
Black Tea Reduces Diet-Induced Obesity in Mice via Modulation of Gut Microbiota and Gene Expression in Host Tissues

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Study
2.2. Extraction of LS
2.3. Preparation of Alkaloids, Polyphenols, and Crude Tea Polysaccharides in LS
2.4. Fecal Microbiota Transplantation
2.5. Biochemical Analyses
2.6. Oil Red O Staining
2.7. HE Staining
2.8. Oral Glucose Tolerance Test
2.9. 16 S rRNA Gene Sequencing
2.10. RNA-Seq Processing and Analysis
2.11. Reduced Representation Bisulfite Sequencing
2.12. Statistical Analysis
3. Results
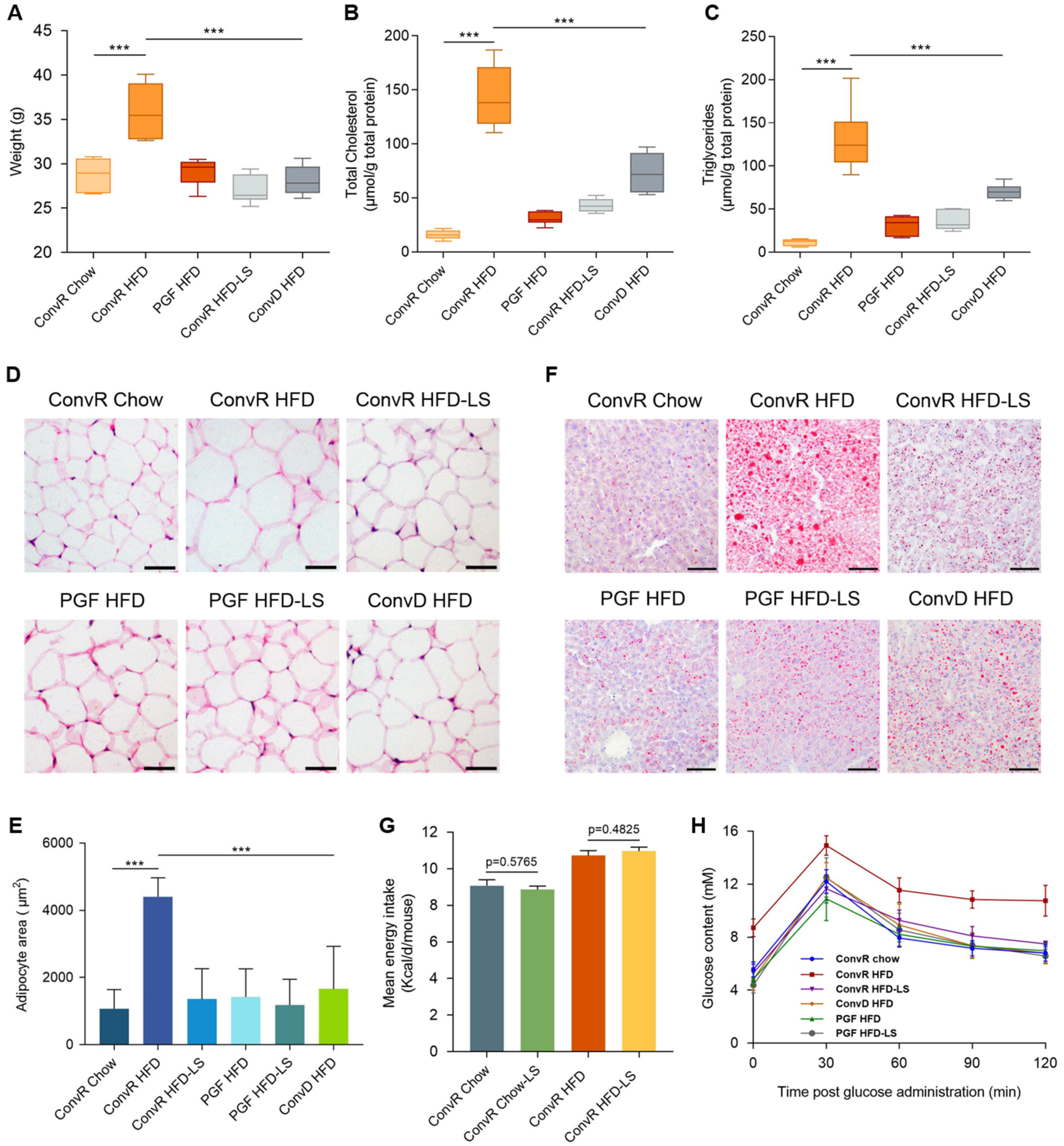
3.1. LS Prevents HFD-Induced Obesity in Mice
3.2. LS-Mediated Prevention of HFD-Induced Obesity in Mice Are Dependent on the Gut Microbiota
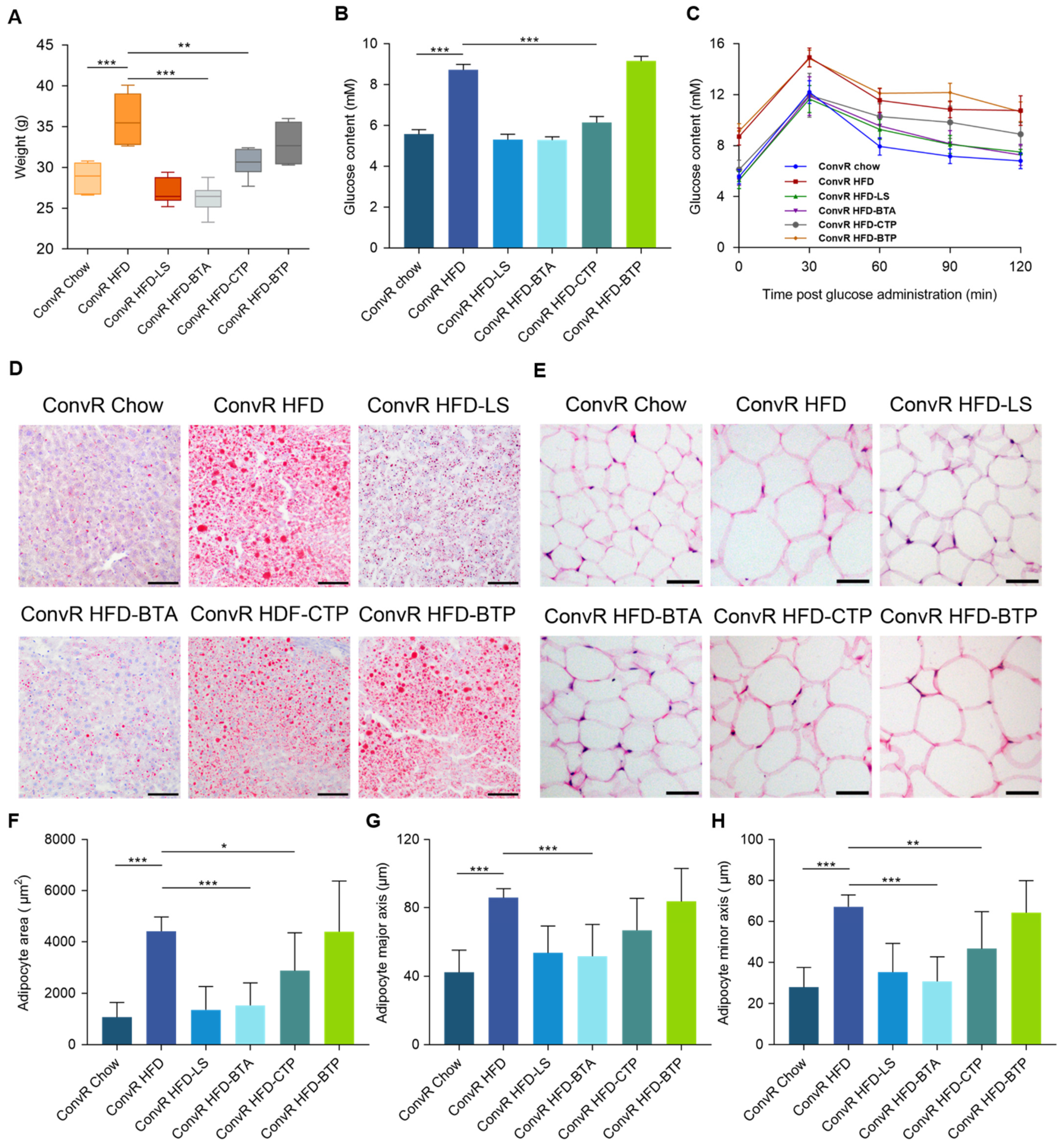
3.3. Alkaloids and Polysaccharides in LS Reduce Obesity
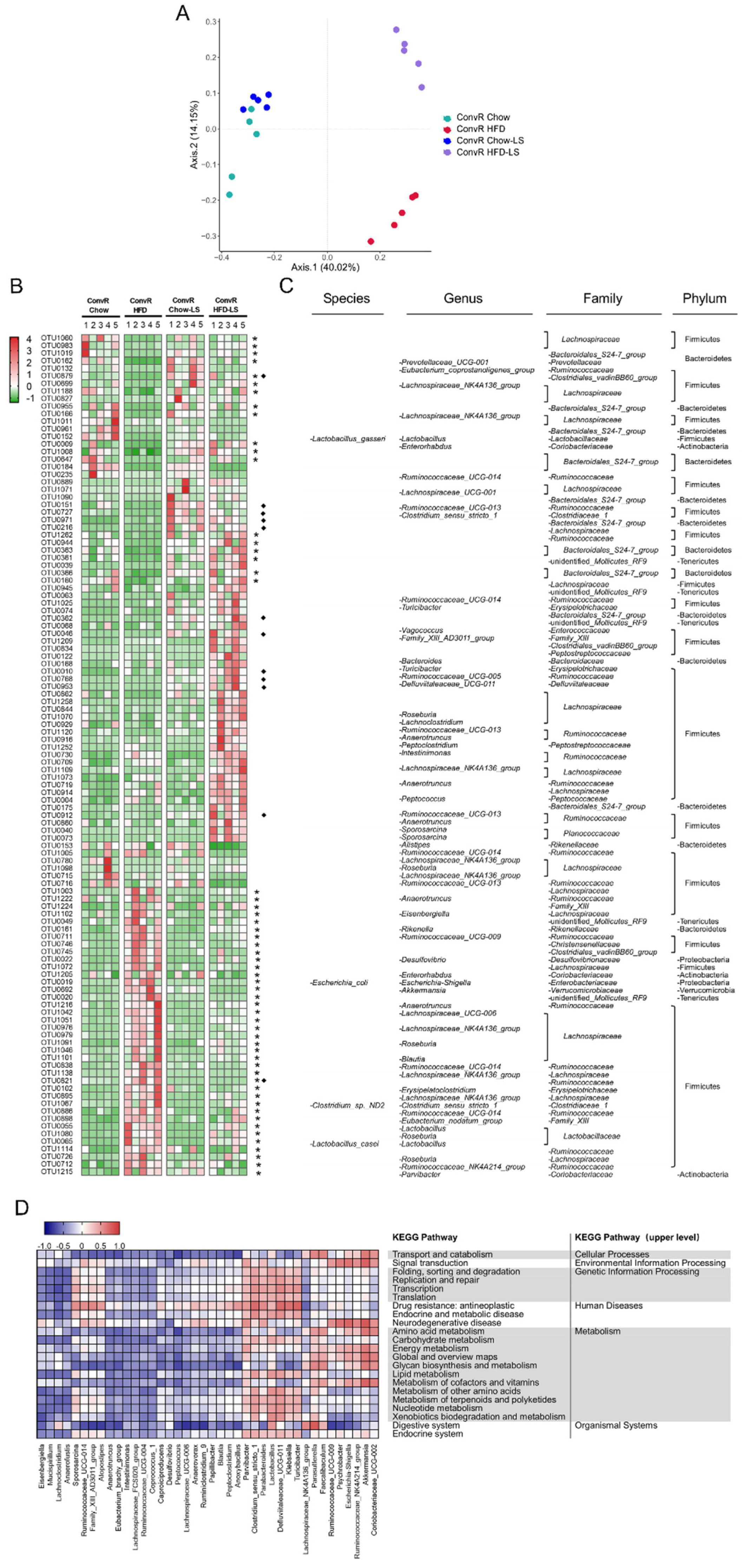
3.4. LS Reverses HFD-Induced Gut Dysbiosis
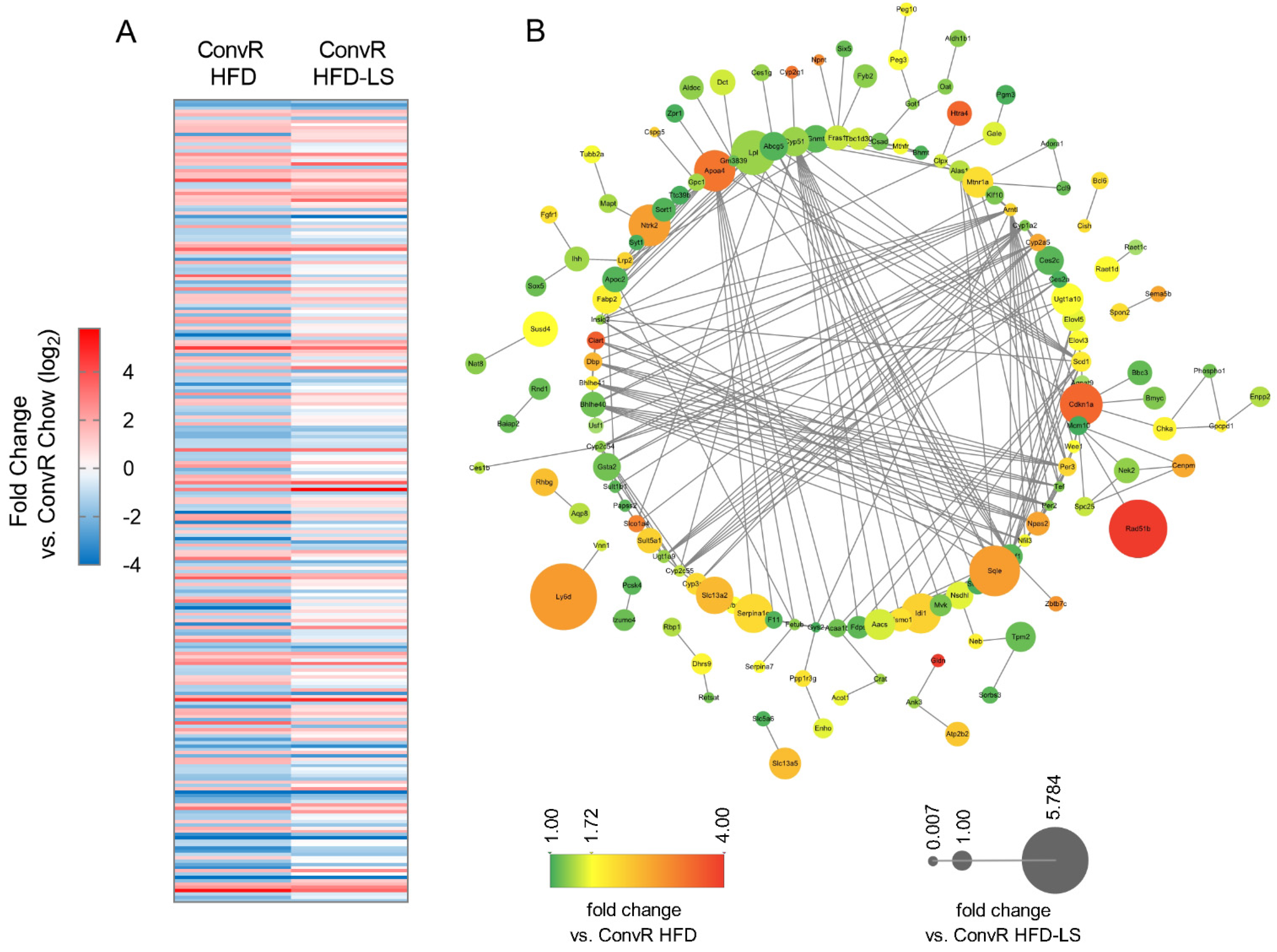
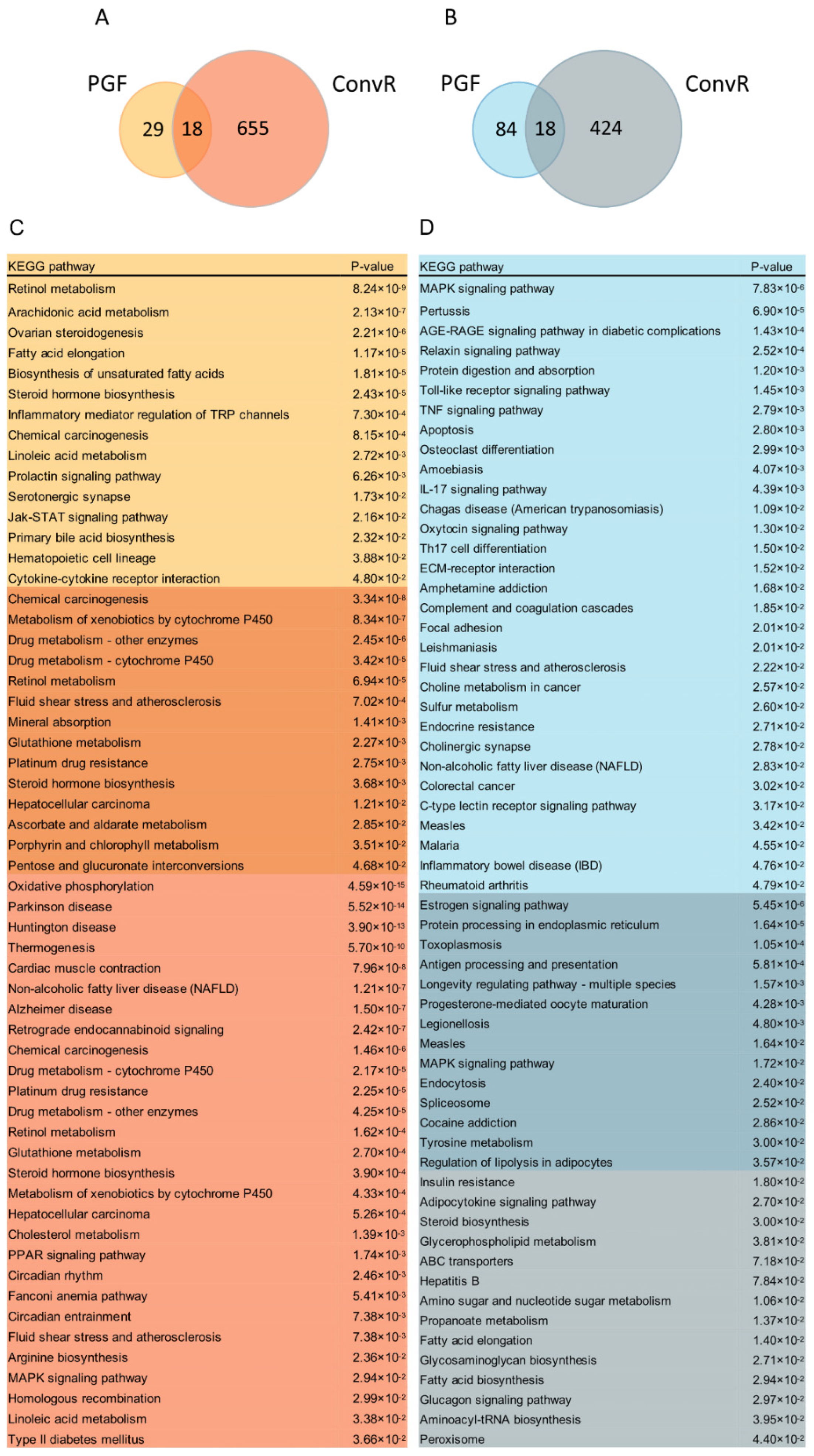
3.5. Co-Regulation of Hepatic Genes Associates with Gut Microbiota Altered by HFD-Fed or LS Treatment
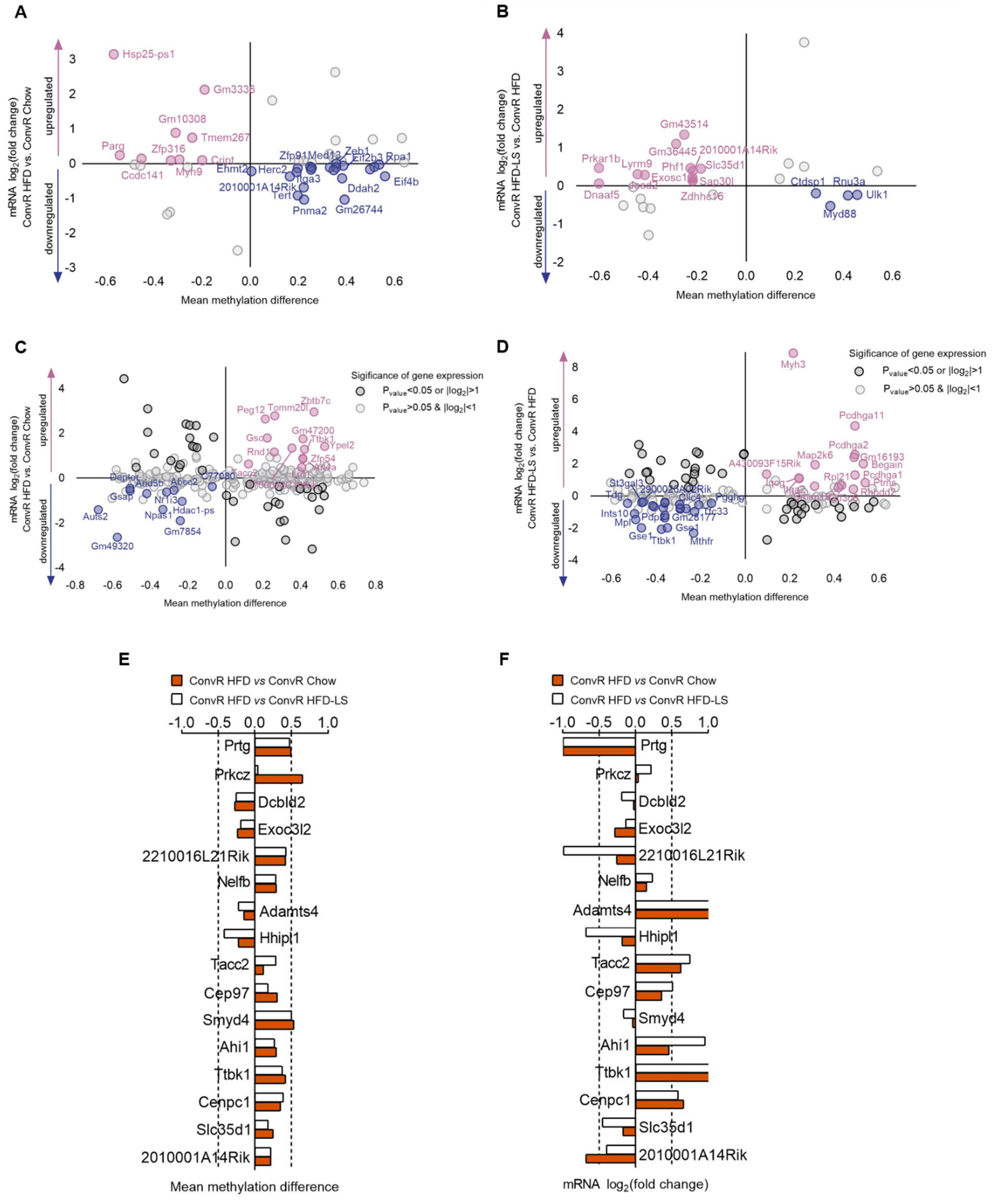
3.6. LS and HFD Feeding Regulated the DNA Methylation Related to Obesity
