
Obesity and Leptin Resistance in the Regulation of the Type I Interferon Early Response and the Increased Risk for Severe COVID-19

 , ,
, ,  and
and
Abstract
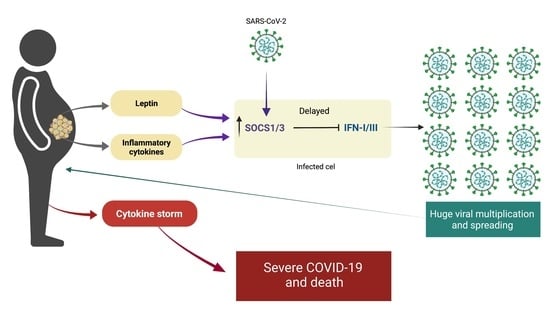
:
1. Introduction
2. Risk Factors for Severe COVID-19
3. Cell Entry by SARS-CoV-2
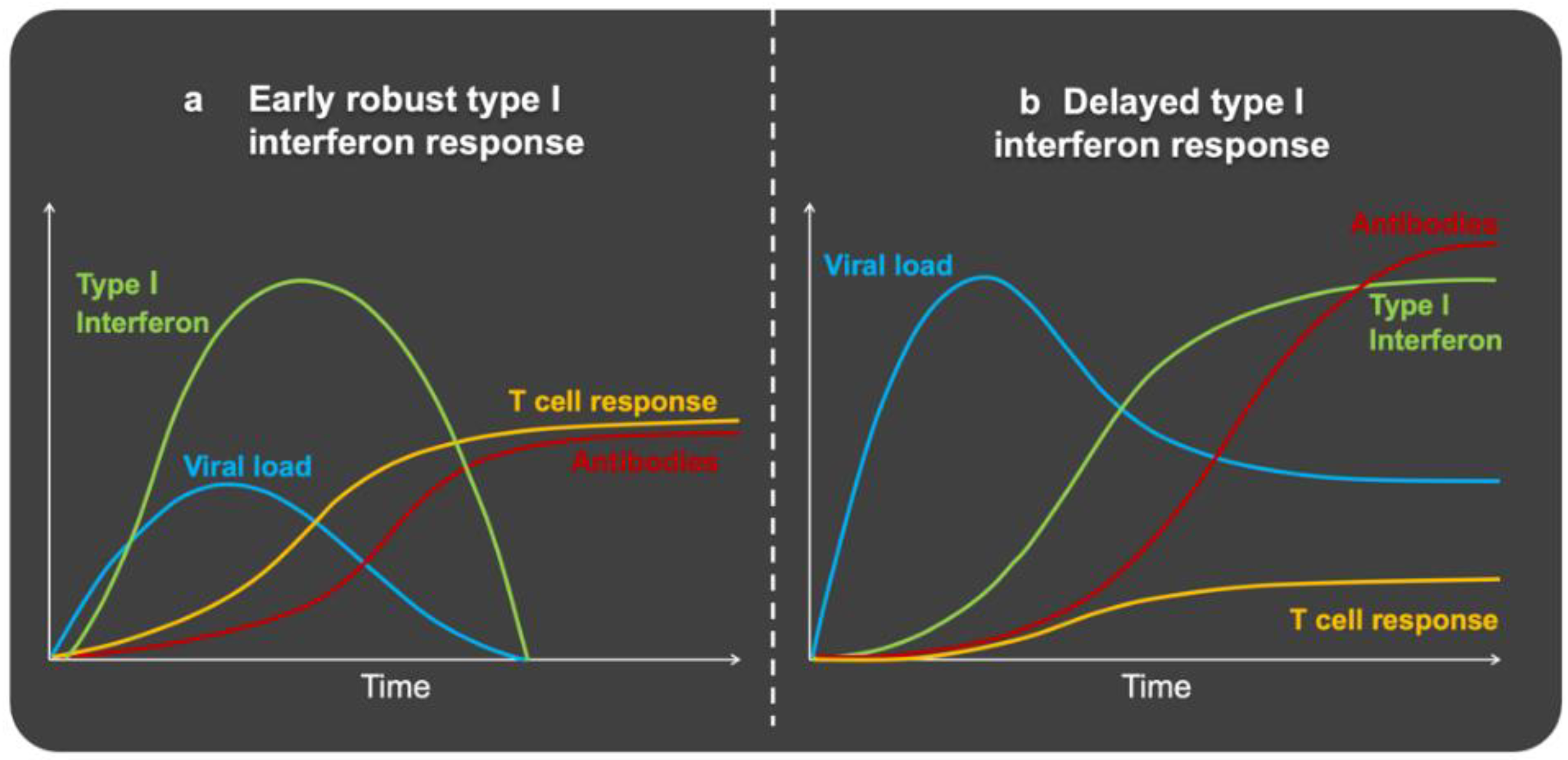
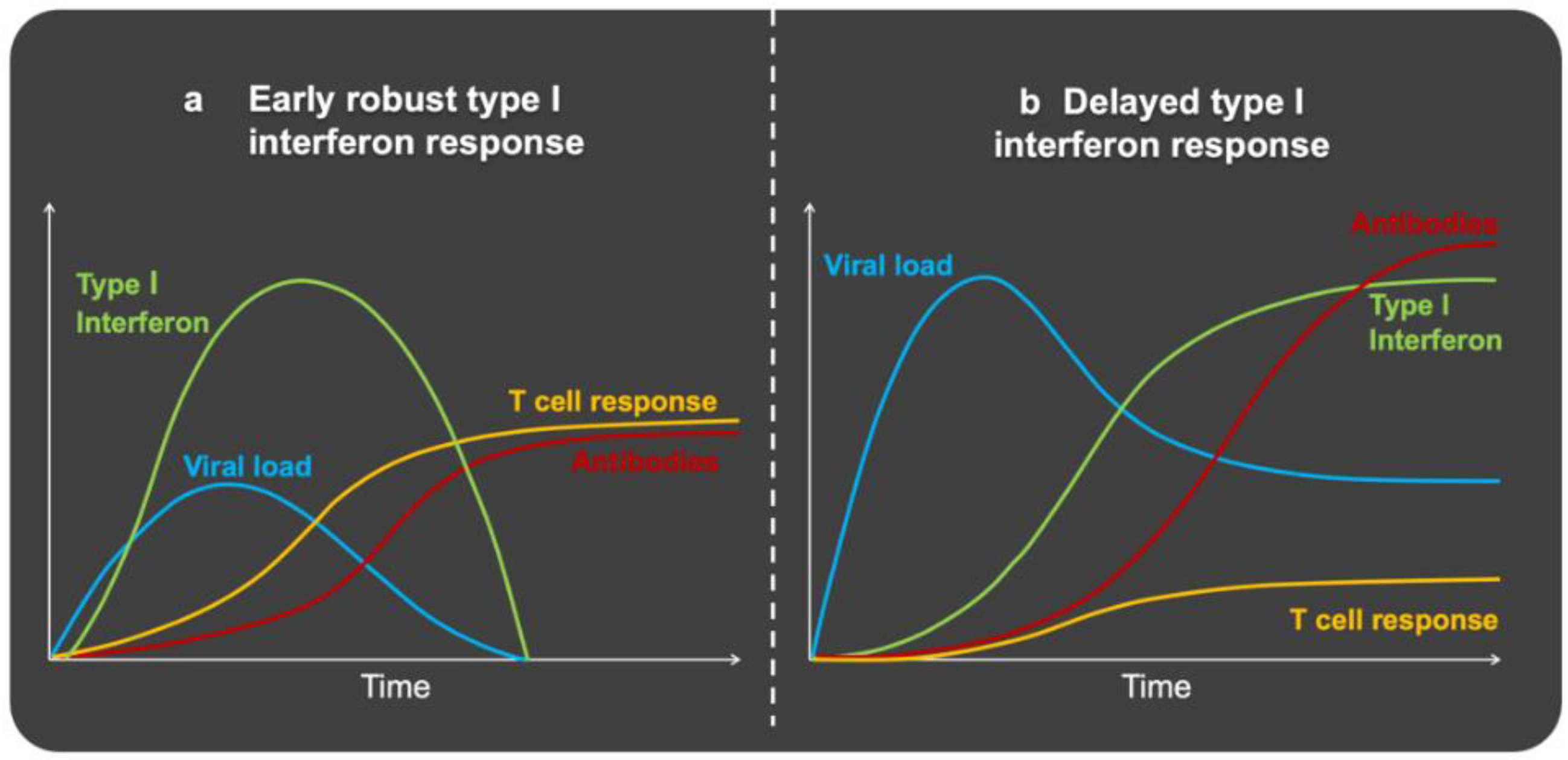
4. Immune Defense Dynamics and Clinical Course
5. Immune Response to a Pathogen in Short
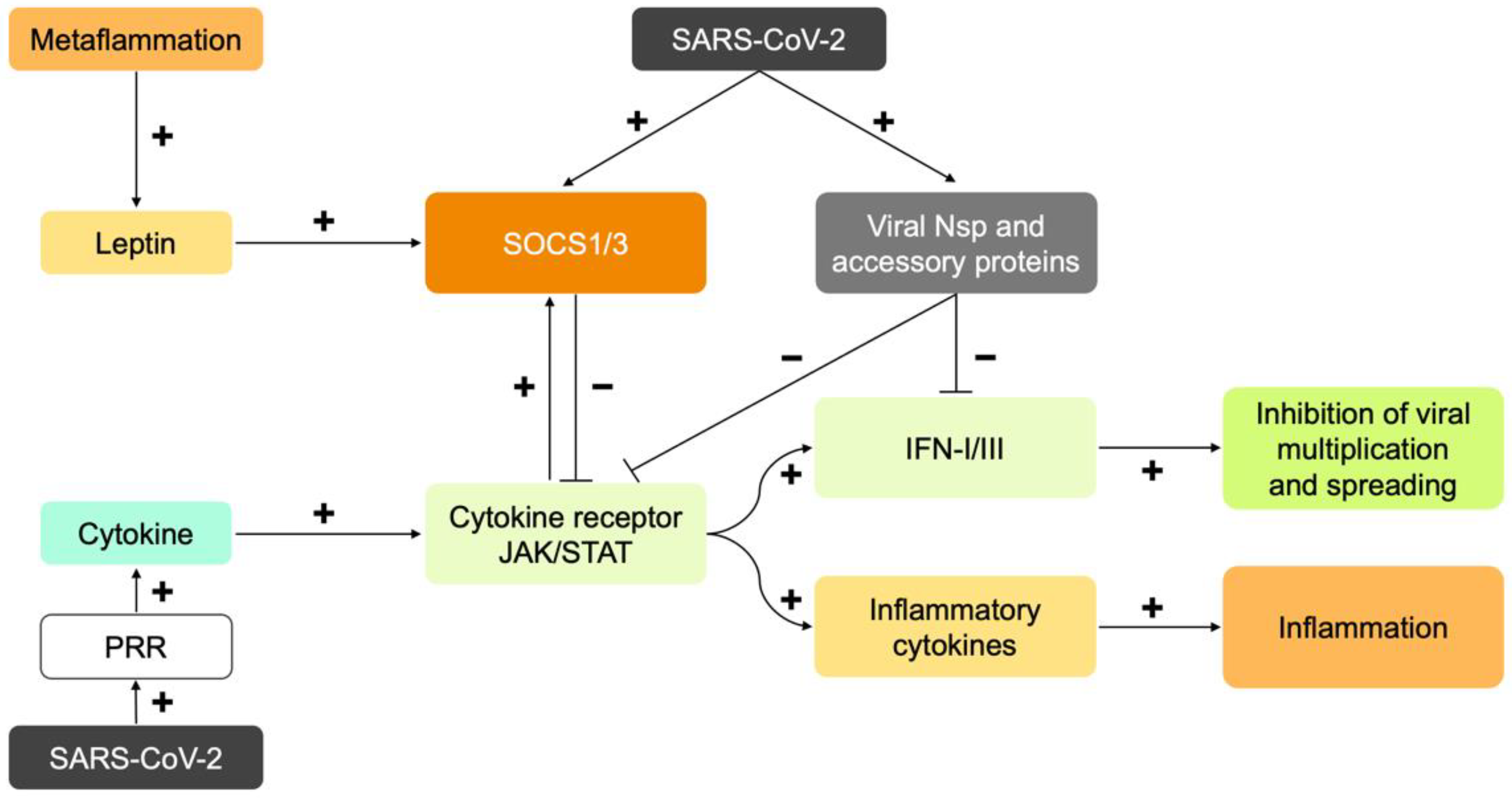
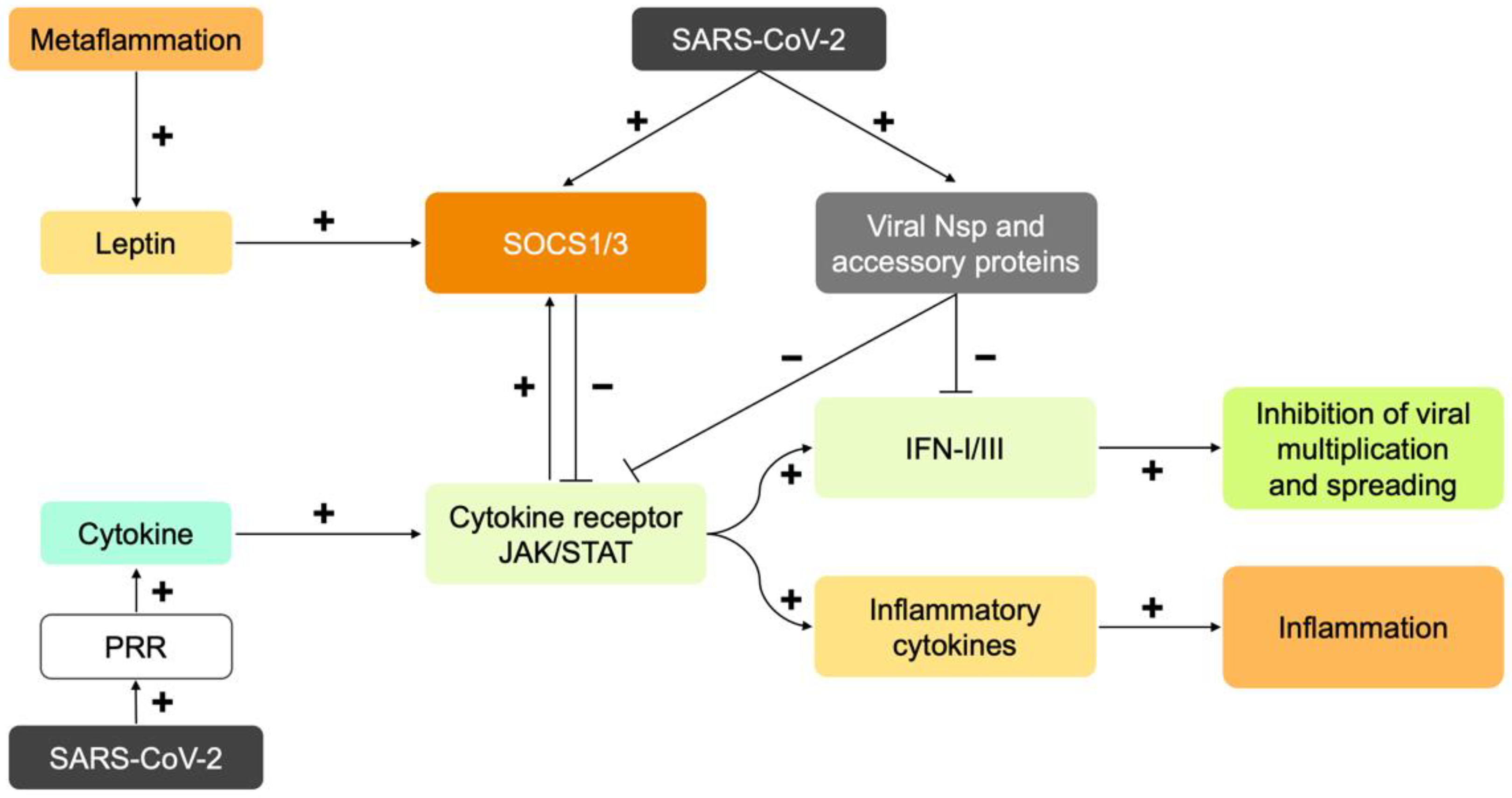
6. SARS-CoV-2 Disrupts the Early Immune Response: Interferons
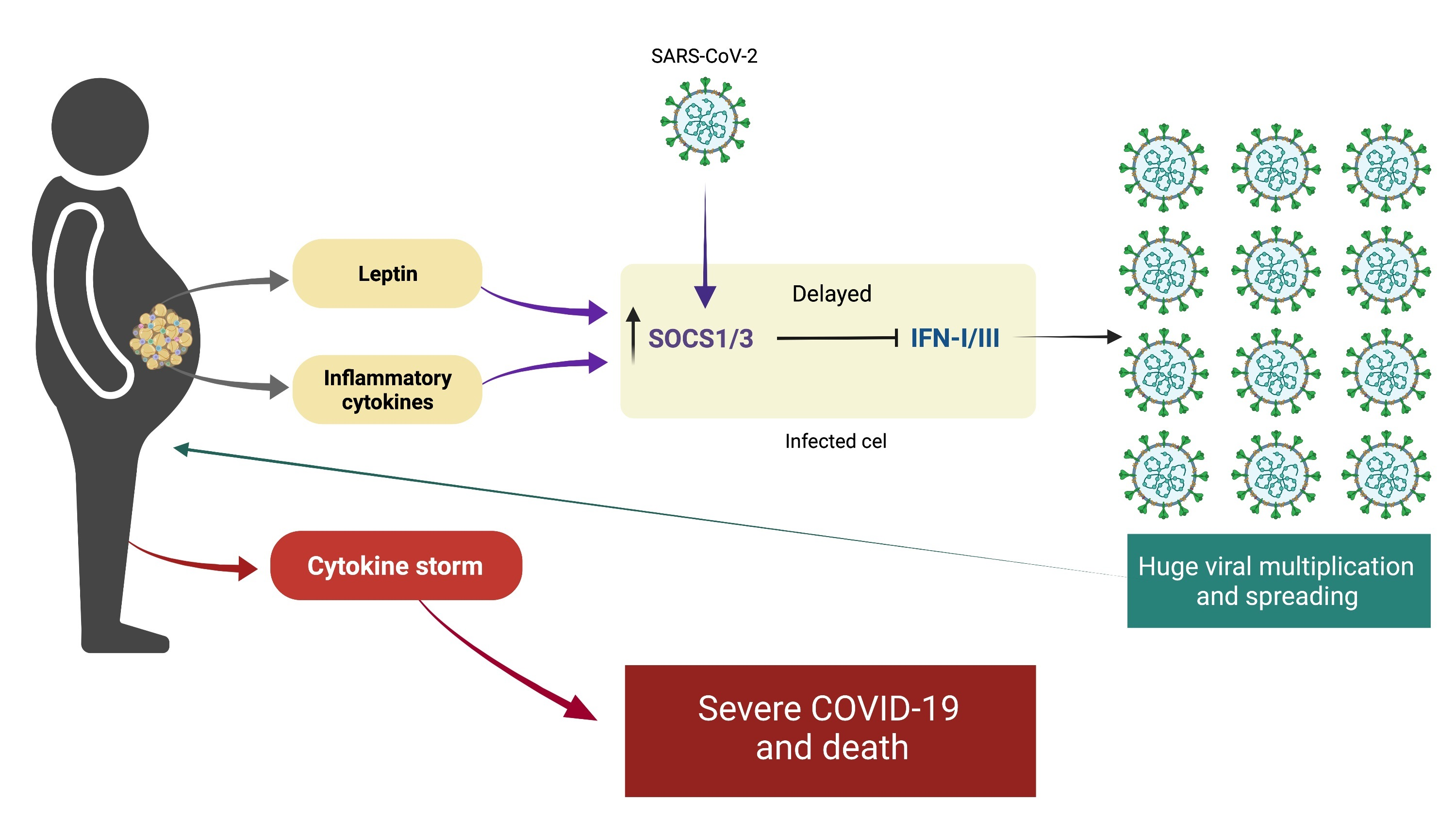
7. Immune Response to SARS-CoV-2 in Obesity: Too Late, Too Weak, and Then Too Strong
8. Role of Insulin and Leptin in the Immune System: Immunometabolism
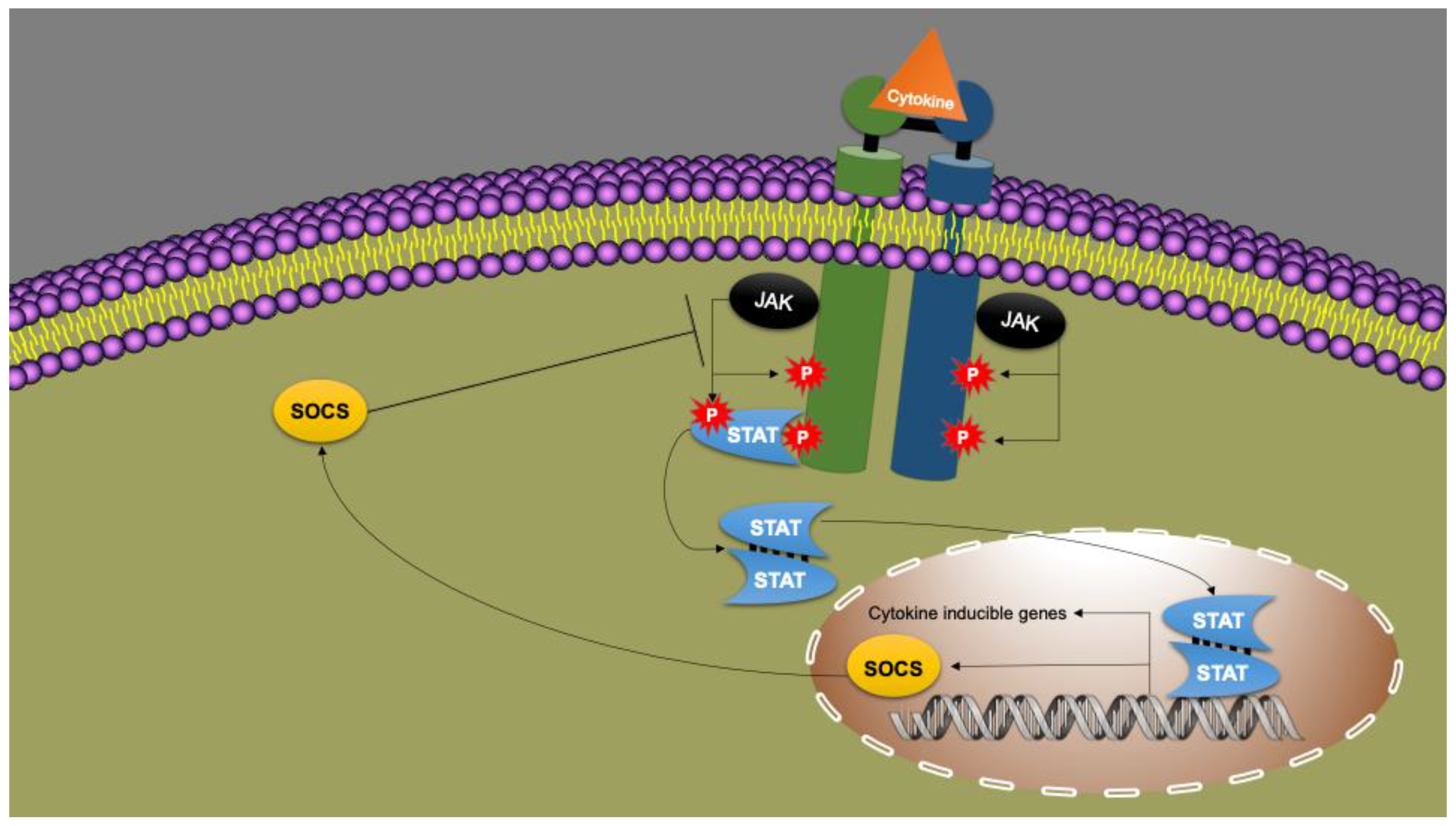
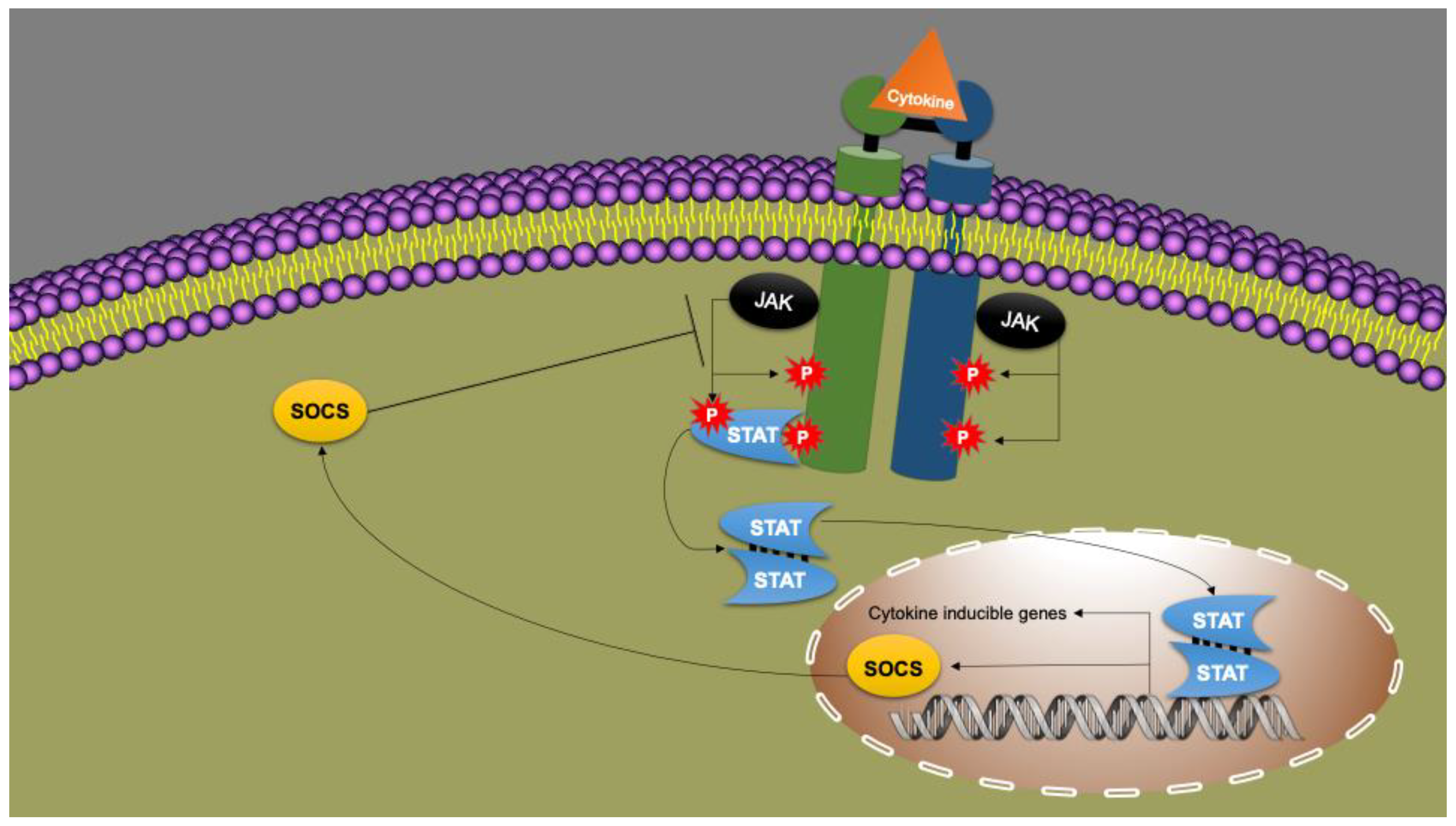
9. Cytokines as Immune System Messengers: JAK/STAT Signal Transduction
10. Obesity and Severe COVID-19
10.1. Obesity, Metaflammation and Combined Leptin and Insulin Resistance
10.2. Leptin and Insulin Resistance as a Cause of Severe COVID-19
10.3. The Core of Severe COVID-19
10.4. Reversing Leptin Resistance
11. Comprehensive Summary and Discussion
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oran, D.P.; Topol, E.J. The Proportion of SARS-CoV-2 Infections That are Asymptomatic: A Systematic Review. Ann. Intern. Med. 2021, 174, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 324, 782. [Google Scholar] [CrossRef] [PubMed]
- Cevik, M.; Kuppalli, K.; Kindrachuk, J.; Peiris, M. Virology, Transmission, and Pathogenesis of SARS-CoV-2. BMJ 2020, 371, m3862. [Google Scholar] [CrossRef]
- Mauvais-Jarvis, F. Aging, Male Sex, Obesity, and Metabolic Inflammation Create the Perfect Storm for COVID-19. Diabetes 2020, 69, 1857–1863. [Google Scholar] [CrossRef]
- Gao, Y.; Ding, M.; Dong, X.; Zhang, J.; Kursat Azkur, A.; Azkur, D.; Gan, H.; Sun, Y.; Fu, W.; Li, W.; et al. Risk Factors for Severe and Critically Ill COVID-19 Patients: A Review. Allergy 2021, 76, 428–455. [Google Scholar] [CrossRef] [PubMed]
- De Frel, D.L.; Atsma, D.E.; Pijl, H.; Seidell, J.C.; Leenen, P.J.M.; Dik, W.A.; van Rossum, E.F.C. The Impact of Obesity and Lifestyle on the Immune System and Susceptibility to Infections Such as COVID-19. Front. Nutr. 2020, 7, 597600. [Google Scholar] [CrossRef]
- De Larochelambert, Q.; Marc, A.; Antero, J.; Le Bourg, E.; Toussaint, J.-F. COVID-19 Mortality: A Matter of Vulnerability among Nations Facing Limited Margins of Adaptation. Front. Public Health 2020, 8, 604339. [Google Scholar] [CrossRef]
- Sudre, C.H.; Murray, B.; Varsavsky, T.; Graham, M.S.; Penfold, R.S.; Bowyer, R.C.; Pujol, J.C.; Klaser, K.; Antonelli, M.; Canas, L.S.; et al. Attributes and Predictors of Long COVID. Nat. Med. 2021, 27, 626–631. [Google Scholar] [CrossRef]
- Nalbandian, A.; Sehgal, K.; Gupta, A.; Madhavan, M.V.; McGroder, C.; Stevens, J.S.; Cook, J.R.; Nordvig, A.S.; Shalev, D.; Sehrawat, T.S.; et al. Post-Acute COVID-19 Syndrome. Nat. Med. 2021, 27, 601–615. [Google Scholar] [CrossRef]
