
Fucoxanthin Attenuates the Reprogramming of Energy Metabolism during the Activation of Hepatic Stellate Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Primary Mouse HSC Isolation and Culture
2.2. FCX Treatment
2.3. Energy Metabolism of Cells
2.4. Quantitative Real-Time PCR (qRT-PCR)
2.5. Statistical Analysis
3. Results
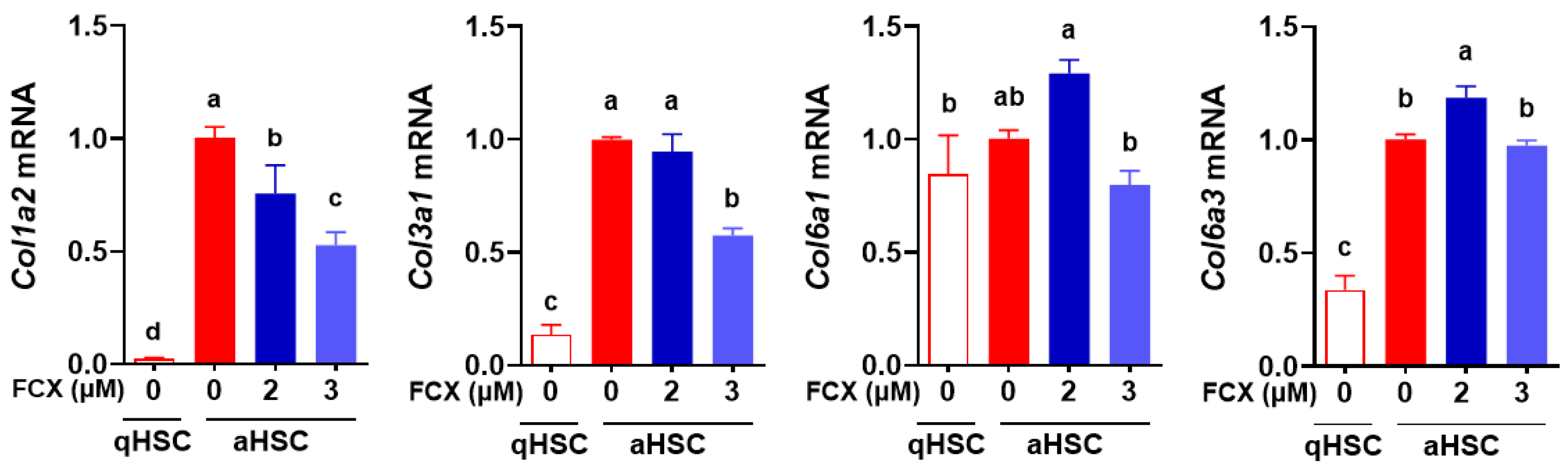
3.1. FCX Abolished the Induction of Collagen Genes during HSC Activation
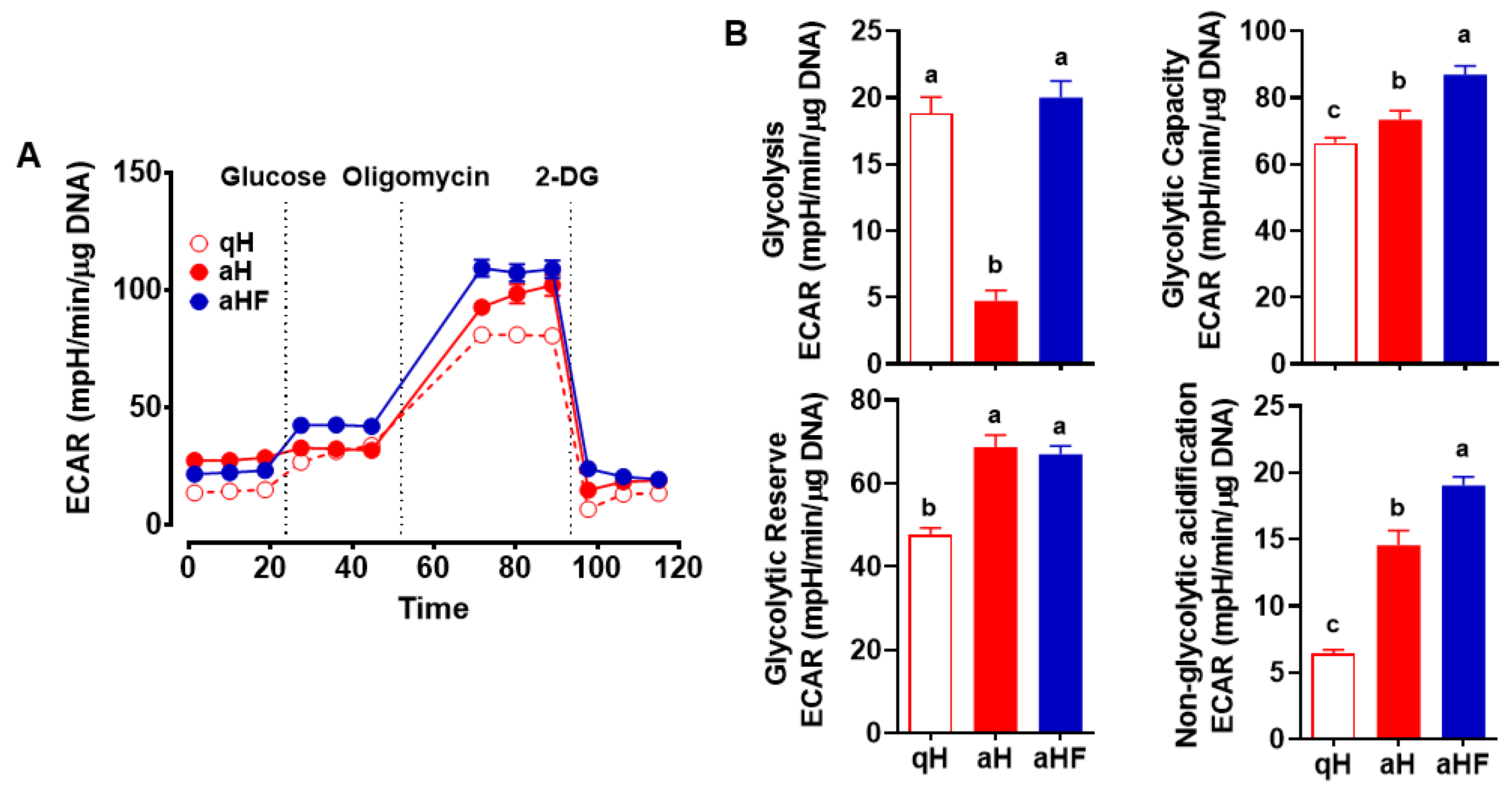
3.2. Decreased Glycolysis in Primary Mouse aHSC was Inhibited by FCX
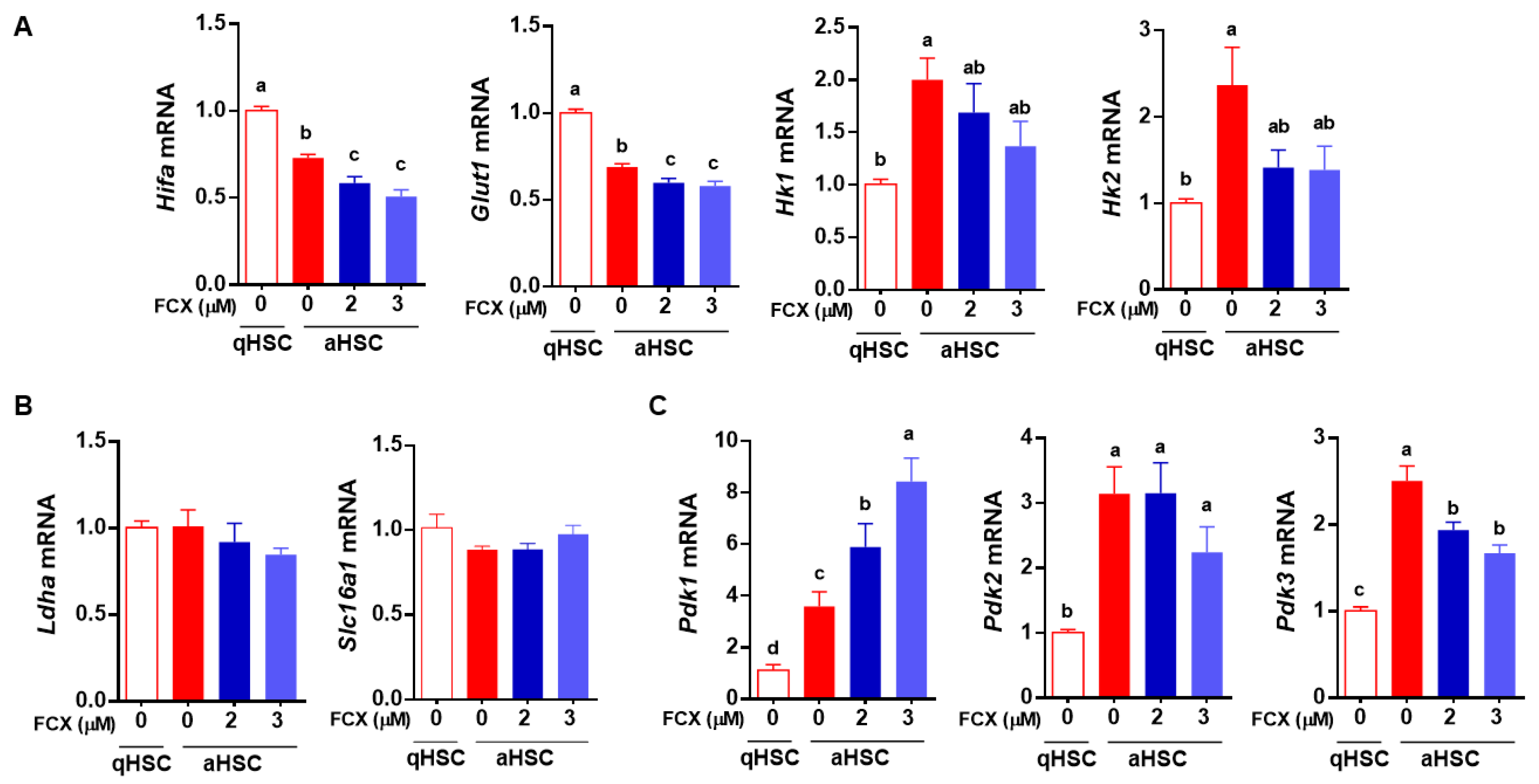
3.3. FCX Partially Attenuated Changes in the Expression of Genes Involved in Glycolysis during HSC Activation
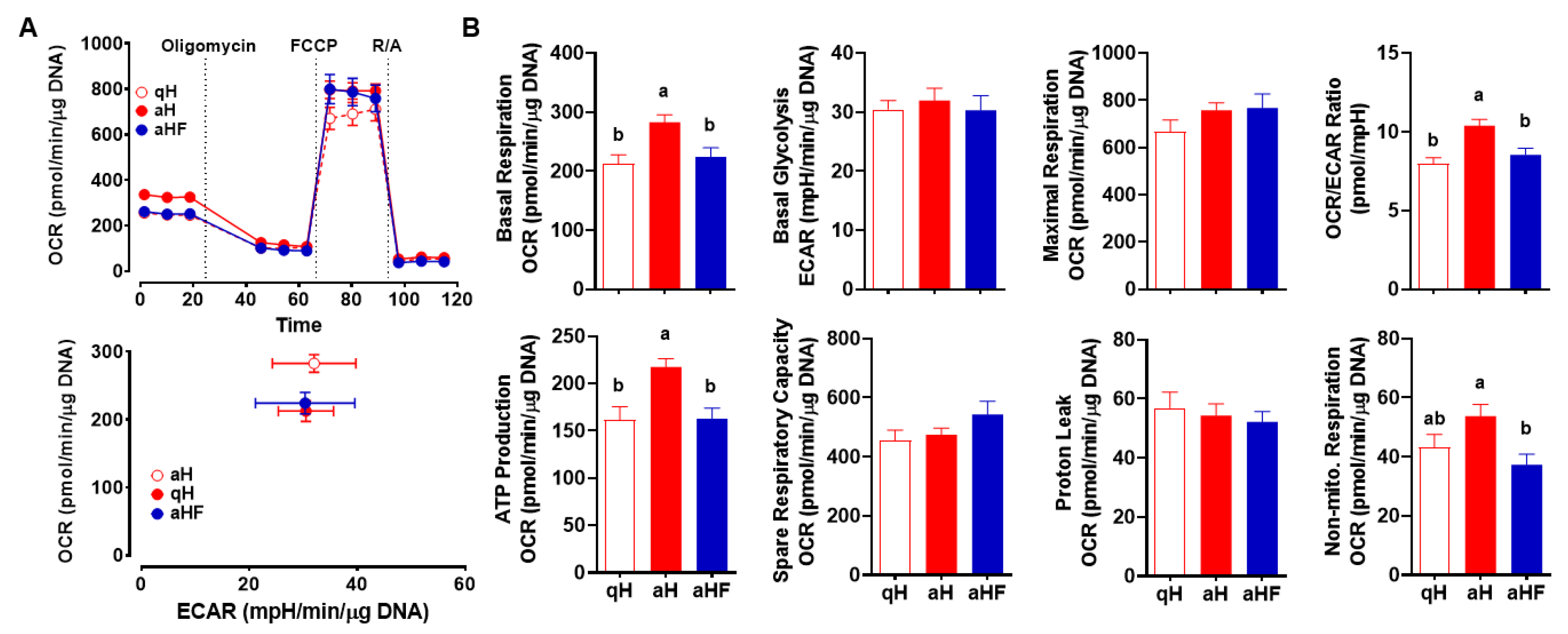
3.4. FCX Inhibited an Increase in Mitochondrial Respiration in Primary Mouse aHSC
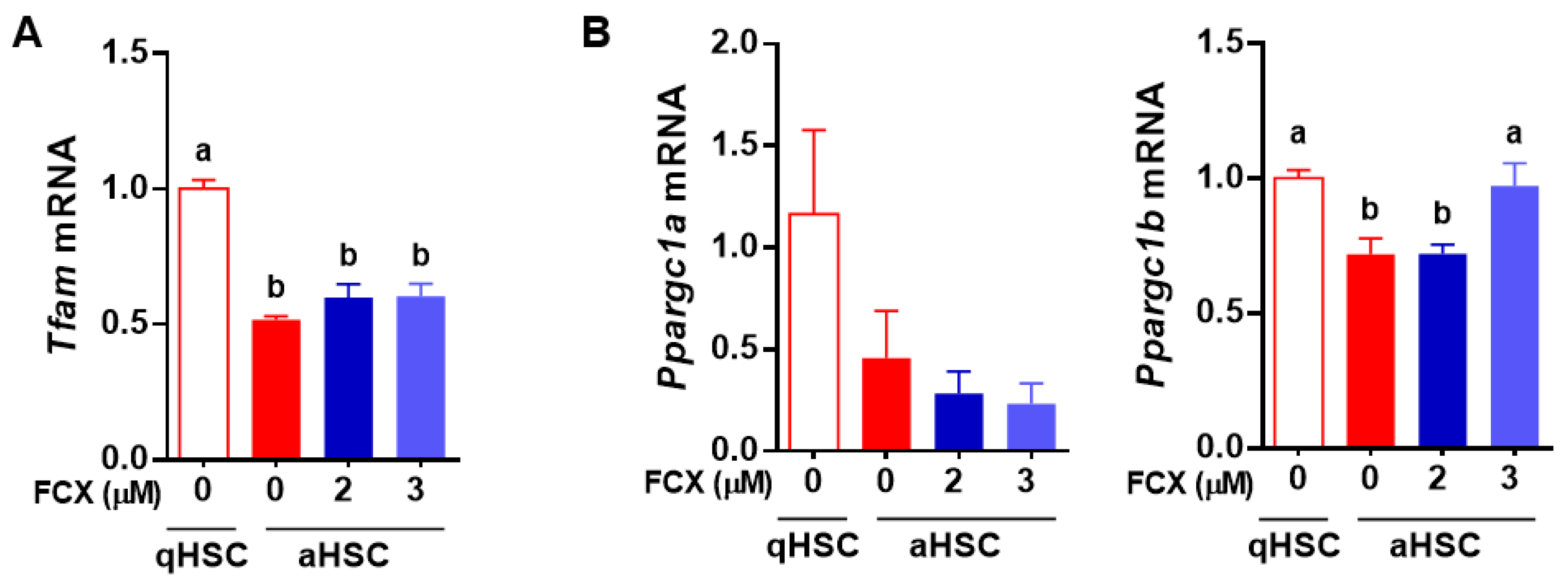
3.5. FCX Prevented a Reduction in Ppargc1a Expression in Primary Mouse aHSC
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Friedman, S.L. Liver fibrosis—From bench to bedside. J. Hepatol. 2003, 38, 38–53. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Crespo Yanguas, S.; Cogliati, B.; Willebrords, J.; Maes, M.; Colle, I.; Van den Bossche, B.; de Oliveira, C.P.M.S.; Andraus, W.; Alves, V.A.; Leclercq, I. Experimental models of liver fibrosis. Arch. Toxicol. 2016, 90, 1025–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef]
