
25(OH)Vitamin D Deficiency and Calcifediol Treatment in Pediatrics


Abstract
:1. Introduction
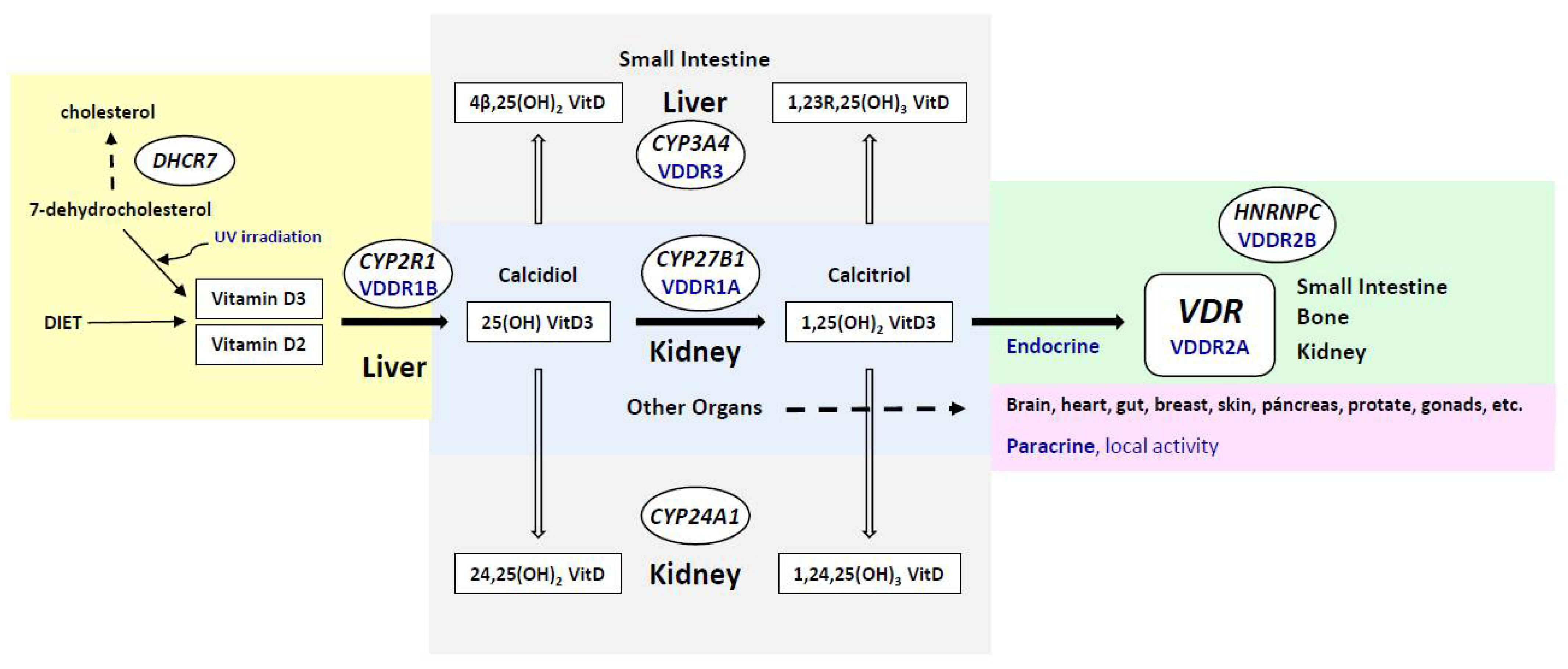
2. Metabolism of the Vitamin D Endocrine System: Role of Genetic Factors
3. Prevalence of Calcifediol Deficiency and Insufficiency in Childhood
4. Risk Factors for Calcifediol Deficiency in Childhood
4.1. Lack of Sun Exposure
4.2. Pregnancy and Lactation
4.3. Prematurity
4.4. Obesity
4.5. Children with Chronic Diseases
- ○
- Kidney disease: calcifediol deficiency is frequent and may be severe in children and adults with chronic kidney disease [37].