- Aminian, A.; Bena, J.; Pantalone, K.M.; Burguera, B. Association of Obesity with Postacute Sequelae of COVID-19. Diabetes Obes. Metab. 2021, 23, 2183–2188. [Google Scholar] [CrossRef]
- Lopez-Leon, S.; Wegman-Ostrosky, T.; Perelman, C.; Sepulveda, R.; Rebolledo, P.A.; Cuapio, A.; Villapol, S. More than 50 Long-Term Effects of COVID-19: A Systematic Review and Meta-Analysis. Sci. Rep. 2021, 11, 16144. [Google Scholar] [CrossRef] [PubMed]
- Honce, R.; Schultz-Cherry, S. Impact of Obesity on Influenza A Virus Pathogenesis, Immune Response, and Evolution. Front. Immunol. 2019, 10, 1071. [Google Scholar] [CrossRef] [PubMed]
- Louie, J.K.; Acosta, M.; Samuel, M.C.; Schechter, R.; Vugia, D.J.; Harriman, K.; Matyas, B.T.; the California Pandemic (H1N1) Working Group. A Novel Risk Factor for a Novel Virus: Obesity and 2009 Pandemic Influenza A (H1N1). Clin. Infect. Dis. 2011, 52, 301–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badawi, A.; Ryoo, S.G. Prevalence of Comorbidities in the Middle East Respiratory Syndrome Coronavirus (MERS-CoV): A Systematic Review and Meta-Analysis. Int. J. Infect. Dis. 2016, 49, 129–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riaz, H.; Khan, M.S.; Siddiqi, T.J.; Usman, M.S.; Shah, N.; Goyal, A.; Khan, S.S.; Mookadam, F.; Krasuski, R.A.; Ahmed, H. Association between Obesity and Cardiovascular Outcomes: A Systematic Review and Meta-Analysis of Mendelian Randomization Studies. JAMA Netw. Open 2018, 1, e183788. [Google Scholar] [CrossRef]
- Blüher, M. Obesity: Global Epidemiology and Pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef]
- Milner, J.J.; Beck, M.A. The Impact of Obesity on the Immune Response to Infection. Proc. Nutr. Soc. 2012, 71, 298–306. [Google Scholar] [CrossRef] [Green Version]
- Green, W.D.; Beck, M.A. Obesity Impairs the Adaptive Immune Response to Influenza Virus. Ann. ATS 2017, 14 (Suppl. S5), 406–409. [Google Scholar] [CrossRef]
- Pérez-Galarza, J.; Prócel, C.; Cañadas, C.; Aguirre, D.; Pibaque, R.; Bedón, R.; Sempértegui, F.; Drexhage, H.; Baldeón, L. Immune Response to SARS-CoV-2 Infection in Obesity and T2D: Literature Review. Vaccines 2021, 9, 102. [Google Scholar] [CrossRef]
- Guglielmi, V.; Colangeli, L.; D’Adamo, M.; Sbraccia, P. Susceptibility and Severity of Viral Infections in Obesity: Lessons from Influenza to COVID-19. Does Leptin Play a Role? Int. J. Mol. Sci. 2021, 22, 3183. [Google Scholar] [CrossRef]
- Zhao, X.; Gang, X.; He, G.; Li, Z.; Lv, Y.; Han, Q.; Wang, G. Obesity Increases the Severity and Mortality of Influenza and COVID-19: A Systematic Review and Meta-Analysis. Front. Endocrinol. 2020, 11, 595109. [Google Scholar] [CrossRef] [PubMed]
- Chiappetta, S.; Sharma, A.M.; Bottino, V.; Stier, C. COVID-19 and the Role of Chronic Inflammation in Patients with Obesity. Int. J. Obes. 2020, 44, 1790–1792. [Google Scholar] [CrossRef] [PubMed]
- Hulme, K.D.; Noye, E.C.; Short, K.R.; Labzin, L.I. Dysregulated Inflammation during Obesity: Driving Disease Severity in Influenza Virus and SARS-CoV-2 Infections. Front. Immunol. 2021, 12, 770066. [Google Scholar] [CrossRef] [PubMed]