- Wells, R.G. Cellular sources of extracellular matrix in hepatic fibrosis. Clin. Liver Dis. 2008, 12, 759–768. [Google Scholar] [CrossRef] [Green Version]
- Hellerbrand, C. Hepatic stellate cells—The pericytes in the liver. Pflügers Arch. Eur. J. Physiol. 2013, 465, 775–778. [Google Scholar] [CrossRef]
- Blomhoff, R.; Blomhoff, H.K. Overview of retinoid metabolism and function. J. Neurobiol. 2006, 66, 606–630. [Google Scholar] [CrossRef]
- Wu, J.; Zern, M.A. Hepatic stellate cells: A target for the treatment of liver fibrosis. J. Gastroenterol. 2000, 35, 665–672. [Google Scholar] [CrossRef]
- Yetkin-Arik, B.; Vogels, I.; Nowak-Sliwinska, P.; Weiss, A.; Houtkooper, R.H.; Van Noorden, C.J.; Klaassen, I.; Schlingemann, R.O. The role of glycolysis and mitochondrial respiration in the formation and functioning of endothelial tip cells during angiogenesis. Sci. Rep. 2019, 9, 12608. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. The metabolism of carcinoma cells. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef] [Green Version]
- van der Windt, G.J.; Pearce, E.L. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol. Rev. 2012, 249, 27–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The metabolic signature of macrophage responses. Front. Immunol. 2019, 1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendoza, R.P.; Fudge, D.H.; Brown, J.M. Cellular Energetics of Mast Cell Development and Activation. Cells 2021, 10, 524. [Google Scholar] [CrossRef] [PubMed]