- ○
- Liver disease: severe chronic liver disease leads to altered synthesis and impaired absorption of vitamin D due to impaired bile acid production or intestinal edema secondary to portal hypertension [36].
- ○
- Inflammatory bowel disease: vitamin D deficiency is very frequent in children with inflammatory bowel disease and other malabsorption bowel diseases such as cystic fibrosis due to malabsorption. In addition, several studies have reported a higher severity of inflammatory bowel disease secondary to vitamin D deficiency [38].
4.6. Others
- ○
- Drugs: some treatments may interfere with vitamin D metabolism, either by increased catabolism of 25(OH)D or 1,25(OH)2D (antiepileptic or antiretroviral drugs), inhibition of intestinal absorption of vitamin D (glucocorticoids), or an increased requirement due to hydroxylation blocking (ketoconazole) [39].
- ○
- Nephrotic syndrome: low serum levels of 25(OH)D can be seen in relation to an increased urinary loss of vitamin D-binding protein and of vitamin D itself [40].
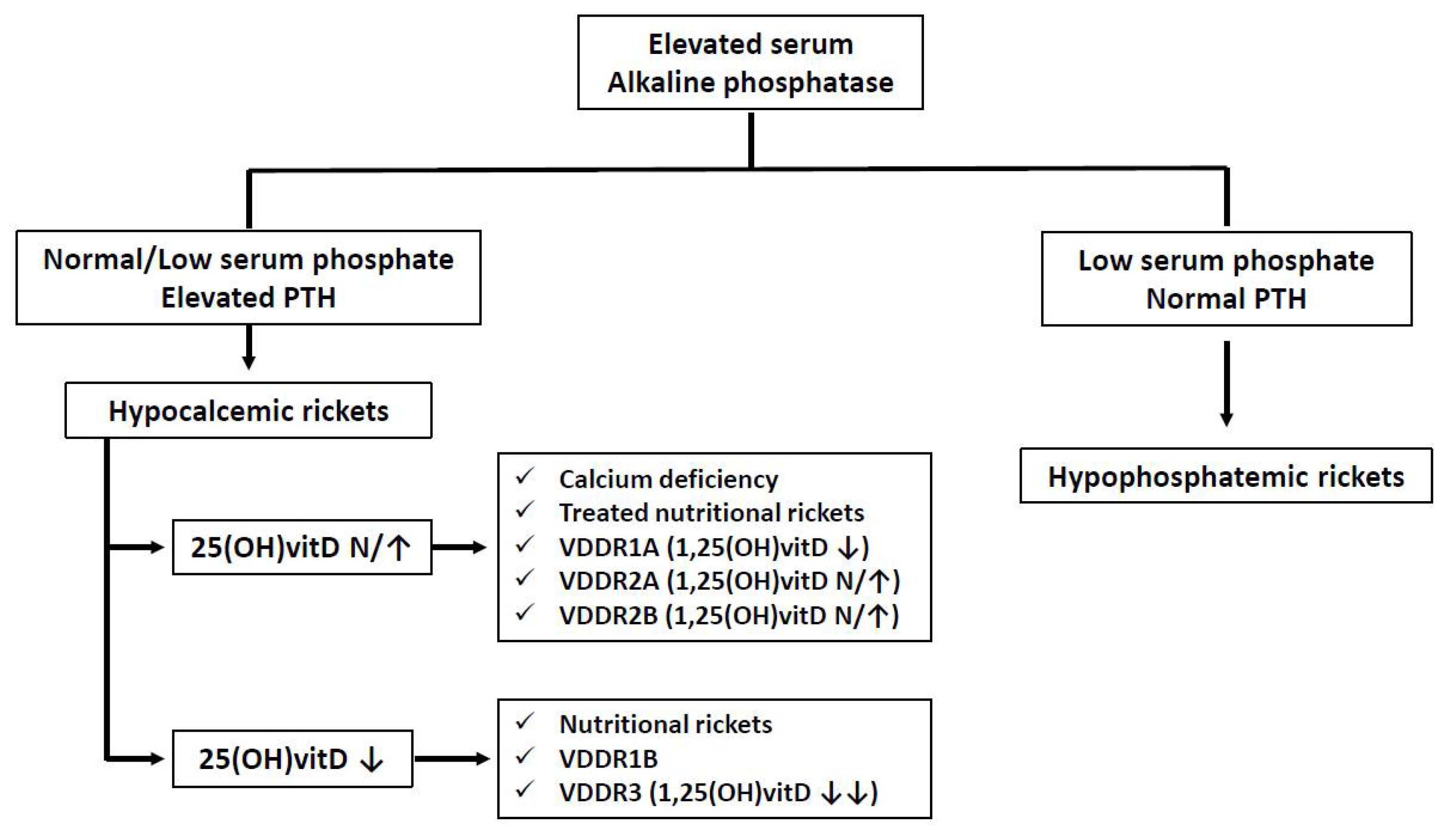
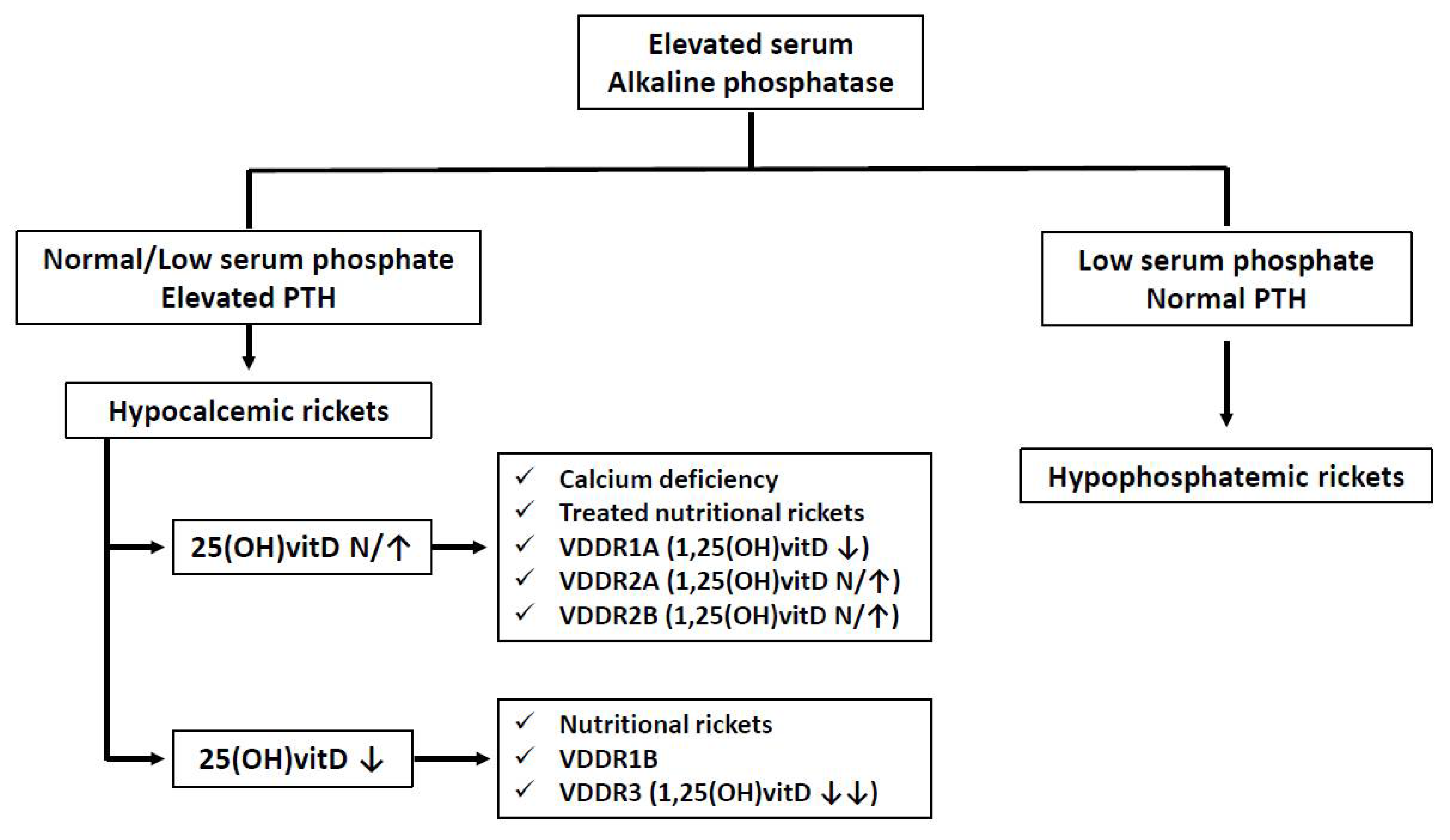
5. Clinical Manifestations of Calcifediol Deficiency in Childhood
5.1. Calcifediol Deficiency and Bone Metabolism in Childhood
5.2. Other Anomalies Associated with Calcifediol Deficiency
6. Treatment of Calcifediol Deficiency in Childhood
6.1. Vitamin D for the Prevention of Nutritional Rickets
6.2. Vitamin D for the Treatment of Nutritional Rickets
6.3. Treatment of Genetic Forms of Rickets
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yamshchikov, A.V.; Desai, N.S.; Blumberg, H.M.; Ziegler, T.R.; Tangpricha, V. Vitamin D for treatment and prevention of infectious diseases: A systematic review of randomized controlled trials. Endocr. Pract. 2009, 15, 438–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantorna, M.T.; Snyder, L.; Lin, Y.D.; Yang, L. Vitamin D and 1,25(OH)2D regulation of T cells. Nutrients 2015, 7, 3011–3021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lifschitz, C. Vitamin D. Ann. Nutr. Metab. 2020, 76 (Suppl. S2), 1–4. [Google Scholar] [CrossRef] [PubMed]