- Domingues, R.; Lippi, A.; Setz, C.; Outeiro, T.F.; Krisko, A. SARS-CoV-2, Immunosenescence and Inflammaging: Partners in the COVID-19 Crime. Aging 2020, 12, 18778–18789. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation, Metaflammation and Immunometabolic Disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Reaven, G.M. The insulin resistance syndrome: Definition and Dietary Approaches to Treatment. Annu. Rev. Nutr. 2005, 25, 391–406. [Google Scholar] [CrossRef] [Green Version]
- Stefan, N.; Birkenfeld, A.L.; Schulze, M.B. Global Pandemics Interconnected—Obesity, Impaired Metabolic Health and COVID-19. Nat. Rev. Endocrinol. 2021, 17, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Denson, J.L.; Gillet, A.S.; Zu, Y.; Brown, M.; Pham, T.; Yoshida, Y.; Mauvais-Jarvis, F.; Douglas, I.S.; Moore, M.; Tea, K.; et al. Metabolic Syndrome and Acute Respiratory Distress Syndrome in Hospitalized Patients with COVID-19. JAMA Netw. Open 2021, 4, e2140568. [Google Scholar] [CrossRef]
- Huang, P.L. A Comprehensive Definition for Metabolic Syndrome. Dis. Models Mech. 2009, 2, 231–237. [Google Scholar] [CrossRef] [Green Version]
- Rebello, C.J.; Kirwan, J.P.; Greenway, F.L. Obesity, the Most Common Comorbidity in SARS-CoV-2: Is Leptin the Link? Int. J. Obes. 2020, 44, 1810–1817. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, S.; Aziz, R.; Al Mahri, S.; Malik, S.S.; Haji, E.; Khan, A.H.; Khatlani, T.S.; Bouchama, A. Obesity and COVID-19: What Makes Obese Host so Vulnerable? Immun. Ageing 2021, 18, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.-C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef] [PubMed]
- Petersen, E.; Koopmans, M.; Go, U.; Hamer, D.H.; Petrosillo, N.; Castelli, F.; Storgaard, M.; Al Khalili, S.; Simonsen, L. Comparing SARS-CoV-2 with SARS-CoV and Influenza Pandemics. Lancet Infect. Dis. 2020, 20, e238–e244. [Google Scholar] [CrossRef]
- Kirkman, M.S.; Briscoe, V.J.; Clark, N.; Florez, H.; Haas, L.B.; Halter, J.B.; Huang, E.S.; Korytkowski, M.T.; Munshi, M.N.; Odegard, P.S.; et al. Diabetes in Older Adults. Diabetes Care 2012, 35, 2650–2664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbieri, M.; Gambardella, A.; Paolisso, G.; Varricchio, M. Metabolic Aspects of the Extreme Longevity. Exp. Gerontol. 2008, 43, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Sell, H.; Habich, C.; Eckel, J. Adaptive Immunity in Obesity and Insulin Resistance. Nat. Rev. Endocrinol. 2012, 8, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Van Bussel, E.F.; Hoevenaar-Blom, M.P.; Poortvliet, R.K.E.; Gussekloo, J.; van Dalen, J.W.; van Gool, W.A.; Richard, E.; Moll van Charante, E.P. Predictive Value of Traditional Risk Factors for Cardiovascular Disease in Older People: A Systematic Review. Prev. Med. 2020, 132, 105986. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Age and Cancer Risk; National Cancer Institute: Bethesda, MD, USA, 2021.