- Negmadjanov, U.; Godic, Z.; Rizvi, F.; Emelyanova, L.; Ross, G.; Richards, J.; Holmuhamedov, E.L.; Jahangir, A. TGF-β1-mediated differentiation of fibroblasts is associated with increased mitochondrial content and cellular respiration. PLoS ONE 2015, 10, e0123046. [Google Scholar]
- Bae, M.; Lee, Y.; Park, Y.-K.; Shin, D.-G.; Joshi, P.; Hong, S.-H.; Alder, N.; Koo, S.I.; Lee, J.-Y. Astaxanthin attenuates the increase in mitochondrial respiration during the activation of hepatic stellate cells. J. Nutr. Biochem. 2019, 71, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Bae, M.; Lee, Y.; Pham, T.X.; Hu, S.; Park, Y.-K.; Lee, J.-Y. Astaxanthin inhibits the reduction of glycolysis during the activation of hepatic stellate cells. Life Sci. 2020, 256, 117926. [Google Scholar] [CrossRef]
- Peng, J.; Yuan, J.-P.; Wu, C.-F.; Wang, J.-H. Fucoxanthin, a marine carotenoid present in brown seaweeds and diatoms: Metabolism and bioactivities relevant to human health. Mar. Drugs 2011, 9, 1806–1828. [Google Scholar] [CrossRef]
- Bae, M.; Kim, M.-B.; Park, Y.-K.; Lee, J.-Y. Health benefits of fucoxanthin in the prevention of chronic diseases. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158618. [Google Scholar] [CrossRef]
- Kim, M.-B.; Bae, M.; Hu, S.; Kang, H.; Park, Y.-K.; Lee, J.-Y. Fucoxanthin exerts anti-fibrogenic effects in hepatic stellate cells. Biochem. Biophys. Res. Commun. 2019, 513, 657–662. [Google Scholar] [CrossRef]