3.7. LS and HFD Feeding Altered the DNA Methylation Level of Imprinted Genes in Spermatozoa of Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yao, S.S.; Guo, W.F.; Lu, Y.; Jiang, Y.X. Flavor characteristics of Lapsang souchong and smoked Lapsang souchong, a special Chinese black tea with pine smoking process. J. Agric. Food Chem. 2005, 53, 8688–8693. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, X.H.; Chen, Q.B.; Luo, L.Y.; Ma, M.J.; Xiao, B.; Zeng, L. Camellia sinensis and Litsea coreana ameliorate intestinal inflammation and modulate gut microbiota in dextran sulfate sodium-induced colitis mice. Mol. Nutr. Food Res. 2020, 64, 1900943. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Prateeksha Rawat, A.K.S.; Bhagat, R.M.; Singh, B.R. Black tea: Phytochemicals, cancer chemoprevention, and clinical studies. Crit. Rev. Food Sci. 2017, 57, 1394–1410. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, J.M.; Croft, K.D. Tea flavonoids and cardiovascular health. Mol. Aspects Med. 2010, 31, 495–502. [Google Scholar] [CrossRef]
- Henning, S.M.; Yang, J.P.; Hsu, M.; Lee, R.P.; Grojean, E.M.; Ly, A.; Tseng, C.H.; Heber, D.; Li, Z. P Decaffeinated green and black tea polyphenols decrease weight gain and alter microbiome populations and function in diet—Induced obese mice. Eur. J. Nutr. 2018, 57, 2759–2769. [Google Scholar] [CrossRef]
- Gao, Y.; Xu, Y.Q.; Yin, J.F. Black tea benefits short-chain fatty acid producers but inhibits genus Lactobacillus in the gut of healthy Sprague-Dawley rats. J. Sci. Food Agric. 2020, 100, 5466–5475. [Google Scholar] [CrossRef]