- Elder, C.J.; Bishop, N.J. Rickets. Lancet 2014, 383, 1665–1676. [Google Scholar] [CrossRef]
- Taylor, S.N. Vitamin D in Toddlers, Preschool Children, and Adolescents. Ann. Nutr. Metab. 2020, 76 (Suppl. S2), 30–40. [Google Scholar] [CrossRef] [PubMed]
- Hilger, J.; Friedel, A.; Herr, R.; Rausch, T. A systematic review of vitamin D status in populations worldwide. Br. J. Nutr. 2014, 111, 23–45. [Google Scholar] [CrossRef] [Green Version]
- Amrein, K.; Scherkl, M.; Hoffmann, M.; Neuwersch-Sommeregger, S.; Köstenberger, M.; Tmava Berisha, A.; Malle, O. Vitamin D deficiency 2.0: An update on the current status worldwide. Eur. J. Clin. Nutr. 2020, 74, 1498–1513. [Google Scholar] [CrossRef]
- Michaëlsson, K.; Baron, J.A.; Snellman, G.; Gedeborg, R. Plasma vitamin D and mortality in older men: A community-based prospective cohort study. Am. J. Clin. Nutr. 2010, 92, 841–848. [Google Scholar] [CrossRef] [Green Version]
- Stolzenberg-Solomon, R.Z.; Jacobs, E.J.; Arslan, A.A.; Qi, D. Circulating 25-hydroxyvitamin D and risk of pancreatic cancer: Cohort Consortium Vitamin D Pooling Project of Rarer Cancers. Am. J. Epidemiol. 2010, 172, 81–93. [Google Scholar] [CrossRef]
- Bouillon, R.; Manousaki, D.; Rosen, C.; Trajanoska, K.; Rivadeneira, F.; Richards, J.B. The health effects of vitamin D supplementation: Evidence from human studies. Nat. Rev. Endocrinol. 2022, 18, 96–110. [Google Scholar] [CrossRef]
- Fronczek, M.; Strzelczyk, J.K.; Biernacki, K.; Salatino, S.; Osadnik, T.; Ostrowska, Z. New Variants of the Cytochrome P450 2R1 (CYP2R1) Gene in Individuals with Severe Vitamin D-Activating Enzyme 25(OH)D Deficiency. Biomolecules 2021, 11, 1867. [Google Scholar] [CrossRef] [PubMed]