- Gombart, A.F.; Pierre, A.; Maggini, S. A Review of Micronutrients and the Immune System–Working in Harmony to Reduce the Risk of Infection. Nutrients 2020, 12, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Araújo Morais, A.H.; de Souza Aquino, J.; da Silva-Maia, J.K.; de Lima Vale, S.H.; Maciel, B.L.L.; Passos, T.S. Nutritional Status, Diet and Viral Respiratory Infections: Perspectives for Severe Acute Respiratory Syndrome Coronavirus 2. Br. J. Nutr. 2021, 125, 851–862. [Google Scholar] [CrossRef]
- Fedele, D.; De Francesco, A.; Riso, S.; Collo, A. Obesity, Malnutrition, and Trace Element Deficiency in the Coronavirus Disease (COVID-19) Pandemic: An Overview. Nutrition 2021, 81, 111016. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Nutrition and Immunity: Lessons for COVID-19. Eur. J. Clin. Nutr. 2021, 75, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Vogel-González, M.; Talló-Parra, M.; Herrera-Fernández, V.; Pérez-Vilaró, G.; Chillón, M.; Nogués, X.; Gómez-Zorrilla, S.; López-Montesinos, I.; Arnau-Barrés, I.; Sorli-Redó, M.L.; et al. Low Zinc Levels at Admission Associates with Poor Clinical Outcomes in SARS-CoV-2 Infection. Nutrients 2021, 13, 562. [Google Scholar] [CrossRef]
- Jothimani, D.; Kailasam, E.; Danielraj, S.; Nallathambi, B.; Ramachandran, H.; Sekar, P.; Manoharan, S.; Ramani, V.; Narasimhan, G.; Kaliamoorthy, I.; et al. COVID-19: Poor Outcomes in Patients with Zinc Deficiency. Int. J. Infect. Dis. 2020, 100, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Heller, R.A.; Sun, Q.; Hackler, J.; Seelig, J.; Seibert, L.; Cherkezov, A.; Minich, W.B.; Seemann, P.; Diegmann, J.; Pilz, M.; et al. Prediction of Survival Odds in COVID-19 by Zinc, Age and Selenoprotein P as Composite Biomarker. Redox Biol. 2021, 38, 101764. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, A.; Heller, R.; Sun, Q.; Seelig, J.; Cherkezov, A.; Seibert, L.; Hackler, J.; Seemann, P.; Diegmann, J.; Pilz, M.; et al. Selenium Deficiency is Associated with Mortality Risk from COVID-19. Nutrients 2020, 12, 2098. [Google Scholar] [CrossRef]
- Wang, Z.; Joshi, A.; Leopold, K.; Jackson, S.; Christensen, S.; Nayfeh, T.; Mohammed, K.; Creo, A.; Tebben, P.; Kumar, S. Association of Vitamin D Deficiency with COVID-19 Infection Severity: Systematic Review and Meta-analysis. Clin. Endocrinol. 2021, 96, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, F.; Luciani, A.; Perego, G.; Dognini, G.; Colombelli, P.L.; Ghidini, A. Therapeutic and Prognostic Role of Vitamin D for COVID-19 Infection: A Systematic Review and Meta-Analysis of 43 Observational Studies. J. Steroid Biochem. Mol. Biol. 2021, 211, 105883. [Google Scholar] [CrossRef]
- Borsche, L.; Glauner, B.; von Mendel, J. COVID-19 Mortality Risk Correlates Inversely with Vitamin D3 Status, and a Mortality Rate Close to Zero Could Theoretically Be Achieved at 50 Ng/ML 25(OH)D3: Results of a Systematic Review and Meta-Analysis. Nutrients 2021, 13, 3596. [Google Scholar] [CrossRef] [PubMed]
- Kaidar-Person, O.; Person, B.; Szomstein, S.; Rosenthal, R.J. Nutritional Deficiencies in Morbidly Obese Patients: A New Form of Malnutrition?: Part A: Vitamins. Obes. Surg. 2008, 18, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Kaidar-Person, O.; Person, B.; Szomstein, S.; Rosenthal, R.J. Nutritional Deficiencies in Morbidly Obese Patients: A New Form of Malnutrition?: Part B: Minerals. Obes. Surg. 2008, 18, 1028–1034. [Google Scholar] [CrossRef]
- Banach, W.; Nitschke, K.; Krajewska, N.; Mongiałło, W.; Matuszak, O.; Muszyński, J.; Skrypnik, D. The Association between Excess Body Mass and Disturbances in Somatic Mineral Levels. Int. J. Mol. Sci. 2020, 21, 7306. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Santos, M.; Costa, P.R.F.; Assis, A.M.O.; Santos, C.A.S.T.; Santos, D.B. Obesity and Vitamin D Deficiency: A Systematic Review and Meta-Analysis: Obesity and Vitamin D. Obes. Rev. 2015, 16, 341–349. [Google Scholar] [CrossRef]
- Malden, S.; Gillespie, J.; Hughes, A.; Gibson, A.; Farooq, A.; Martin, A.; Summerbell, C.; Reilly, J.J. Obesity in Young Children and Its Relationship with Diagnosis of Asthma, Vitamin D Deficiency, Iron Deficiency, Specific Allergies and Flat-footedness: A Systematic Review and Meta-analysis. Obes. Rev. 2021, 22, e13129. [Google Scholar] [CrossRef]
- Pepersack, T.; Rotsaert, P.; Benoit, F.; Willems, D.; Fuss, M.; Bourdoux, P.; Duchateau, J. Prevalence of Zinc Deficiency and Its Clinical Relevance among Hospitalised Elderly. Arch. Gerontol. Geriatr. 2001, 33, 243–253. [Google Scholar] [CrossRef]
- Savarino, L.; Granchi, D.; Ciapetti, G.; Cenni, E.; Ravaglia, G.; Forti, P.; Maioli, F.; Mattioli, R. Serum Concentrations of Zinc and Selenium in Elderly People: Results in Healthy Nonagenarians/Centenarians. Exp. Gerontol. 2001, 36, 327–339. [Google Scholar] [CrossRef]
- Yasuda, H.; Tsutsui, T. Infants and Elderlies are Susceptible to Zinc Deficiency. Sci. Rep. 2016, 6, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastian, R.S.; Cleveland, L.E.; Goldman, J.D.; Moshfegh, A.J. Older Adults Who Use Vitamin/Mineral Supplements Differ from Nonusers in Nutrient Intake Adequacy and Dietary Attitudes. J. Am. Diet. Assoc. 2007, 107, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Smit, E.; Winters-Stone, K.M.; Loprinzi, P.D.; Tang, A.M.; Crespo, C.J. Lower Nutritional Status and Higher Food Insufficiency in Frail Older US Adults. Br. J. Nutr. 2013, 110, 172–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fantacone, M.L.; Lowry, M.B.; Uesugi, S.L.; Michels, A.J.; Choi, J.; Leonard, S.W.; Gombart, S.K.; Gombart, J.S.; Bobe, G.; Gombart, A.F. The Effect of a Multivitamin and Mineral Supplement on Immune Function in Healthy Older Adults: A Double-Blind, Randomized, Controlled Trial. Nutrients 2020, 12, 2447. [Google Scholar] [CrossRef] [PubMed]
- Haraj, N.E.; El Aziz, S.; Chadli, A.; Dafir, A.; Mjabber, A.; Aissaoui, O.; Barrou, L.; El Kettani El Hamidi, C.; Nsiri, A.; AL Harrar, R.; et al. Nutritional Status Assessment in Patients with COVID-19 after Discharge from the Intensive Care Unit. Clin. Nutr. ESPEN 2021, 41, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Elham, A.S.; Azam, K.; Azam, J.; Mostafa, L.; Nasrin, B.; Marzieh, N. Serum Vitamin D, Calcium, and Zinc Levels in Patients with COVID-19. Clin. Nutr. ESPEN 2021, 43, 276–282. [Google Scholar] [CrossRef]
- Tomasa-Irriguible, T.-M.; Bielsa-Berrocal, L.; Bordejé-Laguna, L.; Tural-Llàcher, C.; Barallat, J.; Manresa-Domínguez, J.-M.; Torán-Monserrat, P. Low Levels of Few Micronutrients May Impact COVID-19 Disease Progression: An Observational Study on the First Wave. Metabolites 2021, 11, 565. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.M.; Morley, J.E. Editorial: COVID-19 in Older Persons: The Role of Nutrition. Curr. Opin. Clin. Nutr. Metab. Care 2021, 24, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Rayman, M.P.; Calder, P.C. Optimising COVID-19 Vaccine Efficacy by Ensuring Nutritional Adequacy. Br. J. Nutr. 2021, 126, 1919–1920. [Google Scholar] [CrossRef]
- Gao, F.; Zheng, K.I.; Wang, X.-B.; Sun, Q.-F.; Pan, K.-H.; Wang, T.-Y.; Chen, Y.-P.; Targher, G.; Byrne, C.D.; George, J.; et al. Obesity is a Risk Factor for Greater COVID-19 Severity. Diabetes Care 2020, 43, 72–74. [Google Scholar] [CrossRef]
- Caussy, C.; Pattou, F.; Wallet, F.; Simon, C.; Chalopin, S.; Telliam, C.; Mathieu, D.; Subtil, F.; Frobert, E.; Alligier, M.; et al. Prevalence of Obesity among Adult Inpatients with COVID-19 in France. Lancet Diabetes Endocrinol. 2020, 8, 562–564. [Google Scholar] [CrossRef]
- Fresán, U.; Guevara, M.; Elía, F.; Albéniz, E.; Burgui, C.; Castilla, J.; for the Working Group for the Study of COVID-19 in Navarra; Martín, C.; Navascués, A.; Portillo, M.E.; et al. Independent Role of Severe Obesity as a Risk Factor for COVID-19 Hospitalization: A Spanish Population-Based Cohort Study. Obesity 2021, 29, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Kass, D.A.; Duggal, P.; Cingolani, O. Obesity Could Shift Severe COVID-19 Disease to Younger Ages. Lancet 2020, 395, 1544–1545. [Google Scholar] [CrossRef]
- Földi, M.; Farkas, N.; Kiss, S.; Zádori, N.; Váncsa, S.; Szakó, L.; Dembrovszky, F.; Solymár, M.; Bartalis, E.; Szakács, Z.; et al. Obesity Is a Risk Factor for Developing Critical Condition in COVID-19 Patients: A Systematic Review and Meta-analysis. Obes. Rev. 2020, 21, e13095. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.; Chou, C.; Chang, L. Effect of Obesity and Body Mass Index on Coronavirus Disease 2019 Severity: A Systematic Review and Meta-analysis. Obes. Rev. 2020, 21, e13089. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Yang, J.; Shi, J.; Zhang, P.; Wang, X. Obesity Is Associated with Increased Severity of Disease in COVID-19 Pneumonia: A Systematic Review and Meta-Analysis. Eur. J. Med. Res. 2020, 25, 1–25. [Google Scholar] [CrossRef]
- Huang, Y.; Lu, Y.; Huang, Y.-M.; Wang, M.; Ling, W.; Sui, Y.; Zhao, H.-L. Obesity in Patients with COVID-19: A Systematic Review and Meta-Analysis. Metabolism 2020, 113, 154378. [Google Scholar] [CrossRef] [PubMed]
- Földi, M.; Farkas, N.; Kiss, S.; Dembrovszky, F.; Szakács, Z.; Balaskó, M.; Erőss, B.; Hegyi, P.; Szentesi, A. Visceral Adiposity Elevates the Risk of Critical Condition in COVID-19: A Systematic Review and Meta-Analysis. Obesity 2021, 29, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Bunnell, K.M.; Thaweethai, T.; Buckless, C.; Shinnick, D.J.; Torriani, M.; Foulkes, A.S.; Bredella, M.A. Body Composition Predictors of Outcome in Patients with COVID-19. Int. J. Obes. 2021, 45, 2238–2243. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Mori, M.; Jingushi, Y.; Matsuzaki, H.; Katahira, K.; Ishimatsu, A.; Enokizu-Ogawa, A.; Taguchi, K.; Moriwaki, A.; Yoshida, M. Impact of Visceral Fat on the Prognosis of Coronavirus Disease 2019: An Observational Cohort Study. BMC Infect. Dis. 2021, 21, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Roncon, L.; Zuin, M.; Rigatelli, G.; Zuliani, G. Diabetic Patients with COVID-19 Infection Are at Higher Risk of ICU Admission and Poor Short-Term Outcome. J. Clin. Virol. 2020, 127, 104354. [Google Scholar] [CrossRef] [PubMed]
- Ghoneim, S.; Butt, M.U.; Hamid, O.; Shah, A.; Asaad, I. The Incidence of COVID-19 in Patients with Metabolic Syndrome and Non-Alcoholic Steatohepatitis: A Population-Based Study. Metab. Open 2020, 8, 100057. [Google Scholar] [CrossRef] [PubMed]
- National Center for Health Statistics. Prevalence of Obesity and Severe Obesity among Adults: United States, 2017–2018; NCHS Data Brief, no 360; National Center for Health Statistics: Hyattsville, MD, USA, 2020.