- Xu, J.L.; Li, M.X.; Zhang, Y.; Chu, S.; Huo, Y.; Zhao, J.; Wan, C.P. Huangjinya black tea alleviates obesity and insulin resistance via modulating fecal metabolome in high-fat diet-fed mice. Mol. Nutr. Food Res. 2020, 64, e2000353. [Google Scholar] [CrossRef]
- Sun, L.L.; Xu, H.R.; Ye, J.H.; Gaikwad, N.W. Comparative effect of black, green, oolong and white tea intake on body weight gain and bile acid metabolism. Nutrition 2019, 65, 208–215. [Google Scholar] [CrossRef]
- Shen, Y.; Xiao, X.J.; Wu, K.L.; Wang, Y.P.; Yuan, Y.J.; Liu, J.W.; Sun, S.M.; Liu, J. Effects and molecular mechanisms of Ninghong black tea extract in nonalcoholic fatty liver disease of rats. J. Food Sci. 2020, 85, 800–807. [Google Scholar] [CrossRef]
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F.; et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014, 384, 746. [Google Scholar] [CrossRef] [Green Version]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Osborn, O.; Olefsky, J.M. The cellular and signaling networks linking the immune system and metabolism in disease. Nat. Med. 2012, 18, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Bleich, S.N.; Cutler, D.; Murray, C.; Adams, A. Why is the developed world obese? Annu. Rev. Public Health 2008, 29, 273–295. [Google Scholar] [CrossRef] [Green Version]
- Obri, A.; Serra, D.; Herrero, D.; Mera, P. The role of epigenetics in the development of obesity. Biochem. Pharm. 2020, 177, 113973. [Google Scholar] [CrossRef] [PubMed]
