and flexible method for genome-wide genotyping. Nat. Methods 2012, 9, 808–810. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Huang, S.; Zhu, P.; Tzehau, L.; Zhao, H.; Lv, J.; Zhang, R.; Zhou, L.; Niu, Q.; Wang, X.; et al. Species-resolved sequencing of low-biomass or degraded microbiomes using 2bRAD-M. Genome Biol. 2022, 23, 36. [Google Scholar] [CrossRef] [PubMed]
- Zerem, E. Treatment of severe acute pancreatitis and its complications. World J. Gastroenterol. 2014, 20, 13879–13892. [Google Scholar] [CrossRef] [PubMed]
- Adak, A.; Khan, M.R. An insight into gut microbiota and its functionalities. Cell. Mol. Life Sci. CMLS 2019, 76, 473–493. [Google Scholar] [CrossRef]
- Martin-Gallausiaux, C.; Marinelli, L.; Blottière, H.M.; Larraufie, P.; Lapaque, N. SCFA: Mechanisms and functional importance in the gut. Proc. Nutr. Soc. 2021, 80, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.D.; Aminzadeh-Gohari, S.; Tulipan, J.; Catalano, L.; Feichtinger, R.G.; Kofler, B. Ketogenic diet in the treatment of cancer—Where do we stand? Mol. Metab. 2020, 33, 102–121. [Google Scholar] [CrossRef]
- Rescigno, M. The intestinal epithelial barrier in the control of homeostasis and immunity. Trends Immunol. 2011, 32, 256–264. [Google Scholar] [CrossRef]
- Linsalata, M.; Russo, F.; Riezzo, G.; D’Attoma, B.; Prospero, L.; Orlando, A.; Ignazzi, A.; Di Chito, M.; Sila, A.; De Nucci, S.; et al. The Effects of a Very-Low-Calorie Ketogenic Diet on the Intestinal Barrier Integrity and Function in Patients with Obesity: A Pilot Study. Nutrients 2023, 15, 2561. [Google Scholar] [CrossRef]
- Zmora, N.; Suez, J.; Elinav, E. You are what you eat: Diet, health and the gut microbiota. Nature reviews. Gastroenterol. Hepatol. 2019, 16, 35–56. [Google Scholar]
- Gudi, R.; Suber, J.; Brown, R.; Johnson, B.M.; Vasu, C. Pretreatment with Yeast-Derived Complex Dietary Polysaccharides Suppresses Gut Inflammation, Alters the Microbiota Composition, and Increases Immune Regulatory Short-Chain Fatty Acid Production in C57BL/6 Mice. J. Nutr. 2020, 150, 1291–1302. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wang, Y.; Yang, G.; Zhang, Q.; Meng, L.; Xin, Y.; Jiang, X. The role of short-chain fatty acids in intestinal barrier function, inflammation, oxidative stress, and colonic carcinogenesis. Pharmacol. Res. 2021, 165, 105420. [Google Scholar] [CrossRef]
- Isenmann, R.; Rau, B.; Beger, H.G. Early severe acute pancreatitis: Characteristics of a new subgroup. Pancreas 2001, 22, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Al Mofleh, I.A. Severe acute pancreatitis: Pathogenetic aspects and prognostic factors. World J. Gastroenterol. 2008, 14, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Bugiantella, W.; Rondelli, F.; Boni, M.; Stella, P.; Polistena, A.; Sanguinetti, A.; Avenia, N. Necrotizing pancreatitis: A review of the interventions. Int. J. Surg. 2016, 28 (Suppl. 1), S163–S171. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Chen, J.; Yi, K.; Peng, L.; Xie, J.; Gou, X.; Peng, T.; Tang, L. Phlorizin ameliorates obesity-associated endotoxemia and insulin resistance in high-fat diet-fed mice by targeting the gut microbiota and intestinal barrier integrity. Gut Microbes 2020, 12, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.K.; Howden, C.W. Prophylactic antibiotic administration reduces sepsis and mortality in acute necrotizing pancreatitis: A meta-analysis. Pancreas 2001, 22, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Petrov, M.S.; Kukosh, M.V.; Emelyanov, N.V. A randomized controlled trial of enteral versus parenteral feeding in patients with predicted severe acute pancreatitis shows a significant reduction in mortality and in infected pancreatic complications with total enteral nutrition. Dig. Surg. 2006, 23, 336–344; discussion 344–345. [Google Scholar] [CrossRef] [PubMed]
- Baron, T.H.; DiMaio, C.J.; Wang, A.Y.; Morgan, K.A. American Gastroenterological Association Clinical Practice Update: Management of Pancreatic Necrosis. Gastroenterology 2020, 158, 67–75.e1. [Google Scholar] [CrossRef]
- Jiang, K.; Huang, W.; Yang, X.N.; Xia, Q. Present and future of prophylactic antibiotics for severe acute pancreatitis. World J. Gastroenterol. 2012, 18, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Dai, W.; Shen, J.; Zhang, M.; Shi, Y.; Jiang, K.; Guo, L. Assessment of Prophylactic Carbapenem Antibiotics Administration for Severe Acute Pancreatitis: An Updated Systematic Review and Meta-Analysis. Digestion 2022, 103, 183–191. [Google Scholar] [CrossRef]