- Thacher, T.D.; Fischer, P.R.; Singh, R.J.; Roizen, J.; Levine, M.A. CYP2R1 Mutations Impair Generation of 25-hydroxyvitamin D and Cause an Atypical Form of Vitamin D Deficiency. J. Clin. Endocrinol. Metab. 2015, 100, E1005–E1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Signorello, L.B.; Shi, J.; Cai, Q.; Zheng, W.; Williams, S.M.; Long, J.; Cohen, S.S. Common variation in vitamin D pathway genes predicts circulating 25-hydroxyvitamin D Levels among African Americans. PLoS ONE 2011, 6, e28623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Hewison, M.; Hu, B.; Adams, J.S. Heterogeneous nuclear ribonucleoprotein (hnRNP) binding to hormone response elements: A cause of vitamin D resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 6109–6114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roizen, J.D.; Li, D.; O’Lear, L.; Javaid, M.K.; Shaw, N.J.; Ebeling, P.R.; Nguyen, H.H. CYP3A4 mutation causes Vitamin D-dependent rickets type 3. J. Clin. Investig. 2018, 128, 1913–1918. [Google Scholar] [CrossRef] [PubMed]
- Kurylowicz, A.; Ramos-Lopez, E.; Bednarczuk, T.; Badenhoop, K. Vitamin D-binding protein (DBP) gene polymorphism is associated with Graves’ disease and the vitamin D status in a Polish population study. Exp. Clin. Endocrinol. Diabetes 2006, 114, 329–335. [Google Scholar] [CrossRef]
- Prabhu, A.V.; Luu, W.; Li, D.; Sharpe, L.J.; Brown, A.J. DHCR7: A vital enzyme switch between cholesterol and vitamin D production. Prog. Lipid. Res. 2016, 64, 138–151. [Google Scholar] [CrossRef]
- Jolliffe, D.A.; Walton, R.T.; Griffiths, C.J.; Martineau, A.R. Single nucleotide polymorphisms in the vitamin D pathway associating with circulating concentrations of vitamin D metabolites and non-skeletal health outcomes: Review of genetic association studies. J. Steroid. Biochem. Mol. Biol. 2016, 164, 18–29. [Google Scholar] [CrossRef]
- Bahrami, A.; Sadeghnia, H.R.; Tabatabaeizadeh, S.A.; Bahrami-Taghanaki, H.; Behboodi, N.; Esmaeili, H.; Avan, A. Genetic and epigenetic factors influencing vitamin D status. J. Cell Physiol. 2018, 233, 4033–4043. [Google Scholar] [CrossRef]
- Jiang, X.; O’Reilly, P.F.; Aschard, H.; Hsu, Y.H.; Richards, J.B.; Dupuis, J.; Ingelsson, E. Genome-wide association study in 79,366 European-ancestry individuals informs the genetic architecture of 25-hydroxyvitamin D levels. Nat. Commun. 2018, 9, 260. [Google Scholar] [CrossRef]