- Waddington, C.H. Canalization of development and the inheritance of acquired characters. Nature 1942, 150, 563–565. [Google Scholar] [CrossRef]
- Eccleston, A.; DeWitt, N.; Gunter, C.; Marte, B.; Nath, D. Epigenetics. Nature 2007, 447, 395. [Google Scholar] [CrossRef] [Green Version]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Fan, J.; Krautkramer, K.A.; Feldman, J.L.; Denu, J.M. Metabolic regulation of histone post-translational modifications. ACS Chem. Biol. 2015, 10, 95–108. [Google Scholar] [CrossRef] [Green Version]
- Blaschke, K.; Ebata, K.T.; Karimi, M.M.; Zepeda-Martínez, J.A.; Goyal, P.; Mahapatra, S.; Tam, A.; Laird, D.J.; Hirst, M.; Rao, A.; et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature 2013, 500, 222–226. [Google Scholar] [CrossRef]
- Yin, R.C.; Mao, S.Q.; Zhao, B.L.; Chong, Z.C.; Yang, Y.; Zhao, C.; Zhang, D.P.; Huang, H.; Gao, J.; Li, Z.; et al. Ascorbic acid enhances Tet-mediated 5—Methylcytosine oxidation and promotes DNA demethylation in mammals. J. Am. Chem. Soc. 2013, 135, 10396–10403. [Google Scholar] [CrossRef]
- Krautkramer, K.A.; Kreznar, J.H.; Romano, K.A.; Vivas, E.I.; Barrett-Wilt, G.A.; Rabaglia, M.E.; Keller, M.P.; Attie, A.D.; Rey, F.E.; Denu, J.M. Diet-microbiota interactions mediate global epigenetic programming in multiple host tissues. Mol. Cell 2016, 64, 