- Zhang, S.; Zhu, H.; Ye, H.; Hu, Y.; Zheng, N.; Huang, Z.; Xiong, Z.; Fu, L.; Cai, T. Risk Factors for Prolonged Virus Shedding of Respiratory Tract and Fecal in Adults with Severe Acute Respiratory Syndrome Coronavirus-2 Infection. J. Clin. Lab. Anal. 2021, 35, e23923. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yan, L.-M.; Wan, L.; Xiang, T.-X.; Le, A.; Liu, J.-M.; Peiris, M.; Poon, L.L.M.; Zhang, W. Viral Dynamics in Mild and Severe Cases of COVID-19. Lancet Infect. Dis. 2020, 20, 656–657. [Google Scholar] [CrossRef] [Green Version]
- Honce, R.; Karlsson, E.A.; Wohlgemuth, N.; Estrada, L.D.; Meliopoulos, V.A.; Yao, J.; Schultz-Cherry, S. Obesity-Related Microenvironment Promotes Emergence of Virulent Influenza Virus Strains. mBio 2020, 11, e03341-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheridan, P.A.; Paich, H.A.; Handy, J.; Karlsson, E.A.; Hudgens, M.G.; Sammon, A.B.; Holland, L.A.; Weir, S.; Noah, T.L.; Beck, M.A. Obesity is Associated with Impaired Immune Response to Influenza Vaccination in Humans. Int. J. Obes. 2012, 36, 1072–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banga, N.; Guss, P.; Banga, A.; Rosenman, K.D. Incidence and Variables Associated with Inadequate Antibody Titers after Pre-Exposure Rabies Vaccination among Veterinary Medical Students. Vaccine 2014, 32, 979–983. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Chen, X.; Shen, C.; Guo, Z.; Dong, C. Hepatitis B Vaccine Response in Obesity: A Meta-Analysis. Vaccine 2016, 34, 4835–4841. [Google Scholar] [CrossRef] [PubMed]
- Malavazos, A.E.; Basilico, S.; Iacobellis, G.; Milani, V.; Cardani, R.; Boniardi, F.; Dubini, C.; Prandoni, I.; Capitanio, G.; Renna, L.V.; et al. Antibody Responses to BNT162b2 MRNA Vaccine: Infection-naïve Individuals with Abdominal Obesity Warrant Attention. Obesity 2021, 30, 606–613. [Google Scholar] [CrossRef]
- Gleeson, L.E.; Roche, H.M.; Sheedy, F.J. Obesity, COVID-19 and Innate Immunometabolism. Br. J. Nutr. 2021, 125, 628–632. [Google Scholar] [CrossRef] [PubMed]
- La Cava, A. Leptin in Inflammation and Autoimmunity. Cytokine 2017, 98, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Francisco, V.; Pino, J.; Campos-Cabaleiro, V.; Ruiz-Fernández, C.; Mera, A.; Gonzalez-Gay, M.A.; Gómez, R.; Gualillo, O. Obesity, Fat Mass and Immune System: Role for Leptin. Front. Physiol. 2018, 9, 640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McEwen, B.S.; Wingfield, J.C. The Concept of Allostasis in Biology and Biomedicine. Horm. Behav. 2003, 43, 2–15. [Google Scholar] [CrossRef]
- Mcewen, B.S. Protection and Damage from Acute and Chronic Stress: Allostasis and Allostatic Overload and Relevance to the Pathophysiology of Psychiatric Disorders. Ann. N. Y. Acad. Sci. 2004, 1032, 1–7. [Google Scholar] [CrossRef]
- Wingfield, J.C. Control of Behavioural Strategies for Capricious Environments. Anim. Behav. 2003, 66, 807–816. [Google Scholar] [CrossRef] [Green Version]
- McEwen, B.S. Physiology and Neurobiology of Stress and Adaptation: Central Role of the Brain. Physiol. Rev. 2007, 87, 873–904. [Google Scholar] [CrossRef] [Green Version]
- Huber, M.; Knottnerus, J.A.; Green, L.; van der Horst, H.; Jadad, A.R.; Kromhout, D.; Leonard, B.; Lorig, K.; Loureiro, M.I.; van der Meer, J.W.M.; et al. How Should We Define Health? BMJ 2011, 343, 4163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Otín, C.; Kroemer, G. Hallmarks of Health. Cell 2021, 184, 33–63. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.; Yang, X.; Yang, D.; Bao, J.; Li, R.; Xiao, Y.; Hou, C.; Wang, H.; Liu, J.; Yang, D.; et al. Role of Angiotensin-Converting Enzyme 2 (ACE2) in COVID-19. Crit. Care 2020, 24, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Clausen, T.M.; Sandoval, D.R.; Spliid, C.B.; Pihl, J.; Perrett, H.R.; Painter, C.D.; Narayanan, A.; Majowicz, S.A.; Kwong, E.M.; McVicar, R.N.; et al. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 2020, 183, 1043–1057.e15. [Google Scholar] [CrossRef] [PubMed]
- Scialo, F.; Daniele, A.; Amato, F.; Pastore, L.; Matera, M.G.; Cazzola, M.; Castaldo, G.; Bianco, A. ACE2: The Major Cell Entry Receptor for SARS-CoV-2. Lung 2020, 198, 867–877. [Google Scholar] [CrossRef]
- Foresta, C.; Rocca, M.S.; Di Nisio, A. Gender Susceptibility to COVID-19: A Review of the Putative Role of Sex Hormones and X Chromosome. J. Endocrinol. Investig. 2021, 44, 951–956. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary Manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032. [Google Scholar] [CrossRef]
- The NHLBI LungMap Consortium; The Human Cell Atlas Lung Biological Network; Muus, C.; Luecken, M.D.; Eraslan, G.; Sikkema, L.; Waghray, A.; Heimberg, G.; Kobayashi, Y.; Vaishnav, E.D.; et al. Single-Cell Meta-Analysis of SARS-CoV-2 Entry Genes across Tissues and Demographics. Nat. Med. 2021, 27, 546–559. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 Entry into Cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Glowacka, I.; Bertram, S.; Muller, M.A.; Allen, P.; Soilleux, E.; Pfefferle, S.; Steffen, I.; Tsegaye, T.S.; He, Y.; Gnirss, K.; et al. Evidence That TMPRSS2 Activates the Severe Acute Respiratory Syndrome Coronavirus Spike Protein for Membrane Fusion and Reduces Viral Control by the Humoral Immune Response. J. Virol. 2011, 85, 4122–4134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senapati, S.; Banerjee, P.; Bhagavatula, S.; Kushwaha, P.P.; Kumar, S. Contributions of Human ACE2 and TMPRSS2 in Determining Host–Pathogen Interaction of COVID-19. J. Genet. 2021, 100, 12. [Google Scholar] [CrossRef] [PubMed]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 Facilitates SARS-CoV-2 Cell Entry and Infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Medina-Enríquez, M.M.; Lopez-León, S.; Carlos-Escalante, J.A.; Aponte-Torres, Z.; Cuapio, A.; Wegman-Ostrosky, T. ACE2: The Molecular Doorway to SARS-CoV-2. Cell Biosci. 2020, 10, 148. [Google Scholar] [CrossRef]
- Emilsson, V.; Gudmundsson, E.F.; Aspelund, T.; Jonsson, B.G.; Gudjonsson, A.; Launer, L.J.; Lamb, J.R.; Gudmundsdottir, V.; Jennings, L.L.; Gudnason, V. Serum Levels of ACE2 Are Higher in Patients with Obesity and Diabetes. Obes. Sci. Pract. 2021, 7, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Al-Benna, S. Association of High Level Gene Expression of ACE2 in Adipose Tissue with Mortality of COVID-19 Infection in Obese Patients. Obes. Med. 2020, 19, 100283. [Google Scholar] [CrossRef]
- Libby, P.; Lüscher, T. COVID-19 is, in the End, an Endothelial Disease. Eur. Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef] [PubMed]
- Loganathan, S.; Kuppusamy, M.; Wankhar, W.; Gurugubelli, K.R.; Mahadevappa, V.H.; Lepcha, L.; Choudhary, A.K. Angiotensin-Converting Enzyme 2 (ACE2): COVID 19 Gate Way to Multiple Organ Failure Syndromes. Respir. Physiol. Neurobiol. 2021, 283, 103548. [Google Scholar] [CrossRef]
- Imazio, M.; Klingel, K.; Kindermann, I.; Brucato, A.; De Rosa, F.G.; Adler, Y.; De Ferrari, G.M. COVID-19 Pandemic and Troponin: Indirect Myocardial Injury, Myocardial Inflammation or Myocarditis? Heart 2020, 106, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Randeva, H.; Chatha, K.; Hall, M.; Spandidos, D.; Karteris, E.; Kyrou, I. Neuropilin-1 as a New Potential SARS-CoV-2 Infection Mediator Implicated in the Neurologic Features and Central Nervous System Involvement of COVID-19. Mol. Med. Rep. 2020, 22, 4221–4226. [Google Scholar] [CrossRef] [PubMed]
- Franke, C.; Ferse, C.; Kreye, J.; Reincke, S.M.; Sanchez-Sendin, E.; Rocco, A.; Steinbrenner, M.; Angermair, S.; Treskatsch, S.; Zickler, D.; et al. High Frequency of Cerebrospinal Fluid Autoantibodies in COVID-19 Patients with Neurological Symptoms. Brain Behav. Immun. 2021, 93, 415–419. [Google Scholar] [CrossRef]
- Yang, A.C.; Kern, F.; Losada, P.M.; Agam, M.R.; Maat, C.A.; Schmartz, G.P.; Fehlmann, T.; Stein, J.A.; Schaum, N.; Lee, D.P.; et al. Dysregulation of Brain and Choroid Plexus Cell Types in Severe COVID-19. Nature 2021, 595, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Douaud, G.; Lee, S.; Alfaro-Almagro, F.; Arthofer, C.; Wang, C.; McCarthy, P.; Lange, F.; Andersson, J.L.R.; Griffanti, L.; Duff, E.; et al. SARS-CoV-2 Is Associated with Changes in Brain Structure in UK Biobank. medRxiv 2022. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Hu, W.; Yu, H.; Zhao, L.; Zhao, Y.; Zhao, X.; Xue, H.; Zhao, Y. Little to No Expression of Angiotensin-converting Enzyme-2 on Most Human Peripheral Blood Immune Cells but Highly Expressed on Tissue Macrophages. Cytometry 2020. [Google Scholar] [CrossRef] [PubMed]
- Abassi, Z.; Knaney, Y.; Karram, T.; Heyman, S.N. The Lung Macrophage in SARS-CoV-2 Infection: A Friend or a Foe? Front. Immunol. 2020, 11, 1312. [Google Scholar] [CrossRef] [PubMed]
- Ryan, P.M.; Caplice, N.M. Is Adipose Tissue a Reservoir for Viral Spread, Immune Activation, and Cytokine Amplification in Coronavirus Disease 2019? Obesity 2020, 28, 1191–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, T. Neurological Infection with SARS-CoV-2—The Story so Far. Nat. Rev. Neurol. 2021, 17, 65–66. [Google Scholar] [CrossRef] [PubMed]
- Hoagland, D.A.; Møller, R.; Uhl, S.A.; Oishi, K.; Frere, J.; Golynker, I.; Horiuchi, S.; Panis, M.; Blanco-Melo, D.; Sachs, D.; et al. Leveraging the Antiviral Type I Interferon System as a First Line of Defense against SARS-CoV-2 Pathogenicity. Immunity 2021, 54, 557–570.e5. [Google Scholar] [CrossRef] [PubMed]
- Matricardi, P.M.; Dal Negro, R.W.; Nisini, R. The First, Holistic Immunological Model of COVID-19: Implications for Prevention, Diagnosis, and Public Health Measures. Pediatr. Allergy Immunol. 2020, 31, 454–470. [Google Scholar] [CrossRef]
- Lau, E.H.Y.; Tsang, O.T.Y.; Hui, D.S.C.; Kwan, M.Y.W.; Chan, W.; Chiu, S.S.; Ko, R.L.W.; Chan, K.H.; Cheng, S.M.S.; Perera, R.A.P.M.; et al. Neutralizing Antibody Titres in SARS-CoV-2 Infections. Nat. Commun. 2021, 12, 63. [Google Scholar] [CrossRef]
- Lucas, C.; Klein, J.; Sundaram, M.E.; Liu, F.; Wong, P.; Silva, J.; Mao, T.; Oh, J.E.; Mohanty, S.; Huang, J.; et al. Delayed Production of Neutralizing Antibodies Correlates with Fatal COVID-19. Nat. Med. 2021, 27, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.D.; de Graaf, E.L.; Sonneveld, M.E.; Plomp, H.R.; Nouta, J.; Hoepel, W.; Chen, H.-J.; Linty, F.; Visser, R.; Brinkhaus, M.; et al. Afucosylated IgG Characterizes Enveloped Viral Responses and Correlates with COVID-19 Severity. Science 2021, 371, eabc8378. [Google Scholar] [CrossRef]
- Štambuk, T.; Klasić, M.; Zoldoš, V.; Lauc, G. N-Glycans as Functional Effectors of Genetic and Epigenetic Disease Risk. Mol. Asp. Med. 2021, 79, 100891. [Google Scholar] [CrossRef]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F.P. The Trinity of COVID-19: Immunity, Inflammation and Intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider Cytokine Storm Syndromes and Immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef]
- Bhaskar, S.; Sinha, A.; Banach, M.; Mittoo, S.; Weissert, R.; Kass, J.S.; Rajagopal, S.; Pai, A.R.; Kutty, S. Cytokine Storm in COVID-19—Immunopathological Mechanisms, Clinical Considerations, and Therapeutic Approaches: The REPROGRAM Consortium Position Paper. Front. Immunol. 2020, 11, 1648. [Google Scholar] [CrossRef] [PubMed]
- Savla, S.R.; Prabhavalkar, K.S.; Bhatt, L.K. Cytokine Storm Associated Coagulation Complications in COVID-19 Patients: Pathogenesis and Management. Expert Rev. Anti-Infect. Ther. 2021, 19, 1397–1413. [Google Scholar] [CrossRef]
- Carvalho, T.; Krammer, F.; Iwasaki, A. The First 12 Months of COVID-19: A Timeline of Immunological Insights. Nat. Rev. Immunol. 2021, 21, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Giamarellos-Bourboulis, E.J.; Domínguez-Andrés, J.; Curtis, N.; van Crevel, R.; van de Veerdonk, F.L.; Bonten, M. Trained Immunity: A Tool for Reducing Susceptibility to and the Severity of SARS-CoV-2 Infection. Cell 2020, 181, 969–977. [Google Scholar] [CrossRef]
- Jarjour, N.N.; Masopust, D.; Jameson, S.C. T Cell Memory: Understanding COVID-19. Immunity 2021, 54, 14–18. [Google Scholar] [CrossRef]
- Dan, J.M.; Mateus, J.; Kato, Y.; Hastie, K.M.; Yu, E.D.; Faliti, C.E.; Grifoni, A.; Ramirez, S.I.; Haupt, S.; Frazier, A.; et al. Immunological Memory to SARS-CoV-2 Assessed for up to 8 Months after Infection. Science 2021, 371, eabf4063. [Google Scholar] [CrossRef] [PubMed]