- Yang, Y.; Kim, B.; Park, Y.K.; Koo, S.I.; Lee, J.Y. Astaxanthin prevents TGFbeta1-induced pro-fibrogenic gene expression by inhibiting Smad3 activation in hepatic stellate cells. Biochim. Biophys. Acta 2015, 1850, 178–185. [Google Scholar] [CrossRef]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Bae, M.; Kim, B.; Park, Y.K.; Koo, S.I.; Lee, J.Y. Astaxanthin prevents and reverses the activation of mouse primary hepatic stellate cells. J. Nutr. Biochem. 2016, 29, 21–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, T.X.; Park, Y.-K.; Bae, M.; Lee, J.-Y. The Potential Role of an Endotoxin Tolerance-Like Mechanism for the Anti-inflammatory Effect of Spirulina platensis Organic Extract in Macrophages. J. Med. Food 2017, 20, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.-K.; Rasmussen, H.E.; Ehlers, S.J.; Blobaum, K.R.; Lu, F.; Schlegal, V.L.; Carr, T.P.; Lee, J.-Y. Repression of proinflammatory gene expression by lipid extract of Nostoc commune var sphaeroides Kützing, a blue-green alga, via inhibition of nuclear factor-κB in RAW 264.7 macrophages. Nutr. Res. 2008, 28, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, H.E.; Blobaum, K.R.; Park, Y.-K.; Ehlers, S.J.; Lu, F.; Lee, J.-Y. Lipid extract of Nostoc commune var. sphaeroides Kützing, a blue-green alga, inhibits the activation of sterol regulatory element binding proteins in HepG2 cells. J. Nutr. 2008, 138, 476–481. [Google Scholar] [CrossRef] [Green Version]