982–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Chirlaque, C.; Aranda, C.J.; Ocón, B.; Capitán-Cañadas, F.; Ortega-González, M.; Carrero, J.J.; Suárez, M.D.; Zarzuelo, A.; de Medina, F.S.; Martínez-Augustin, O. Germ-free and antibiotic-treated mice are highly susceptible to epithelial injury in DSS colitis. J. Crohn’s Colitis 2016, 10, 1324–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipide extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Hirschhorn, J.N.; Brown, S.A.; Clark, C.D.; Winston, F. Evidence that SNF2/SWI2 and SNF5 activate transcription in yeast by altering chromatin structure. Gene Dev. 1992, 6, 2288–2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cote, J.; Quinn, J.; Workman, J.L.; Peterson, C.L. Stimulation of GAL4 derivative binding to nucleosomal DNA by the yeast SWI/SNF complex. Science 1994, 265, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Rots, D.; Chater-Diehl, E.; Dingemans, A.J.M.; Goodman, S.J.; Siu, M.T.; Cytrynbaum, C.; Choufani, S.; Hoang, N.; Walker, S.; Awamleh, Z.; et al. Truncating SRCAP variants outside the Floating-Harbor syndrome locus cause a distinct neurodevelopmental disorder with a specific DNA methylation signature. Am. J. Hum. Genet. 2021, 108, 1053–1068. [Google Scholar] [CrossRef]
- Tsukada, Y.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006, 439, 811–816. [Google Scholar] [CrossRef]