- Munns, C.F.; Shaw, N.; Kiely, M.; Specker, B.L.; Thacher, T.D.; Ozono, K.; Michigami, T. Global consensus recommendations on prevention and management of nutritional rickets. Horm. Res. Paediatr. 2016, 85, 83–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Public Health England. National Diet and Nutrition Survey Results from Years 1, 2, 3 and 4 (combined) of the Rolling Programme. About Public Health Engl. 2012, 4, 1–158. [Google Scholar]
- Blarduni Cardón, E.; Cardón, B.E.; Ugarte, A.H.; Etxebarria, U.I.; González, C.L.; Goivide, G. La dieta como factor de riesgo de hipovitaminosis D en la población pediátrica española. Rev. Osteoporos. Metab. Miner. 2021, 4, 122–129. [Google Scholar]
- Gordon, C.M.; Feldman, H.A.; Williams, A.L.; Kleinman, P.K.; Perez-Rossello, J.; Cox, J.E. Prevalence of vitamin D deficiency among healthy infants and toddlers. Arch. Pediatr. Adolesc. Med. 2008, 162, 505–512. [Google Scholar] [CrossRef] [Green Version]
- Saintonge, S.; Bang, H.; Gerber, L.M. Implications of a new definition of vitamin D deficiency in a multiracial us adolescent population: The National Health and Nutrition Examination Survey III. Pediatrics 2009, 123, 797–803. [Google Scholar] [CrossRef] [Green Version]
- Mansbach, J.M.; Ginde, A.A.; Camargo, C.A. Serum 25-Hydroxyvitamin D Levels Among US Children Aged 1 to 11 Years: Do Children Need More Vitamin D? Pediatrics 2009, 124, 1404. [Google Scholar] [CrossRef] [Green Version]
- Frost, P. Vitamin D deficiency among northern Native Peoples: A real or apparent problem? Int. J. Circumpolar Health 2012, 71, 18001. [Google Scholar] [CrossRef] [Green Version]
- Misra, M. Vitamin D insufficiency and deficiency in children and adolescents—UpToDate. Uptodate 2019, 1–26. Available online: https://www.uptodate.com/contents/vitamin-d-insufficiency-and-deficiency-in-children-and-adolescents#! (accessed on 2 April 2021).