- Rogers, A.W.L.; Tsolis, R.M.; Bäumler, A.J. Salmonella versus the Microbiome. Microbiol. Mol. Biol. Rev. MMBR 2021, 85, e00027-19. [Google Scholar] [CrossRef]
- Tang, X.; Wang, W.; Hong, G.; Duan, C.; Zhu, S.; Tian, Y.; Han, C.; Qian, W.; Lin, R.; Hou, X. Gut microbiota-mediated lysophosphatidylcholine generation promotes colitis in intestinal epithelium-specific Fut2 deficiency. J. Biomed. Sci. 2021, 28, 20. [Google Scholar] [CrossRef]
- Macfarlane, S.; Macfarlane, G.T. Regulation of short-chain fatty acid production. Proc. Nutr. Soc. 2003, 62, 67–72. [Google Scholar] [CrossRef]
- Blaak, E.E.; Canfora, E.E.; Theis, S.; Frost, G.; Groen, A.K.; Mithieux, G.; Nauta, A.; Scott, K.; Stahl, B.; van Harsselaar, J.; et al. Short chain fatty acids in human gut and metabolic health. Benef. Microbes 2020, 11, 411–455. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, F.; Wu, W.; Sun, M.; Bilotta, A.J.; Yao, S.; Xiao, Y.; Huang, X.; Eaves-Pyles, T.D.; Golovko, G.; et al. GPR43 mediates microbiota metabolite SCFA regulation of antimicrobial peptide expression in intestinal epithelial cells via activation of mTOR and STAT3. Mucosal Immunol. 2018, 11, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Li, Z.R.; Green, R.S.; Holzman, I.R.; Lin, J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J. Nutr. 2009, 139, 1619–1625. [Google Scholar] [CrossRef]
- Nikolaus, S.; Schulte, B.; Al-Massad, N.; Thieme, F.; Schulte, D.M.; Bethge, J.; Rehman, A.; Tran, F.; Aden, K.; Häsler, R.; et al. Increased Tryptophan Metabolism Is Associated with Activity of Inflammatory Bowel Diseases. Gastroenterology 2017, 153, 1504–1516.e2. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, M.; Mukherjee, S.; Wang, H.; Li, H.; Sun, K.; Benechet, A.P.; Qiu, Z.; Maher, L.; Redinbo, M.R.; Phillips, R.S.; et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity 2014, 41, 296–310. [Google Scholar] [CrossRef]
- Beam, A.; Clinger, E.; Hao, L. Effect of Diet and Dietary Components on the Composition of the Gut Microbiota. Nutrients 2021, 13, 2795. [Google Scholar] [CrossRef]
- Holscher, H.D. Dietary fiber and prebiotics and the gastrointestinal microbiota. Gut Microbes 2017, 8, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, F.; Cechova, K.; Amlerova, J.; Hort, J. Antibiotics, gut microbiota, and Alzheimer’s disease. J. Neuroinflammation 2019, 16, 108. [Google Scholar] [CrossRef]
- Weingarden, A.R.; Vaughn, B.P. Intestinal microbiota, fecal microbiota transplantation, and inflammatory bowel disease. Gut Microbes 2017, 8, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Wieërs, G.; Belkhir, L.; Enaud, R.; Leclercq, S.; Philippart de Foy, J.M.; Dequenne, I.; de Timary, P.; Cani, P.D. How Probiotics Affect the Microbiota. Front. Cell. Infect. Microbiol. 2019, 9, 454. [Google Scholar] [CrossRef]
- Li, H.Y.; Zhou, D.D.; Gan, R.Y.; Huang, S.Y.; Zhao, C.N.; Shang, A.; Xu, X.Y.; Li, H.B. Effects and Mechanisms of Probiotics, Prebiotics, Synbiotics, and Postbiotics on Metabolic Diseases Targeting Gut Microbiota: A Narrative Review. Nutrients 2021, 13, 3211. [Google Scholar] [CrossRef]
- Villatoro, E.; Mulla, M.; Larvin, M. Antibiotic therapy for prophylaxis against infection of pancreatic necrosis in acute pancreatitis. Cochrane Database Syst. Rev. 2010, 2010, Cd002941. [Google Scholar] [CrossRef] [PubMed]
- Jafri, N.S.; Mahid, S.S.; Idstein, S.R.; Hornung, C.A.; Galandiuk, S. Antibiotic prophylaxis is not protective in severe acute pancreatitis: A systematic review and meta-analysis. Am. J. Surg. 2009, 197, 806–813. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, H.; Guo, J.; Shen, J.; Jiang, S.; Han, S.; Li, L. Ketogenic Diet Exacerbates L-Arginine-Induced Acute Pancreatitis and Reveals the Therapeutic Potential of Butyrate. Nutrients 2023, 15, 4427. https://doi.org/10.3390/nu15204427
Xia H, Guo J, Shen J, Jiang S, Han S, Li L. Ketogenic Diet Exacerbates L-Arginine-Induced Acute Pancreatitis and Reveals the Therapeutic Potential of Butyrate. Nutrients. 2023; 15(20):4427. https://doi.org/10.3390/nu15204427
Chicago/Turabian StyleXia, He, Jing Guo, Jian Shen, Shiman Jiang, Shengyi Han, and Lanjuan Li. 2023. "Ketogenic Diet Exacerbates L-Arginine-Induced Acute Pancreatitis and Reveals the Therapeutic Potential of Butyrate" Nutrients 15, no. 20: 4427. https://doi.org/10.3390/nu15204427
APA StyleXia, H., Guo, J., Shen, J., Jiang, S., Han, S., & Li, L. (2023). Ketogenic Diet Exacerbates L-Arginine-Induced Acute Pancreatitis and Reveals the Therapeutic Potential of Butyrate. Nutrients, 15(20), 4427. https://doi.org/10.3390/nu15204427