- Thomson, J.P.; Skene, P.J.; Selfridge, J.; Clouaire, T.; Guy, J.; Webb, S.; Kerr, A.R.W.; Deaton, A.; Andrews, R.; James, K.D.; et al. CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature 2010, 464, 1082–1086. [Google Scholar] [CrossRef] [Green Version]
- Blackledge, N.P.; Zhou, J.C.; Tolstorukov, M.Y.; Farcas, A.M.; Park, P.J.; Klose, R.J. CpG Islands Recruit a Histone H3 Lysine 36 Demethylase. Mol. Cell 2010, 38, 179–190. [Google Scholar] [CrossRef]
- Tucci, V.; Isles, A.R.; Kelsey, G.; Ferguson-Smith, A.C. Genomic imprinting and physiological processes in mammals. Cell 2019, 176, 952–965. [Google Scholar] [CrossRef] [Green Version]
- Ahuja, N.; Sharma, A.R.; Baylin, S.B. Epigenetic therapeutics: A new weapon in the war against cancer. Annu. Rev. Med. 2016, 67, 73–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlov, S.V.; Bogenpohl, J.W.; Howell, M.P.; Wevrick, R.; Panda, S.; Hogenesch, J.B.; Muglia, L.J.; Gelder, R.N.V.; Herzog, E.D.; Stewart, C.L. The imprinted gene Magel2 regulates normal circadian output. Nat. Genet. 2007, 39, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Sujit, K.M.; Sarkar, S.; Singh, V.; Pandey, R.; Agrawal, N.K.; Trivedi, S.; Singh, K.; Gupta, G.; Rajender, S. Genome-wide differential methylation analyses identifies methylation signatures of male infertility. Hum. Reprod. 2018, 33, 2256–2267. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Obesity Andover Weight. 2022. Available online: http://www.who.int/mediacentre/factsheets/fs311/en/ (accessed on 13 April 2022).