- Havervall, S.; Ng, H.; Jernbom Falk, A.; Greilert-Norin, N.; Månberg, A.; Marking, U.; Laurén, I.; Gabrielsson, L.; Salomonsson, A.; Aguilera, K.; et al. Robust Humoral and Cellular Immune Responses and Low Risk for Reinfection at Least 8 Months Following Asymptomatic to Mild COVID-19. J. Intern. Med. 2022, 291, 72–80. [Google Scholar] [CrossRef]
- Sadarangani, M.; Marchant, A.; Kollmann, T.R. Immunological Mechanisms of Vaccine-Induced Protection against COVID-19 in Humans. Nat. Rev. Immunol. 2021, 21, 475–484. [Google Scholar] [CrossRef]
- Ieronymaki, E.; Daskalaki, M.G.; Lyroni, K.; Tsatsanis, C. Insulin Signaling and Insulin Resistance Facilitate Trained Immunity in Macrophages Through Metabolic and Epigenetic Changes. Front. Immunol. 2019, 10, 1330. [Google Scholar] [CrossRef] [PubMed]
- Moorlag, S.J.C.F.M.; van Deuren, R.C.; van Werkhoven, C.H.; Jaeger, M.; Debisarun, P.; Taks, E.; Mourits, V.P.; Koeken, V.A.C.M.; de Bree, L.C.J.; ten Doesschate, T.; et al. Safety and COVID-19 Symptoms in Individuals Recently Vaccinated with BCG: A Retrospective Cohort Study. Cell Rep. Med. 2020, 1, 100073. [Google Scholar] [CrossRef]
- Macintyre, A.N.; Gerriets, V.A.; Nichols, A.G.; Michalek, R.D.; Rudolph, M.C.; Deoliveira, D.; Anderson, S.M.; Abel, E.D.; Chen, B.J.; Hale, L.P.; et al. The Glucose Transporter Glut1 Is Selectively Essential for CD4 T Cell Activation and Effector Function. Cell Metab. 2014, 20, 61–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, C.S.; Palchaudhuri, R.; Albargy, H.; Abdel-Mohsen, M.; Crowe, S.M. Exploiting Immune Cell Metabolic Machinery for Functional HIV Cure and the Prevention of Inflammaging. F1000Research 2018, 7, 125. [Google Scholar] [CrossRef] [Green Version]
- Sa Ribero, M.; Jouvenet, N.; Dreux, M.; Nisole, S. Interplay between SARS-CoV-2 and the Type I Interferon Response. PLoS Pathog. 2020, 16, e1008737. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Shi, P.-Y. Antagonism of Type I Interferon by Severe Acute Respiratory Syndrome Coronavirus 2. J. Interferon Cytokine Res. 2020, 40, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Lopez, L.; Sang, P.C.; Tian, Y.; Sang, Y. Dysregulated Interferon Response Underlying Severe COVID-19. Viruses 2020, 12, 1433. [Google Scholar] [CrossRef]
- Suryawanshi, R.K.; Koganti, R.; Agelidis, A.; Patil, C.D.; Shukla, D. Dysregulation of Cell Signaling by SARS-CoV-2. Trends Microbiol. 2021, 29, 224–237. [Google Scholar] [CrossRef]
- Samuel, C.E. Antiviral Actions of Interferons. Clin. Microbiol. Rev. 2001, 14, 778–809. [Google Scholar] [CrossRef] [Green Version]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The Molecular Details of Cytokine Signaling via the JAK/STAT Pathway: Cytokine Signaling via the JAK/STAT Pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Gong, M.; Zhao, F.; Shao, J.; Xie, Y.; Zhang, Y.; Chang, H. Type I Interferons: Distinct Biological Activities and Current Applications for Viral Infection. Cell Physiol. Biochem. 2018, 51, 2377–2396. [Google Scholar] [CrossRef]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the Eye of the Cytokine Storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meffre, E.; Iwasaki, A. Interferon Deficiency Can Lead to Severe COVID. Nature 2020, 587, 374–376. [Google Scholar] [CrossRef]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.-H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Béziat, V.; et al. Autoantibodies against Type I IFNs in Patients with Life-Threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef] [PubMed]
- Dotan, A.; Muller, S.; Kanduc, D.; David, P.; Halpert, G.; Shoenfeld, Y. The SARS-CoV-2 as an Instrumental Trigger of Autoimmunity. Autoimmun. Rev. 2021, 20, 102792. [Google Scholar] [CrossRef] [PubMed]
- Novelli, L.; Motta, F.; De Santis, M.; Ansari, A.A.; Gershwin, M.E.; Selmi, C. The JANUS of Chronic Inflammatory and Autoimmune Diseases Onset during COVID-19—A Systematic Review of the Literature. J. Autoimmun. 2021, 117, 102592. [Google Scholar] [CrossRef]
- Frasca, D.; Reidy, L.; Romero, M.; Diaz, A.; Cray, C.; Kahl, K.; Blomberg, B.B. The Majority of SARS-CoV-2-Specific Antibodies in COVID-19 Patients with Obesity Are Autoimmune and Not Neutralizing. Int. J. Obes. Lond. 2021. [Google Scholar] [CrossRef]
- Liu, S.; Yan, R.; Chen, B.; Pan, Q.; Chen, Y.; Hong, J.; Zhang, L.; Liu, W.; Wang, S.; Chen, J.-L. Influenza Virus-Induced Robust Expression of SOCS3 Contributes to Excessive Production of IL-6. Front. Immunol. 2019, 10, 1843. [Google Scholar] [CrossRef] [Green Version]
- Johnson, H.M.; Lewin, A.S.; Ahmed, C.M. SOCS, Intrinsic Virulence Factors, and Treatment of COVID-19. Front. Immunol. 2020, 11, 582102. [Google Scholar] [CrossRef]
- Alti, D.; Sambamurthy, C.; Kalangi, S.K. Emergence of Leptin in Infection and Immunity: Scope and Challenges in Vaccines Formulation. Front. Cell. Infect. Microbiol. 2018, 8, 147. [Google Scholar] [CrossRef]
- Akhtar, L.N.; Benveniste, E.N. Viral Exploitation of Host SOCS Protein Functions. J. Virol. 2011, 85, 1912–1921. [Google Scholar] [CrossRef] [Green Version]
- He, B.; Tran, J.T.; Sanchez, D.J. Manipulation of Type I Interferon Signaling by HIV and AIDS-Associated Viruses. J. Immunol. Res. 2019, 2019, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Hou, M.; Liu, X.; Li, Z.; Yang, Y.; Zhang, W. Induction of SOCS Expression by EV71 Infection Promotes EV71 Replication. BioMed Res. Int. 2020, 2020, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Liu, K.; Cheng, A.; Wang, M.; Cui, M.; Huang, J.; Zhu, D.; Chen, S.; Liu, M.; Zhao, X.; et al. SOCS Proteins Participate in the Regulation of Innate Immune Response Caused by Viruses. Front. Immunol. 2020, 11, 558341. [Google Scholar] [CrossRef]
- Xie, J.; Wang, M.; Cheng, A.; Jia, R.; Zhu, D.; Liu, M.; Chen, S.; Zhao, X.; Yang, Q.; Wu, Y.; et al. The Role of SOCS Proteins in the Development of Virus- Induced Hepatocellular Carcinoma. Virol. J. 2021, 18, 74. [Google Scholar] [CrossRef]
- Duncan, S.A.; Baganizi, D.R.; Sahu, R.; Singh, S.R.; Dennis, V.A. SOCS Proteins as Regulators of Inflammatory Responses Induced by Bacterial Infections: A Review. Front. Microbiol. 2017, 8, 2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.-F.; Peng, W.-M.; Schlee, M.; Barchet, W.; Eis-Hübinger, A.M.; Kolanus, W.; Geyer, M.; Schmitt, S.; Steinhagen, F.; Oldenburg, J.; et al. SOCS1 and SOCS3 Target IRF7 Degradation To Suppress TLR7-Mediated Type I IFN Production of Human Plasmacytoid Dendritic Cells. J. Immunol. 2018, 200, 4024–4035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Y.; Yang, F.; Wang, Q.; Xu, N.; Xie, Y.; Chen, S.; Qin, T.; Peng, D. Influenza a Virus Antagonizes Type I and Type II Interferon Responses via SOCS1-Dependent Ubiquitination and Degradation of JAK1. Virol. J. 2020, 17, 74. [Google Scholar] [CrossRef]
- Shen-Orr, S.S.; Furman, D.; Kidd, B.A.; Hadad, F.; Lovelace, P.; Huang, Y.-W.; Rosenberg-Hasson, Y.; Mackey, S.; Grisar, F.A.G.; Pickman, Y.; et al. Defective Signaling in the JAK-STAT Pathway Tracks with Chronic Inflammation and Cardiovascular Risk in Aging Humans. Cell Syst. 2016, 3, 374–384.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, D.; Giles, J.R.; Baxter, A.E.; Oldridge, D.A.; Greenplate, A.R.; Wu, J.E.; Alanio, C.; Kuri-Cervantes, L.; Pampena, M.B.; D’Andrea, K.; et al. Deep Immune Profiling of COVID-19 Patients Reveals Distinct Immunotypes with Therapeutic Implications. Science 2020, 369, eabc8511. [Google Scholar] [CrossRef]
- Iba, T.; Levy, J.H.; Levi, M.; Thachil, J. Coagulopathy in COVID-19. J. Thromb. Haemost. 2020, 18, 2103–2109. [Google Scholar] [CrossRef] [PubMed]
- Loof, T.G.; Mörgelin, M.; Johansson, L.; Oehmcke, S.; Olin, A.I.; Dickneite, G.; Norrby-Teglund, A.; Theopold, U.; Herwald, H. Coagulation, an Ancestral Serine Protease Cascade, Exerts a Novel Function in Early Immune Defense. Blood 2011, 118, 2589–2598. [Google Scholar] [CrossRef]
- Rapkiewicz, A.V.; Mai, X.; Carsons, S.E.; Pittaluga, S.; Kleiner, D.E.; Berger, J.S.; Thomas, S.; Adler, N.M.; Charytan, D.M.; Gasmi, B.; et al. Megakaryocytes and Platelet-Fibrin Thrombi Characterize Multi-Organ Thrombosis at Autopsy in COVID-19: A Case Series. EClinicalMedicine 2020, 24, 100434. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.E.; Akmatbekov, A.; Harbert, J.L.; Li, G.; Quincy Brown, J.; Vander Heide, R.S. Pulmonary and Cardiac Pathology in African American Patients with COVID-19: An Autopsy Series from New Orleans. Lancet Respir. Med. 2020, 8, 681–686. [Google Scholar] [CrossRef]
- Wichmann, D.; Sperhake, J.-P.; Lütgehetmann, M.; Steurer, S.; Edler, C.; Heinemann, A.; Heinrich, F.; Mushumba, H.; Kniep, I.; Schröder, A.S.; et al. Autopsy Findings and Venous Thromboembolism in Patients With COVID-19: A Prospective Cohort Study. Ann. Intern. Med. 2020, 173, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Belen-Apak, F.B.; Sarıalioğlu, F. Pulmonary Intravascular Coagulation in COVID-19: Possible Pathogenesis and Recommendations on Anticoagulant/Thrombolytic Therapy. J. Thromb. Thrombolysis 2020, 50, 278–280. [Google Scholar] [CrossRef]
- Jamilloux, Y.; Henry, T.; Belot, A.; Viel, S.; Fauter, M.; El Jammal, T.; Walzer, T.; François, B.; Sève, P. Should We Stimulate or Suppress Immune Responses in COVID-19? Cytokine and Anti-Cytokine Interventions. Autoimmun. Rev. 2020, 19, 102567. [Google Scholar] [CrossRef]
- Smith, A.G.; Sheridan, P.A.; Harp, J.B.; Beck, M.A. Diet-Induced Obese Mice Have Increased Mortality and Altered Immune Responses When Infected with Influenza Virus. J. Nutr. 2007, 137, 1236–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, A.J.X.; To, K.K.W.; Li, C.; Lau, C.C.Y.; Poon, V.K.M.; Chan, C.C.S.; Zheng, B.-J.; Hung, I.F.N.; Lam, K.S.L.; Xu, A.; et al. Leptin Mediates the Pathogenesis of Severe 2009 Pandemic Influenza A(H1N1) Infection Associated With Cytokine Dysregulation in Mice With Diet-Induced Obesity. J. Infect. Dis. 2013, 207, 1270–1280. [Google Scholar] [CrossRef]
- Cohen, S.; Danzaki, K.; MacIver, N.J. Nutritional Effects on T-Cell Immunometabolism: Highlights. Eur. J. Immunol. 2017, 47, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S.; Erbay, E. Nutrient Sensing and Inflammation in Metabolic Diseases. Nat. Rev. Immunol. 2008, 8, 923–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straub, R.H.; Cutolo, M.; Buttgereit, F.; Pongratz, G. Energy Regulation and Neuroendocrine-Immune Control in Chronic Inflammatory Diseases. J. Intern. Med. 2010, 267, 543–560. [Google Scholar] [CrossRef]
- Straub, R.H. The Brain and Immune System Prompt Energy Shortage in Chronic Inflammation and Ageing. Nat. Rev. Rheumatol. 2017, 13, 743–751. [Google Scholar] [CrossRef]
- Cavaillon, J.-M. Molecular Mediators: Cytokines. In Reviews in Cell Biology and Molecular Medicine; Meyers, R.A., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015; pp. 1–37. ISBN 978-3-527-60090-8. [Google Scholar]
- Fischer, H.J.; Sie, C.; Schumann, E.; Witte, A.-K.; Dressel, R.; van den Brandt, J.; Reichardt, H.M. The Insulin Receptor Plays a Critical Role in T Cell Function and Adaptive Immunity. J. Immunol. 2017, 198, 1910–1920. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.; Clemente-Casares, X.; Zhou, A.C.; Lei, H.; Ahn, J.J.; Chan, Y.T.; Choi, O.; Luck, H.; Woo, M.; Dunn, S.E.; et al. Insulin Receptor-Mediated Stimulation Boosts T Cell Immunity during Inflammation and Infection. Cell Metab. 2018, 28, 922–934.e4. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Liu, C.; Mao, Z.; Xiao, M.; Wang, L.; Qi, S.; Zhou, F. Predictive Values of Neutrophil-to-Lymphocyte Ratio on Disease Severity and Mortality in COVID-19 Patients: A Systematic Review and Meta-Analysis. Crit. Care 2020, 24, 647. [Google Scholar] [CrossRef] [PubMed]
- Lagunas-Rangel, F.A. Neutrophil-to-lymphocyte Ratio and Lymphocyte-to-C-reactive Protein Ratio in Patients with Severe Coronavirus Disease 2019 (COVID-19): A Meta-analysis. J. Med. Virol. 2020, 92, 1733–1734. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-C.; Ko, H.-J.; Liu, W.-S.; Hung, C.-L.; Hu, K.-C.; Yu, L.-Y.; Shih, S.-C. Neutrophil-to-Lymphocyte Ratio as a Predictive Marker of Metabolic Syndrome. Medicine 2019, 98, e17537. [Google Scholar] [CrossRef]