- Hollis, B.W. Circulating 25-hydroxyvitamin D levels indicative of vitamin D sufficiency: Implications for establishing a new effective dietary intake recommendation for vitamin D. J. Nutr. 2005, 135, 317–322. [Google Scholar] [CrossRef] [Green Version]
- Blarduni, E.; Arrospide, A.; Galar, M.; Castaño, L.; Mar, J. Factors associated with the prevalence of hypovitaminosis D in pregnant women and their newborns. An. Pediatría. 2019, 91, 96–104. [Google Scholar] [CrossRef]
- Mohamed, H.J.J.; Rowan, A.; Fong, B.; Loy, S.L. Maternal serum and breast milk vitamin D levels: Findings from the Universiti Sains Malaysia Pregnancy Cohort Study. PLoS ONE 2014, 9, e100705. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, M.L.; Felton, S.K.; Riek, A.E.; Bernal-Mizrachi, C. Implications of vitamin D deficiency in pregnancy and lactation. Am. J. Obstet Gynecol. 2010, 202, 429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrams, S.A. Vitamin D in Preterm and Full-Term Infants. Ann. Nutr. Metab. 2020, 76 (Suppl. S2), 6–14. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.L.; Hollis, B.W. Early-Life Effects of Vitamin D: A Focus on Pregnancy and Lactation. Ann. Nutr. Metab. 2020, 76 (Suppl. S2), 16–28. [Google Scholar] [CrossRef]
- Greer, F.R. Fat-soluble vitamin supplements for enterally fed preterm infants. Neonatal Netw. 2001, 20, 7–11. [Google Scholar] [CrossRef]
- Targher, G.; Bertolini, L.; Scala, L.; Cigolini, M.; Zenari, L.; Falezza, G.; Arcaro, G. Associations between serum 25-hydroxyvitamin D3 concentrations and liver histology in patients with non-alcoholic fatty liver disease. Nutr. Metab. Cardiovasc. Dis. 2007, 17, 517–524. [Google Scholar] [CrossRef]
- LaClair, R.E.; Hellman, R.N.; Karp, S.L.; Kraus, K.M. Prevalence of calcidiol deficiency in CKD: A cross-sectional study across latitudes in the United States. Am. J. Kidney Dis. 2005, 45, 1026–1033. [Google Scholar] [CrossRef]
- Rigterink, T.; Appleton, L.; Day, A.S. Vitamin D therapy in children with inflammatory bowel disease: A systematic review. World J. Clin. Pediatr. 2019, 8, 1–14. [Google Scholar] [CrossRef]
- Lehmann, B.; Genehr, T.; Knuschke, P.; Pietzsch, J.; Meurer, M. UVB-induced conversion of 7-dehydrocholesterol to 1α,25- dihydroxyvitamin D3 in an in vitro human skin equivalent model. J. Investig. Dermatol. 2001, 117, 1179–1185. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.P.; Ong, L.; Loh, T.P.; Chua, H.R.; Tham, C.; Meng, K.C.; Pin, L. Calcium, Vitamin D, and Bone Derangement in Nephrotic Syndrome. J. ASEAN Fed. Endocr. Soc. 2021, 36, 50–55. [Google Scholar] [CrossRef]
- Ward, L.M.; Gaboury, I.; Ladhani, M.; Zlotkin, S. Vitamin D-deficiency rickets among children in Canada. CMAJ 2007, 177, 161–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallo, S.; Comeau, K.; Vanstone, C.; Agellon, S.; Sharma, A.; Jones, G.; Weiler, H. Effect of different dosages of oral vitamin D supplementation on vitamin D status in healthy, breastfed infants: A randomized trial. JAMA 2013, 309, 1785–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Best, C.M.; Xu, J.; Patchen, B.K.; Cassano, P.A. Vitamin D supplementation in pregnant or breastfeeding women or young children for preventing asthma. Cochrane Database Syst. Rev. 2019, 8, CD013396. [Google Scholar] [CrossRef]