- Hill, J.O.; Peters, J.C. Environmental contributions to the obesity epidemic. Science 1998, 280, 1371–1374. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.P. The gut microbiota and obesity: From correlation to causality. Nat. Rev. Microbiol. 2013, 11, 639–647. [Google Scholar] [CrossRef]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet–induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.Y.; Zhao, C.N.; Xu, X.Y.; Tang, G.Y.; Corke, H.; Gan, R.Y.; Li, H.B. Dietary plants, gut microbiota, and obesity: Effects and mechanisms. Trends Food Sci. Technol. 2019, 92, 194–204. [Google Scholar] [CrossRef]
- Everard, A.; Lazarevic, V.; Derrien, M.; Girard, M.; Muccioli, G.G.; Neyrinck, A.M.; Possemiers, S.; Holle, A.V.; François, P.; de Vos, W.M.; et al. Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes 2011, 60, 2775–2786. [Google Scholar] [CrossRef] [Green Version]
- Silveira, A.K.; Moresco, K.S.; Gomes, H.M.; Morrone, M.D.; Grun, L.K.; Gelain, D.P.; de Mattos Pereira, L.; Giongo, A.; De Oliveira, R.R.; Moreira, J.C.F. Guarana (Paullinia cupana Mart.) alters gut microbiota and modulates redox status, partially via caffeine in Wistar rats. Phytother. Res. 2018, 32, 2466–2474. [Google Scholar] [CrossRef]
- Jing, N.N.; Liu, X.X.; Jin, M.L.; Yang, X.B.; Hu, X.; Li, C.Y.; Zhao, K. Fubrick tea attenuates high-fat diet induced fat deposition and metabolic disorder by regulating gut microbiota and caffeine metabolism. Food Funct. 2020, 11, 6971–6986. [Google Scholar] [CrossRef]
- Chang, C.J.; Lin, C.S.; Lu, C.C.; Martel, J.; Ko, Y.F.; Ojcius, D.M.; Tseng, S.F.; Wu, T.R.; Chen, Y.Y.M.; Young, J.D. Ganoderma lucidum reduces obesity in mice by modulating the composition of the gut microbiota. Nat. Commun. 2015, 6, 7489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.T.; Liu, A.B.; Sun, S.L.; Ajami, N.J.; Ross, M.C.; Wang, H.; Zhang, L.; Reuhl, K.; Kobayashi, K.; Onishi, J.C.; et al. Green tea polyphenols modify the gut microbiome in db/db Mice as co-abundance groups correlating with the blood glucose lowering effect. Mol. Nutr. Food Res. 2019, 63, 1801064. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, A.; Aleya, L.; Kamel, M. The link among microbiota, epigenetics, and disease development. Environ. Sci. Pollut. Res. 2021, 28, 28926–28964. [Google Scholar] [CrossRef] [PubMed]
- den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuel, B.S.; Shaito, A.; Motoike, T.; Rey, F.E.; Backhed, F.; Manchester, J.K.; Hammer, R.E.; Williams, S.C.; Crowley, J.; Yanagisawa, M.; et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc. Natl. Acad. Sci. USA 2008, 105, 16767–16772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, S.H.; Holtrop, G.; Lobley, G.E.; Calder, A.G.; Stewart, C.S.; Flint, H.J. Contribution of acetate to butyrate formation by human faecal bacteria. Brit. J. Nutr. 2004, 91, 915–923. [Google Scholar] [CrossRef] [Green Version]
- Bai, L.C.; Merchant, J.L. Role for CITED2, a CBP/p300 interacting protein, in colon cancer cell invasion. FEBS Lett. 2007, 581, 5904–5910. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.C.; Lockhart, S.M.; Rathjen, T.; Albadawi, H.; Sørensen, D.; O’Neill, B.T.; Dwivedi, N.; Preil, S.R.; Beck, H.C.; Dunwoodie, S.L.; et al. Insulin downregulates the transcriptional coregulator CITED2, an inhibitor of proangiogenic function in endothelial cells. Diabetes 2016, 65, 3680–3690. [Google Scholar] [CrossRef] [Green Version]
- Qiao, S.S.; Bao, L.; Wang, K.; Sun, S.S.; Liao, M.F.; Liu, C.; Zhou, N.; Ma, K.; Zhang, Y.W.; Chen, Y.H.; et al. Activation of a specific gut bacteroides-folate-liver axis benefits for the alleviation of nonalcoholic hepatic steatosis. Cell Rep. 2020, 32, 108005. [Google Scholar] [CrossRef]
- Pompei, A.; Cordisco, L.; Amaretti, A.; Zanoni, S.; Raimondi, S.; Matteuzzi, D.; Rossi, M. Administration of folate-producing bifidobacteria enhances folate status in Wistar rats. J. Nutr. 2007, 137, 2742–2746. [Google Scholar] [CrossRef]
- Hong, S.M.; Woo, H.W.; Kim, M.K.; Kim, S.Y.; Lee, Y.H.; Shin, D.H.; Shin, M.H.; Chun, B.Y.; Choi, B.Y. A prospective association between dietary folate intake and type 2 diabetes risk among Korean adults aged 40 years or older: The Korean multi-rural communities cohort (MRCohort) study. Brit. J. Nutr. 2017, 118, 1078–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, E.; Matte, A.; Perfilyev, A.; de Mello, V.D.; Käkelä, P.; Pihlajamäki, J.; Ling, C. Epigenetic alterations in human liver from subjects with type 2 diabetes in parallel with reduced folate levels. J. Clin. Endocr. Metab. 2015, 100, E1491–E1501. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yan, T.; Yang, L.P.; Zhang, K.X.; Jia, J.M. Monitoring the consistency quality and antioxidant activity of Da Hong Pao teas by HPLC fingerprinting. J. Chromatogr. Sci. 2017, 55, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Dincer, Y.; Yuksel, S. Antiobesity effects of phytochemicals from an epigenetic perspective. Nutrition 2021, 84, 111119. [Google Scholar] [CrossRef] [PubMed]