- Van Wijk, R.; Van Solinge, W.W. The energy-less red blood cell is lost: Erythrocyte enzyme abnormalities of glycolysis. Blood 2005, 106, 4034–4042. [Google Scholar] [CrossRef]
- Krieg, A.F.; Rosenblum, L.J.; Henry, J.B. Lactate dehydrogenase isoenzymes: A comparison of pyruvate-to-lactate and lactate-to-pyruvate assays. Clin. Chem. 1967, 13, 196–203. [Google Scholar] [CrossRef]
- Bosshart, P.D.; Kalbermatter, D.; Bonetti, S.; Fotiadis, D. Mechanistic basis of L-lactate transport in the SLC16 solute carrier family. Nat. Commun. 2019, 10, 2649. [Google Scholar] [CrossRef]
- Patel, M.S.; Korotchkina, L.G. The biochemistry of the pyruvate dehydrogenase complex. Biochem. Mol. Biol. Educ. 2003, 31, 5–15. [Google Scholar] [CrossRef]
- Ngo, H.B.; Lovely, G.A.; Phillips, R.; Chan, D.C. Distinct structural features of TFAM drive mitochondrial DNA packaging versus transcriptional activation. Nat. Commun. 2014, 5, 3077. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Mikami, K.; Hosokawa, M. Biosynthetic pathway and health benefits of fucoxanthin, an algae-specific xanthophyll in brown seaweeds. Int. J. Mol. Sci. 2013, 14, 13763–13781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Takatani, N.; Kono, Y.; Beppu, F.; Okamatsu-Ogura, Y.; Yamano, Y.; Miyashita, K.; Hosokawa, M. Fucoxanthin inhibits hepatic oxidative stress, inflammation, and fibrosis in diet-induced non-alcoholic steatohepatitis model mice. Biochem. Biophys. Res. Commun. 