- Yakoob, M.Y.; Salam, R.A.; Khan, F.R.; Bhutta, Z.A. Vitamin D supplementation for preventing infections in children under five years of age. Cochrane Database Syst. Rev. 2016, 11, CD008824. [Google Scholar] [CrossRef] [Green Version]
- Huey, S.L.; Acharya, N.; Silver, A.; Sheni, R.; Yu, E.A.; Peña-Rosas, J.P.; Mehta, S. Effects of oral vitamin D supplementation on linear growth and other health outcomes among children under five years of age. Cochrane Database Syst. Rev. 2020, 12, CD012875. [Google Scholar]
- Specker, B.L.; Ho, M.L.; Oestreich, A.; Yin, A.; Shui, Q.M.; Chen, X.C.; Tsang, R.T. Prospective study of vitamin D supplementation and rickets in China. J. Pediatr. 1992, 120, 733–739. [Google Scholar] [CrossRef]
- Tripkovic, L.; Lambert, H.; Hart, K.; Smith, C.P.; Bucca, G.; Penson, S.; Chope, G. Comparison of vitamin D2 and vitamin D3 supplementation in raising serum 25-hydroxyvitamin D status: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2012, 95, 1357–1364. [Google Scholar] [CrossRef] [Green Version]
- Saggese, G.; Vierucci, F.; Prodam, F.; Cardinale, F.; Cetin, I.; Chiappini, E.; Corsello, G. Vitamin D in pediatric age: Consensus of the Italian Pediatric Society and the Italian Society of Preventive and Social Pediatrics, jointly with the Italian Federation of Pediatricians. Ital. J. Pediatr. 2018, 44, 51. [Google Scholar] [CrossRef] [Green Version]
- Ross, A.C.; Manson, J.A.E.; Abrams, S.A.; Aloia, J.F.; Brannon, P.M.; Clinton, S.K.; Durazo-Arvizu, R.A. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: What clinicians need to know. J. Clin. Endocrinol. Metab. 2011, 96, 53–58. [Google Scholar] [CrossRef]
- Thacher, T.; Glew, R.H.; Isichei, C.; Lawson, J.O.; Scariano, J.K.; Hollis, B.W.; VanderJagt, D.J. Rickets in Nigerian children: Response to calcium supplementation. J. Trop. Pediatr. 1999, 45, 202–207. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, V.; Seth, A.; Aneja, S.; Sharma, B.; Sonkar, P.; Singh, S.; Marwaha, R.K. Role of calcium deficiency in development of nutritional rickets in Indian children: A case control study. J. Clin. Endocrinol. Metab. 2012, 97, 3461–3466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwyer, J.T.; Dietz, W.H.; Hass, G.; Suskind, R. Risk of Nutritional Rickets Among Vegetarian Children. Am. J. Dis. Child. 1979, 133, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Miyako, K.; Kinjo, S.; Kohno, H. Vitamin D deficiency rickets caused by improper lifestyle in Japanese children. Pediatr. Int. 2005, 47, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Arabi, A.; El Rassi, R.; El-Hajj Fuleihan, G. Hypovitaminosis D in developing countries-prevalence, risk factors and outcomes. Nat. Rev. Endocrinol. 2010, 6, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Chapman, T.; Sugar, N.; Done, S.; Marasigan, J.; Wambold, N.; Feldman, K. Fractures in infants and toddlers with rickets. Pediatr. Radiol. 2010, 40, 1184–1189. [Google Scholar] [CrossRef]