- Brodin, P. Immune Determinants of COVID-19 Disease Presentation and Severity. Nat. Med. 2021, 27, 28–33. [Google Scholar] [CrossRef]
- Piątkiewicz, P.; Czech, A.; Tatoń, J. Glucose Transport in Human Peripheral Blood Lymphocytes Influenced by Type 2 Diabetes Mellitus. Arch. Immunol. Ther. Exp. 2007, 55, 119–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaszczak, A.M.; Jalilvand, A.; Hsueh, W.A. Adipocytes, Innate Immunity and Obesity: A Mini-Review. Front. Immunol. 2021, 12, 650768. [Google Scholar] [CrossRef] [PubMed]
- Green, W.D.; Beck, M.A. Obesity Altered T Cell Metabolism and the Response to Infection. Curr. Opin. Immunol. 2017, 46, 1–7. [Google Scholar] [CrossRef]
- Viardot, A.; Grey, S.T.; Mackay, F.; Chisholm, D. Potential Antiinflammatory Role of Insulin via the Preferential Polarization of Effector T Cells toward a T Helper 2 Phenotype. Endocrinology 2007, 148, 346–353. [Google Scholar] [CrossRef] [Green Version]
- Ieronymaki, E.; Theodorakis, E.M.; Lyroni, K.; Vergadi, E.; Lagoudaki, E.; Al-Qahtani, A.; Aznaourova, M.; Neofotistou-Themeli, E.; Eliopoulos, A.G.; Vaporidi, K.; et al. Insulin Resistance in Macrophages Alters Their Metabolism and Promotes an M2-Like Phenotype. J. Immunol. 2019, 202, 1786–1797. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain Energy Rescue: An Emerging Therapeutic Concept for Neurodegenerative Disorders of Ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef]
- Femminella, G.D.; Livingston, N.R.; Raza, S.; van der Doef, T.; Frangou, E.; Love, S.; Busza, G.; Calsolaro, V.; Carver, S.; Holmes, C.; et al. Does Insulin Resistance Influence Neurodegeneration in Non-Diabetic Alzheimer’s Subjects? Alz. Res. Ther. 2021, 13, 47. [Google Scholar] [CrossRef]
- Saucillo, D.C.; Gerriets, V.A.; Sheng, J.; Rathmell, J.C.; MacIver, N.J. Leptin Metabolically Licenses T Cells for Activation To Link Nutrition and Immunity. J. Immunol. 2014, 192, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Palmer, C.S.; Hussain, T.; Duette, G.; Weller, T.J.; Ostrowski, M.; Sada-Ovalle, I.; Crowe, S.M. Regulators of Glucose Metabolism in CD4+ and CD8+ T Cells. Int. Rev. Immunol. 2016, 35, 477–488. [Google Scholar] [CrossRef]
- Pearce, E.L.; Pearce, E.J. Metabolic Pathways in Immune Cell Activation and Quiescence. Immunity 2013, 38, 633–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dielen, F.; van’t Veer, C.; Schols, A.; Soeters, P.; Buurman, W.; Greve, J. Increased Leptin Concentrations Correlate with Increased Concentrations of Inflammatory Markers in Morbidly Obese Individuals. Int. J. Obes. 2001, 25, 1759–1766. [Google Scholar] [CrossRef] [Green Version]
- Koester-Weber, T.; Valtueña, J.; Breidenassel, C.; Beghin, L.; Plada, M.; Moreno, S.; Huybrechts, I.; Palacios, G.; Gómez-Martínez, S.; Albers, U.; et al. Reference Values for Leptin, Cortisol, Insulin and Glucose, among European Adolescents and Their Association with Adiposity: The HELENA Study. Nutr. Hosp. 2014, 30, 1181–1190. [Google Scholar] [CrossRef]
- Abella, V.; Scotece, M.; Conde, J.; Pino, J.; Gonzalez-Gay, M.A.; Gómez-Reino, J.J.; Mera, A.; Lago, F.; Gómez, R.; Gualillo, O. Leptin in the Interplay of Inflammation, Metabolism and Immune System Disorders. Nat. Rev. Rheumatol. 2017, 13, 100–109. [Google Scholar] [CrossRef]
- Pérez-Pérez, A.; Vilariño-García, T.; Fernández-Riejos, P.; Martín-González, J.; Segura-Egea, J.J.; Sánchez-Margalet, V. Role of Leptin as a Link between Metabolism and the Immune System. Cytokine Growth Factor Rev. 2017, 35, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, Y.; Zhang, X.; Wang, S.; Peng, Z.; Guo, J.; Jiang, H.; Liu, J.; Xie, Y.; Wang, J.; et al. Leptin Correlates with Monocytes Activation and Severe Condition in COVID-19 Patients. J. Leukoc. Biol. 2021, 110, 9–20. [Google Scholar] [CrossRef]
- Recchiuti, A. As a Matter of Fat: Leptin, Monocyte Hyperactivation, and COVID-19: A Commentary to “Leptin Correlates with Monocytes Activation and Severe Condition in COVID-19 Patients”. J. Leukoc. Biol. 2021, 110, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Naylor, C.; Petri, W.A. Leptin Regulation of Immune Responses. Trends Mol. Med. 2016, 22, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Pérez, A.; Sánchez-Jiménez, F.; Vilariño-García, T.; Sánchez-Margalet, V. Role of Leptin in Inflammation and Vice Versa. Int. J. Mol. Sci. 2020, 21, 5887. [Google Scholar] [CrossRef]
- Chen, W.; Daines, M.O.; Khurana Hershey, G.K. Turning off Signal Transducer and Activator of Transcription (STAT): The Negative Regulation of STAT Signaling. J. Allergy Clin. Immunol. 2004, 114, 476–489. [Google Scholar] [CrossRef] [PubMed]
- Andersen, C.J.; Murphy, K.E.; Fernandez, M.L. Impact of Obesity and Metabolic Syndrome on Immunity. Adv. Nutr. 2016, 7, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Hur, S.J.; Kim, D.H.; Chun, S.C.; Lee, S.K. Effect of Adenovirus and Influenza Virus Infection on Obesity. Life Sci. 2013, 93, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Popkin, B.M.; Du, S.; Green, W.D.; Beck, M.A.; Algaith, T.; Herbst, C.H.; Alsukait, R.F.; Alluhidan, M.; Alazemi, N.; Shekar, M. Individuals with Obesity and COVID-19: A Global Perspective on the Epidemiology and Biological Relationships. Obes. Rev. 2020, 21, e13128. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J. Diabetes, Obesity, Metabolism, and SARS-CoV-2 Infection: The End of the Beginning. Cell Metab. 2021, 33, 479–498. [Google Scholar] [CrossRef]
- On behalf of the PanSurg Collaborative; Zakka, K.; Chidambaram, S.; Mansour, S.; Mahawar, K.; Salminen, P.; Almino, R.; Schauer, P.; Kinross, J.; Purkayastha, S. SARS-CoV-2 and Obesity: “CoVesity”—A Pandemic Within a Pandemic. Obes. Surg. 2021, 31, 1745–1754. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, V.; Gadi, N.; Spihlman, A.P.; Wu, S.C.; Choi, C.H.; Moulton, V.R. Aging, Immunity, and COVID-19: How Age Influences the Host Immune Response to Coronavirus Infections? Front. Physiol. 2020, 11, 571416. [Google Scholar] [CrossRef] [PubMed]
- Mahase, E. COVID-19: Why Are Age and Obesity Risk Factors for Serious Disease? BMJ 2020, 371, m4130. [Google Scholar] [CrossRef]
- Aguilar, E.G.; Murphy, W.J. Obesity Induced T Cell Dysfunction and Implications for Cancer Immunotherapy. Curr. Opin. Immunol. 2018, 51, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Aiello, A.; Farzaneh, F.; Candore, G.; Caruso, C.; Davinelli, S.; Gambino, C.M.; Ligotti, M.E.; Zareian, N.; Accardi, G. Immunosenescence and Its Hallmarks: How to Oppose Aging Strategically? A Review of Potential Options for Therapeutic Intervention. Front. Immunol. 2019, 10, 2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawelec, G. The Human Immunosenescence Phenotype: Does It Exist? Semin. Immunopathol. 2020, 42, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Pawelec, G.; Bronikowski, A.; Cunnane, S.C.; Ferrucci, L.; Franceschi, C.; Fülöp, T.; Gaudreau, P.; Gladyshev, V.N.; Gonos, E.S.; Gorbunova, V.; et al. The Conundrum of Human Immune System “Senescence”. Mech. Ageing Dev. 2020, 192, 111357. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic Inflammation in the Etiology of Disease across the Life Span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Bencivenga, L.; Rengo, G.; Varricchi, G. Elderly at Time of COronaVIrus Disease 2019 (COVID-19): Possible Role of Immunosenescence and Malnutrition. GeroScience 2020, 42, 1089–1092. [Google Scholar] [CrossRef]
- Steven, S.; Hollingsworth, K.G.; Al-Mrabeh, A.; Avery, L.; Aribisala, B.; Caslake, M.; Taylor, R. Very Low-Calorie Diet and 6 Months of Weight Stability in Type 2 Diabetes: Pathophysiological Changes in Responders and Nonresponders. Diabetes Care 2016, 39, 808–815. [Google Scholar] [CrossRef] [Green Version]
- Appari, M.; Channon, K.M.; McNeill, E. Metabolic Regulation of Adipose Tissue Macrophage Function in Obesity and Diabetes. Antioxid. Redox Signal. 2018, 29, 297–312. [Google Scholar] [CrossRef]
- Tanti, J.-F.; Ceppo, F.; Jager, J.; Berthou, F. Implication of Inflammatory Signaling Pathways in Obesity-Induced Insulin Resistance. Front. Endocrinol. 2013, 3, 181. [Google Scholar] [CrossRef] [Green Version]
- Ross, R.; Neeland, I.J.; Yamashita, S.; Shai, I.; Seidell, J.; Magni, P.; Santos, R.D.; Arsenault, B.; Cuevas, A.; Hu, F.B.; et al. Waist Circumference as a Vital Sign in Clinical Practice: A Consensus Statement from the IAS and ICCR Working Group on Visceral Obesity. Nat. Rev. Endocrinol. 2020, 16, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Lindhorst, A.; Raulien, N.; Wieghofer, P.; Eilers, J.; Rossi, F.M.V.; Bechmann, I.; Gericke, M. Adipocyte Death Triggers a Pro-Inflammatory Response and Induces Metabolic Activation of Resident Macrophages. Cell Death Dis. 2021, 12, 579. [Google Scholar] [CrossRef] [PubMed]
- Pranata, R.; Lim, M.A.; Huang, I.; Yonas, E.; Henrina, J.; Vania, R.; Lukito, A.A.; Nasution, S.A.; Alwi, I.; Siswanto, B.B. Visceral Adiposity, Subcutaneous Adiposity, and Severe Coronavirus Disease-2019 (COVID-19): Systematic Review and Meta-Analysis. Clin. Nutr. ESPEN 2021, 43, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in Inflammation and Metabolic Disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Frühbeck, G.; Catalán, V.; Rodríguez, A.; Ramírez, B.; Becerril, S.; Salvador, J.; Colina, I.; Gómez-Ambrosi, J. Adiponectin-Leptin Ratio Is a Functional Biomarker of Adipose Tissue Inflammation. Nutrients 2019, 11, 454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganji, V.; Kafai, M.R.; McCarthy, E. Serum Leptin Concentrations Are Not Related to Dietary Patterns but Are Related to Sex, Age, Body Mass Index, Serum Triacylglycerol, Serum Insulin, and Plasma Glucose in the US Population. Nutr. Metab. Lond. 2009, 6, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gijón-Conde, T.; Graciani, A.; Guallar-Castillón, P.; Aguilera, M.T.; Rodríguez-Artalejo, F.; Banegas, J.R. Leptin Reference Values and Cutoffs for Identifying Cardiometabolic Abnormalities in the Spanish Population. Rev. Esp. Cardiol. Engl. Ed. 2015, 68, 672–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Könner, A.C.; Brüning, J.C. Selective Insulin and Leptin Resistance in Metabolic Disorders. Cell Metab. 2012, 16, 144–152. [Google Scholar] [CrossRef] [Green Version]
- Izquierdo, A.G.; Crujeiras, A.B.; Casanueva, F.F.; Carreira, M.C. Leptin, Obesity, and Leptin Resistance: Where Are We 25 Years Later? Nutrients 2019, 11, 2704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanik, M.H.; Xu, Y.; Skrha, J.; Dankner, R.; Zick, Y.; Roth, J. Insulin Resistance and Hyperinsulinemia: Is Hyperinsulinemia the Cart or the Horse? Diabetes Care 2008, 31 (Suppl. S2), 262–268. [Google Scholar] [CrossRef] [Green Version]
- Andreoli, M.F.; Donato, J.; Cakir, I.; Perello, M. Leptin Resensitisation: A Reversion of Leptin-Resistant States. J. Endocrinol. 2019, 241, 81–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadden, D.R.; McLaughlin, C. Normal and Abnormal Maternal Metabolism during Pregnancy. Semin. Fetal Neonatal Med. 2009, 14, 66–71. [Google Scholar] [CrossRef]
- Wang, S.; Ma, P.; Zhang, S.; Song, S.; Wang, Z.; Ma, Y.; Xu, J.; Wu, F.; Duan, L.; Yin, Z.; et al. Fasting Blood Glucose at Admission Is an Independent Predictor for 28-Day Mortality in Patients with COVID-19 without Previous Diagnosis of Diabetes: A Multi-Centre Retrospective Study. Diabetologia 2020, 63, 2102–2111. [Google Scholar] [CrossRef]
- Cai, Y.; Shi, S.; Yang, F.; Yi, B.; Chen, X.; Li, J.; Wen, Z. Fasting Blood Glucose Level Is a Predictor of Mortality in Patients with COVID-19 Independent of Diabetes History. Diabetes Res. Clin. Pract. 2020, 169, 108437. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, N.; Reddy, M.A.; Guha, M.; Natarajan, R. High Glucose-Induced Expression of Proinflammatory Cytokine and Chemokine Genes in Monocytic Cells. Diabetes 2003, 52, 1256–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dandona, P.; Chaudhuri, A.; Ghanim, H.; Mohanty, P. Proinflammatory Effects of Glucose and Anti-Inflammatory Effect of Insulin: Relevance to Cardiovascular Disease. Am. J. Cardiol. 2007, 99, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Fang, P.; He, R.; Li, M.; Yu, H.; Zhou, L.; Yi, Y.; Wang, F.; Rong, Y.; Zhang, Y.; et al. O-GlcNAc Transferase Promotes Influenza A Virus-Induced Cytokine Storm by Targeting Interferon Regulatory Factor-5. Sci. Adv. 2020, 6, eaaz7086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Codo, A.C.; Davanzo, G.G.; de Monteiro, L.B.; de Souza, G.F.; Muraro, S.P.; Virgilio-da-Silva, J.V.; Prodonoff, J.S.; Carregari, V.C.; de Biagi Junior, C.A.O.; Crunfli, F.; et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and Monocyte Response through a HIF-1α/Glycolysis-Dependent Axis. Cell Metab. 2020, 32, 437–446.e5. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.D.; Matta, B.; Barnes, B.J. Therapeutic Targeting of IRFs: Pathway-Dependence or Structure-Based? Front. Immunol. 2018, 9, 2622. [Google Scholar] [CrossRef] [PubMed]