2020, 528, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Giralt, A.; Fajas, L. Metabolic Adaptation to Cell Growth and Proliferation in Normal and Pathological Conditions. Front. Endocrinol. 2017, 8, 362. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Caputa, G.; Castoldi, A.; Pearce, E.J. Metabolic adaptations of tissue-resident immune cells. Nat. Immunol. 2019, 20, 793–801. [Google Scholar] [CrossRef]
- Chaudhry, R.; Varacallo, M. Biochemistry, Glycolysis; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Mookerjee, S.A.; Nicholls, D.G.; Brand, M.D. Determining maximum glycolytic capacity using extracellular flux measurements. PLoS ONE 2016, 11, e0152016. [Google Scholar] [CrossRef] [Green Version]
- Liang, Q.; Donthi, R.V.; Kralik, P.M.; Epstein, P.N. Elevated hexokinase increases cardiac glycolysis in transgenic mice. Cardiovasc. Res. 2002, 53, 423–430. [Google Scholar] [CrossRef]
- Roberts, D.; Miyamoto, S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2015, 22, 248–257. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P. The monocarboxylate transporter family—Structure and functional characterization. IUBMB Life 2012, 64, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, S.; Potter, J.J.; Geschwind, J.-F.; Sur, S.; Hamilton, J.P.; Vogelstein, B.; Kinzler, K.W.; Mezey, E.; Ganapathy-Kanniappan, S. Deregulation of energy metabolism promotes anti-fibrotic effects in human hepatic stellate cells and prevents liver fibrosis in a mouse model. Biochem. Biophys. Res. Commun. 