- Cesur, Y.; Çaksen, H.; Gündem, A.; Kirimi, E.; Odabaş, D. Comparison of low and high dose of vitamin D treatment in nutritional vitamin D deficiency rickets. J. Pediatr. Endocrinol. Metab. 2003, 16, 1105–1109. [Google Scholar] [CrossRef]
- Markestad, T.; Halvorsen, S.; Halvorsen, K.S.; Aksnes, L.; Aarskog, D. Plasma concentrations of vitamin D metabolites before and during treatment of vitamin D deficiency rickets in children. Acta. Paediatr. Scand. 1984, 73, 225–231. [Google Scholar] [CrossRef]
- Quesada-Gomez, J.M.; Bouillon, R. Is calcifediol better than cholecalciferol for vitamin D supplementation? Osteoporos. Int. 2018, 29, 1697–1711. [Google Scholar]
- Kruse, K. Pathophysiology of calcium metabolism in children with vitamin D-deficiency rickets. J. Pediatr. 1995, 126, 736–741. [Google Scholar] [CrossRef]
- Takeda, E.; Yamamoto, H.; Taketani, Y.; Miyamoto, K.I. Vitamin D-dependent rickets type I and type II. Acta. Paediatr. Jpn. 1997, 39, 508–513. [Google Scholar]
- Levine, M.A. Diagnosis and Management of Vitamin D Dependent Rickets. Front. Pediatr. 2020, 8, 315. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| 25(OH)D Deficiency nmol/L | 25(OH)D Sufficiency nmol/L | |
|---|---|---|
| Institute of Medicine | <30 | >50 |
| American Academy of Pediatrics | <50 | |
| Endocrine Society | <25 (severe) | >75 |
| European Society of Pediatric Endocrinology | <30 (severe) | >50 |
| European Calcified Tissue Society | <20 (severe) <50 (deficiency) | >50 |
| Stage | I | II | III |
|---|---|---|---|
| Serum calcium | ↓ | Normal | ↓ |
| Serum phosphate | N-↓ | ↓ | ↓ |
| Serum alkaline phosphatase | ↑ | ↑↑ | ↑↑↑ |
| Calciuria | ↓ | ↓ | ↓ |
| Parathyroid hormone | ↑ | ↑↑ | ↑↑↑ |
| 25(OH)D * | ↓ | ↓ | ↓ |
| 1,25(OH)D | N-↓ | N-↓ | ↓ |
| Radiologic findings | None | Low bone density | Yes |
| Age | Calcium (mg/d) | Vitamin D (IU/d) |
|---|---|---|
| 0–6 months | 200 | 400 |
| 6–12 months | 260 | 400 |
| 1–3 years | 700 | 600 |
| 4–8 years | 1000 | 600 |
| 9–13 years | 1300 | 600 |
| 14–18 years | 1300 | 600 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castano, L.; Madariaga, L.; Grau, G.; García-Castaño, A. 25(OH)Vitamin D Deficiency and Calcifediol Treatment in Pediatrics. Nutrients 2022, 14, 1854. https://doi.org/10.3390/nu14091854
Castano L, Madariaga L, Grau G, García-Castaño A. 25(OH)Vitamin D Deficiency and Calcifediol Treatment in Pediatrics. Nutrients. 2022; 14(9):1854. https://doi.org/10.3390/nu14091854
Chicago/Turabian StyleCastano, Luis, Leire Madariaga, Gema Grau, and Alejandro García-Castaño. 2022. "25(OH)Vitamin D Deficiency and Calcifediol Treatment in Pediatrics" Nutrients 14, no. 9: 1854. https://doi.org/10.3390/nu14091854
APA StyleCastano, L., Madariaga, L., Grau, G., & García-Castaño, A. (2022). 25(OH)Vitamin D Deficiency and Calcifediol Treatment in Pediatrics. Nutrients, 14(9), 1854. https://doi.org/10.3390/nu14091854