- Link, A.; Balaguer, F.; Goel, A. Cancer chemoprevention by dietary polyphenols: Promising role for epigenetics. Biochem. Pharmacol. 2010, 80, 1771–1792. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.J.; Su, S.Y.; Barnes, V.A.; Miguel, C.D.; Pollock, J.; Ownby, D.; Shi, H.D.; Zhu, H.D.; Snieder, H.; Wang, X.L. A genome-wide methylation study on obesity Differential variability and differential methylation. Epigenetics 2013, 8, 522–533. [Google Scholar] [CrossRef] [Green Version]
- Demerath, E.W.; Guan, W.H.; Grove, M.L.; Aslibekyan, S.; Mendelson, M.; Zhou, Y.H.; Hedman, Å.K.; Sandling, J.K.; Li, L.A.; Irvin, M.R.; et al. Epigenome-wide association study (EWAS) of BMI, BMI change and waist circumference in African American adults identifies multiple replicated loci. Hum. Mol. Genet. 2015, 24, 4464–4479. [Google Scholar] [CrossRef] [Green Version]
- Sayols-Baixeras, S.; Subirana, I.; Fernández-Sanlés, A.; Sentí, M.; Lluís-Ganella, C.; Marrugat, J.; Elosua, R. DNA methylation and obesity traits: An epigenome-wide association study. The REGICOR study. Epigenetics 2017, 12, 909–916. [Google Scholar] [CrossRef]
- Dayeh, T.; Volkov, P.; Salo, S.; Hall, E.; Nilsson, E.; Olsson, A.H.; Kirkpatrick, C.L.; Wollheim, C.B.; Eliasson, L.; Ronn, T.; et al. Genome-Wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet. 2014, 10, e1004160. [Google Scholar] [CrossRef]
- Nilsson, E.; Jansson, P.A.; Perfilyev, A.; Volkov, P.; Pedersen, M.; Svensson, M.K.; Poulsen, P.; Ribel-Madsen, R.; Pedersen, N.L.; Almgren, P.; et al. Altered DNA methylation and differential expression of genes influencing metabolism and inflammation in adipose tissue from subjects with type 2 diabetes. Diabetes 2014, 63, 2962–2976. [Google Scholar] [CrossRef] [Green Version]
- Wahl, S.; Drong, A.; Lehne, B.; Loh, M.; Scott, W.R.; Kunze, S.; Tsai, P.C.; Ried, J.S.; Zhang, W.H.; Yang, Y.W.; et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature 2017, 541, 81–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendelson, M.M.; Marioni, R.E.; Joehanes, R.; Liu, C.Y.; Hedman, Å.K.; Aslibekyan, S.; Demerath, E.W.; Guan, W.H.; Zhi, D.G.; Yao, C.; et al. Association of body mass index with DNA methylation and gene expression in blood cells and relations to cardiometabolic disease: A mendelian randomization approach. PLoS Med. 2017, 14, e1002215. [Google Scholar] [CrossRef] [PubMed]
- Lassi, G.; Ball, S.T.; Maggi, S.; Colonna, G.; Nieus, T.; Cero, C.; Bartolomucci, A.; Peters, J.; Tucci, V. Loss of Gnas imprinting differentially affects REM/NREM sleep and cognition in mice. PLoS Genet. 2012, 8, e1002706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dent, C.L.; Humby, T.; Lewis, K.; Ward, A.; Fischer-Colbrie, R.; Wilkinson, L.S.; Wilkins, J.F.; Isles, A.R. Impulsive choice in mice lacking paternal expression of Grb10 suggests intragenomic conflict in behavior. Genetics 2018, 209, 233–239. [Google Scholar] [CrossRef] [Green Version]
- Garfield, A.S.; Cowley, M.F.; Smith, M.; Moorwood, K.; Stewart-Cox, J.E.; Gilroy, K.; Baker, S.; Xia, J.; Dalley, J.W.; Hurst, L.D.; et al. Distinct physiological and behavioural functions for parental alleles of imprinted Grb10. Nature 2011, 469, 534–538. [Google Scholar] [CrossRef] [Green Version]
- Hughes, J.; Surakhy, M.; Can, S.; Ducker, M.; Davies, N.; Szele, F.; Bühnemann, C.; Carter, E.; Trikin, R.; Crump, M.P.; et al. Maternal transmission of an Igf2r domain 11: IGF2 binding mutant allele (Igf2r (I1565A)) results in partial lethality, overgrowth and intestinal adenoma progression. Sci. Rep. 2019, 9, 11388. [Google Scholar] [CrossRef]
- Wang, X.M.; Sterr, M.; Ansarullah; Burtscher, I.; Böttcher, A.; Beckenbauer, J.; Siehler, J.; Meitinger, T.; Häring, H.U.; Staiger, H.; et al. Point mutations in the PDX1 transactivation domain impair human beta-cell development and function. Mol. Metab. 2019, 24, 80–97. [Google Scholar] [CrossRef]
- Millership, S.J.; Tunster, S.J.; Van de Pette, M.; Choudhury, A.I.; Irvine, E.E.; Christian, M.; Fisher, A.G.; John, R.M.; Scott, J.; Withers, D.J. Neuronatin deletion causes postnatal growth restriction and adult obesity in 129S2/Sv mice. Mol. Metab. 2018, 18, 97–106. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Hu, G.; Wang, A.; Long, G.; Yang, Y.; Wang, D.; Zhong, N.; Jia, J. Black Tea Reduces Diet-Induced Obesity in Mice via Modulation of Gut Microbiota and Gene Expression in Host Tissues. Nutrients 2022, 14, 1635. https://doi.org/10.3390/nu14081635
Liu X, Hu G, Wang A, Long G, Yang Y, Wang D, Zhong N, Jia J. Black Tea Reduces Diet-Induced Obesity in Mice via Modulation of Gut Microbiota and Gene Expression in Host Tissues. Nutrients. 2022; 14(8):1635. https://doi.org/10.3390/nu14081635
Chicago/Turabian StyleLiu, Xuanli, Gaosheng Hu, Anhua Wang, Guoqing Long, Yongcheng Yang, Dongdong Wang, Nanfang Zhong, and Jingming Jia. 2022. "Black Tea Reduces Diet-Induced Obesity in Mice via Modulation of Gut Microbiota and Gene Expression in Host Tissues" Nutrients 14, no. 8: 1635. https://doi.org/10.3390/nu14081635
APA StyleLiu, X., Hu, G., Wang, A., Long, G., Yang, Y., Wang, D., Zhong, N., & Jia, J. (2022). Black Tea Reduces Diet-Induced Obesity in Mice via Modulation of Gut Microbiota and Gene Expression in Host Tissues. Nutrients, 14(8), 1635. https://doi.org/10.3390/nu14081635