- Fardini, Y.; Dehennaut, V.; Lefebvre, T.; Issad, T. O-GlcNAcylation: A New Cancer Hallmark? Front. Endocrinol. 2013, 4, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoy, N. Involvement of Interleukin-1 Receptor-Associated Kinase 4 and Interferon Regulatory Factor 5 in the Immunopathogenesis of SARS-CoV-2 Infection: Implications for the Treatment of COVID-19. Front. Immunol. 2021, 12, 638446. [Google Scholar] [CrossRef]
- Alzaid, F.; Julla, J.-B.; Diedisheim, M.; Potier, C.; Potier, L.; Velho, G.; Gaborit, B.; Manivet, P.; Germain, S.; Vidal-Trecan, T.; et al. Monocytopenia, Monocyte Morphological Anomalies and Hyperinflammation Characterise Severe COVID-19 in Type 2 Diabetes. EMBO Mol. Med. 2020, 12, e13038. [Google Scholar] [CrossRef] [PubMed]
- De Candia, P.; Prattichizzo, F.; Garavelli, S.; Alviggi, C.; La Cava, A.; Matarese, G. The Pleiotropic Roles of Leptin in Metabolism, Immunity, and Cancer. J. Exp. Med. 2021, 218, e20191593. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Riejos, P.; Najib, S.; Santos-Alvarez, J.; Martín-Romero, C.; Pérez-Pérez, A.; González-Yanes, C.; Sánchez-Margalet, V. Role of Leptin in the Activation of Immune Cells. Mediat. Inflamm. 2010, 2010, 568343. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, K.; MacIver, N.J. The Role of the Adipokine Leptin in Immune Cell Function in Health and Disease. Front. Immunol. 2020, 11, 622468. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Youm, Y.-H.; Vandanmagsar, B.; Rood, J.; Kumar, K.G.; Butler, A.A.; Dixit, V.D. Obesity Accelerates Thymic Aging. Blood 2009, 114, 3803–3812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantzoros, C.S.; Magkos, F.; Brinkoetter, M.; Sienkiewicz, E.; Dardeno, T.A.; Kim, S.-Y.; Hamnvik, O.-P.R.; Koniaris, A. Leptin in Human Physiology and Pathophysiology. Am. J. Physiol. Endocrinol. Metab. 2011, 301, 567–584. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Liu, J.; Feng, X.; Salazar Hernández, M.A.; Mucka, P.; Ibi, D.; Choi, J.W.; Ozcan, U. Withaferin A is a Leptin Sensitizer with Strong Antidiabetic Properties in Mice. Nat. Med. 2016, 22, 1023–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Münzberg, H.; Flier, J.S.; Bjørbaek, C. Region-Specific Leptin Resistance within the Hypothalamus of Diet-Induced Obese Mice. Endocrinology 2004, 145, 4880–4889. [Google Scholar] [CrossRef] [Green Version]
- Howard, J.K.; Flier, J.S. Attenuation of Leptin and Insulin Signaling by SOCS Proteins. Trends Endocrinol. Metab. 2006, 17, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhu, Y.; Schultz, R.D.; Li, N.; He, Z.; Zhang, Z.; Caron, A.; Zhu, Q.; Sun, K.; Xiong, W.; et al. Partial Leptin Reduction as an Insulin Sensitization and Weight Loss Strategy. Cell Metab. 2019, 30, 706–719.e6. [Google Scholar] [CrossRef] [PubMed]
- Terán-Cabanillas, E.; Hernández, J. Role of Leptin and SOCS3 in Inhibiting the Type I Interferon Response during Obesity. Inflammation 2017, 40, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Ueki, K.; Kondo, T.; Kahn, C.R. Suppressor of Cytokine Signaling 1 (SOCS-1) and SOCS-3 Cause Insulin Resistance through Inhibition of Tyrosine Phosphorylation of Insulin Receptor Substrate Proteins by Discrete Mechanisms. Mol. Cell. Biol. 2004, 24, 5434–5446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Hulver, M.; McMillan, R.P.; Cai, L.; Kershaw, E.E.; Yu, L.; Xue, B.; Shi, H. Regulation of Insulin and Leptin Signaling by Muscle Suppressor of Cytokine Signaling 3 (SOCS3). PLoS ONE 2012, 7, e47493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wunderlich, C.M.; Hövelmeyer, N.; Wunderlich, F.T. Mechanisms of Chronic JAK-STAT3-SOCS3 Signaling in Obesity. JAK-STAT 2013, 2, e23878. [Google Scholar] [CrossRef] [PubMed]
- Carow, B.; Rottenberg, M.E. SOCS3, a Major Regulator of Infection and Inflammation. Front. Immunol. 2014, 5, 58. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Chen, L.; Yang, K.; Jiang, H.; Xu, W.; Luan, J. SOCS Molecules: The Growing Players in Macrophage Polarization and Function. Oncotarget 2017, 8, 60710–60722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calay, E.S.; Hotamisligil, G.S. Turning off the Inflammatory, but Not the Metabolic, Flames. Nat. Med. 2013, 19, 265–267. [Google Scholar] [CrossRef] [PubMed]
- Warren, K.J.; Olson, M.M.; Thompson, N.J.; Cahill, M.L.; Wyatt, T.A.; Yoon, K.J.; Loiacono, C.M.; Kohut, M.L. Exercise Improves Host Response to Influenza Viral Infection in Obese and Non-Obese Mice through Different Mechanisms. PLoS ONE 2015, 10, e0129713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Q.; Cui, G.; Chen, J.; Gao, H.; Wei, Y.; Uede, T.; Chen, Z.; Diao, H. Regular Exercise Enhances the Immune Response Against Microbial Antigens Through Up-Regulation of Toll-like Receptor Signaling Pathways. Cell Physiol. Biochem. 2015, 37, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Fedewa, M.V.; Hathaway, E.D.; Ward-Ritacco, C.L. Effect of Exercise Training on C Reactive Protein: A Systematic Review and Meta-Analysis of Randomised and Non-Randomised Controlled Trials. Br. J. Sports Med. 2017, 51, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Iaccarino, G.; Franco, D.; Sorriento, D.; Strisciuglio, T.; Barbato, E.; Morisco, C. Modulation of Insulin Sensitivity by Exercise Training: Implications for Cardiovascular Prevention. J. Cardiovasc. Transl. Res. 2021, 14, 256–270. [Google Scholar] [CrossRef] [PubMed]
- Sallis, R.; Young, D.R.; Tartof, S.Y.; Sallis, J.F.; Sall, J.; Li, Q.; Smith, G.N.; Cohen, D.A. Physical Inactivity Is Associated with a Higher Risk for Severe COVID-19 Outcomes: A Study in 48,440 Adult Patients. Br. J. Sports Med. 2021, 55, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Bouassida, A.; Chamari, K.; Zaouali, M.; Feki, Y.; Zbidi, A.; Tabka, Z. Review on Leptin and Adiponectin Responses and Adaptations to Acute and Chronic Exercise. Br. J. Sports Med. 2010, 44, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Varkaneh Kord, H.; M Tinsley, G.; O Santos, H.; Zand, H.; Nazary, A.; Fatahi, S.; Mokhtari, Z.; Salehi-Sahlabadi, A.; Tan, S.C.; Rahmani, J.; et al. The Influence of Fasting and Energy-Restricted Diets on Leptin and Adiponectin Levels in Humans: A Systematic Review and Meta-Analysis. Clin. Nutr. 2021, 40, 1811–1821. [Google Scholar] [CrossRef]
- Pedroso, J.A.B.; Ramos-Lobo, A.M.; Donato, J. SOCS3 as a Future Target to Treat Metabolic Disorders. Horm. Athens 2019, 18, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Morales, J.S.; Valenzuela, P.L.; Castillo-García, A.; Butragueño, J.; Jiménez-Pavón, D.; Carrera-Bastos, P.; Lucia, A. The Exposome and Immune Health in Times of the COVID-19 Pandemic. Nutrients 2021, 14, 24. [Google Scholar] [CrossRef] [PubMed]
- Van der Voort, P.H.J.; Moser, J.; Zandstra, D.F.; Muller Kobold, A.C.; Knoester, M.; Calkhoven, C.F.; Hamming, I.; van Meurs, M. Leptin Levels in SARS-CoV-2 Infection Related Respiratory Failure: A Cross-Sectional Study and a Pathophysiological Framework on the Role of Fat Tissue. Heliyon 2020, 6, e04696. [Google Scholar] [CrossRef]
- Singh, R.; Hemati, H.; Bajpai, M.; Yadav, P.; Maheshwari, A.; Kumar, S.; Agrawal, S.; Sevak, J.K.; Islam, M.; Mars, J.S.; et al. Sustained Expression of Inflammatory Monocytes and Activated T Cells in COVID-19 Patients and Recovered Convalescent Plasma Donors. Immun. Inflamm. Dis. 2021, 9, 1279–1290. [Google Scholar] [CrossRef]
- Larsson, A.; Lipcsey, M.; Hultström, M.; Frithiof, R.; Eriksson, M. Plasma Leptin Is Increased in Intensive Care Patients with COVID-19-An Investigation Performed in the PronMed-Cohort. Biomedicines 2021, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Lindeberg, S.; Söderberg, S.; Ahrén, B.; Olsson, T. Large Differences in Serum Leptin Levels between Nonwesternized and Westernized Populations: The Kitava Study. J. Intern. Med. 2001, 249, 553–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jönsson, T.; Olsson, S.; Ahrén, B.; Bøg-Hansen, T.C.; Dole, A.; Lindeberg, S. Agrarian Diet and Diseases of Affluence—Do Evolutionary Novel Dietary Lectins Cause Leptin Resistance? BMC Endocr. Disord. 2005, 5, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindeberg, S.; Eliasson, M.; Lindahl, B.; Ahrén, B. Low Serum Insulin in Traditional Pacific Islanders—The Kitava Study. Metab. Clin. Exp. 1999, 48, 1216–1219. [Google Scholar] [CrossRef]
- Carrera-Bastos, P.; Fontes-Villalba, M.; Gurven, M.; Muskiet, F.A.J.; Åkerfeldt, T.; Lindblad, U.; Råstam, L.; Frostegård, J.; Shapira, Y.; Shoenfeld, Y.; et al. C-Reactive Protein in Traditional Melanesians on Kitava. BMC Cardiovasc. Disord. 2020, 20, 524. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Country, Author | Sample population | Conclusions |
|---|---|---|
| China | ||
| Wang, J.; et al. [202] | * 12 healthy subjects (mean age: 48 ± 15.7 years; 50% males; BMI: 23.8 ± 2.9 kg/m2) * 21 mild COVID-19 hospitalized patients (mean age: 47.2 ± 15.7 years; 61.9% males; BMI: 23.69 ± 2.7 kg/m2) * 10 severe COVID-19 patients (mean age: 46.9 ± 16.5 years; 70% males; BMI: 25.3 ± 2.6 kg/m2) | Leptin was associated with greater monocyte activation, systemic inflammation, and disease progression in COVID-19 cases |
| The Netherlands | ||
| van der Voort, P.H.J.; et al. [270] | * 31 COVID-19 patients in ICU requiring ventilation (mean BMI: 31 kg/m2 [range 24.8–48.4]) * 8 critically ill non-infected controls (mean BMI: 26 kg/m2 [range 22.4–33.5]) | Higher serum leptin levels in patients with COVID-19, in comparison with controls |
| India | ||
| Singh, R.; et al. [271] | * 10 healthy subjects (mean age: 48.14 ± 9.05 years; 74% males; BMI: 26.28 ± 2.52 kg/m2) * 15 individuals who recovered from mild COVID-19 (mean age: 38.07 ± 7.7 years; 100% males; BMI: 26.84 ± 2.63 kg/m2) * 34 moderate COVID-19 hospitalized patients (mean age: 48.21 ± 9.79 years; 75.86% males; BMI: 26.31 ± 2.29 kg/m2) | Increased leptin levels in moderate COVID-19 patients, compared to healthy subjects and individuals who recovered from mild COVID-19 |
| Sweden | ||
| Larsson, A.; et al. [272] | * 25 healthy subjects (median age: 57 years [range 47–68 years]; 76% males) * 222 severe COVID-19 hospitalized patients (median age: 64 years [range 24–86 years]; 79.28% males) | Leptin levels was higher in patients with COVID-19 at ICU admission, but it wasn’t associated with mortality |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muskiet, F.A.J.; Carrera-Bastos, P.; Pruimboom, L.; Lucia, A.; Furman, D. Obesity and Leptin Resistance in the Regulation of the Type I Interferon Early Response and the Increased Risk for Severe COVID-19. Nutrients 2022, 14, 1388. https://doi.org/10.3390/nu14071388
Muskiet FAJ, Carrera-Bastos P, Pruimboom L, Lucia A, Furman D. Obesity and Leptin Resistance in the Regulation of the Type I Interferon Early Response and the Increased Risk for Severe COVID-19. Nutrients. 2022; 14(7):1388. https://doi.org/10.3390/nu14071388
Chicago/Turabian StyleMuskiet, Frits A. J., Pedro Carrera-Bastos, Leo Pruimboom, Alejandro Lucia, and David Furman. 2022. "Obesity and Leptin Resistance in the Regulation of the Type I Interferon Early Response and the Increased Risk for Severe COVID-19" Nutrients 14, no. 7: 1388. https://doi.org/10.3390/nu14071388
APA StyleMuskiet, F. A. J., Carrera-Bastos, P., Pruimboom, L., Lucia, A., & Furman, D. (2022). Obesity and Leptin Resistance in the Regulation of the Type I Interferon Early Response and the Increased Risk for Severe COVID-19. Nutrients, 14(7), 1388. https://doi.org/10.3390/nu14071388