2016, 469, 463–469. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.; Liu, Y.; Liu, X.; Zhu, L.; Cui, Y.; Cui, A.; Qiao, A.; Kong, X.; Liu, Y.; Chen, Q. PGC-1β-regulated mitochondrial biogenesis and function in myotubes is mediated by NRF-1 and ERRα. Mitochondrion 2010, 10, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.S.; Nemeria, N.S.; Furey, W.; Jordan, F. The pyruvate dehydrogenase complexes: Structure-based function and regulation. J. Biol. Chem. 2014, 289, 16615–16623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bae, M.; Kim, M.-B.; Lee, J.-Y. Fucoxanthin Attenuates the Reprogramming of Energy Metabolism during the Activation of Hepatic Stellate Cells. Nutrients 2022, 14, 1902. https://doi.org/10.3390/nu14091902
Bae M, Kim M-B, Lee J-Y. Fucoxanthin Attenuates the Reprogramming of Energy Metabolism during the Activation of Hepatic Stellate Cells. Nutrients. 2022; 14(9):1902. https://doi.org/10.3390/nu14091902
Chicago/Turabian StyleBae, Minkyung, Mi-Bo Kim, and Ji-Young Lee. 2022. "Fucoxanthin Attenuates the Reprogramming of Energy Metabolism during the Activation of Hepatic Stellate Cells" Nutrients 14, no. 9: 1902. https://doi.org/10.3390/nu14091902
APA StyleBae, M., Kim, M.-B., & Lee, J.-Y. (2022). Fucoxanthin Attenuates the Reprogramming of Energy Metabolism during the Activation of Hepatic Stellate Cells. Nutrients, 14(9), 1902. https://doi.org/10.3390/nu14091902