
Food-Related Carbonyl Stress in Cardiometabolic and Cancer Risk Linked to Unhealthy Modern Diet

 , ,
, ,  and
and 
Highlights
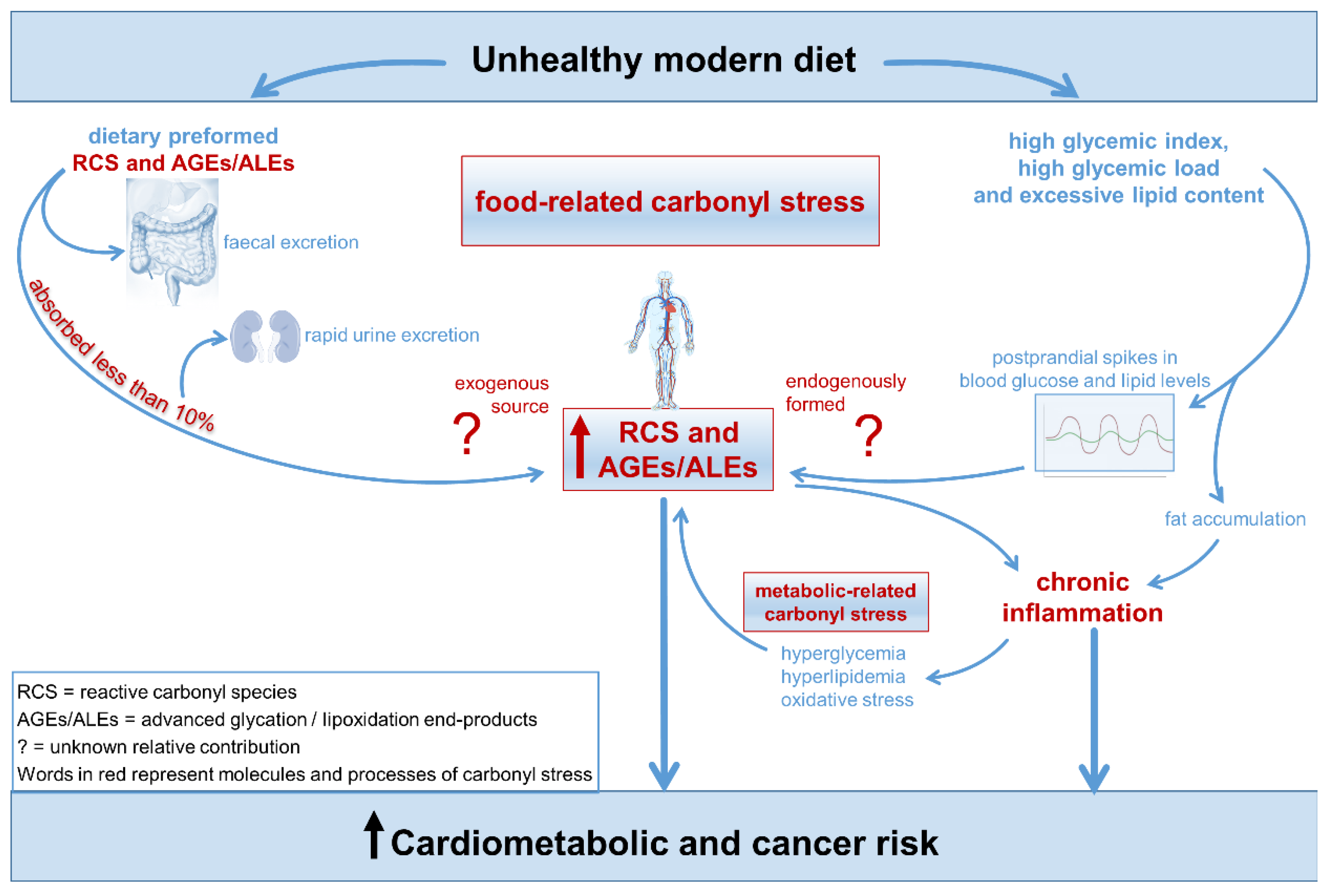
- Carbonyl stress results from elevated levels of reactive carbonyl species (RCS) generated by the oxidative cleavage and metabolism of lipids and sugars. This process leads to the accumulation of advanced glycation end-products (AGEs).
- RCS and AGEs can damage proteins, nucleic acids, and lipids, contributing to inflammation-related diseases such as diabetes, cardiovascular diseases, and cancer.
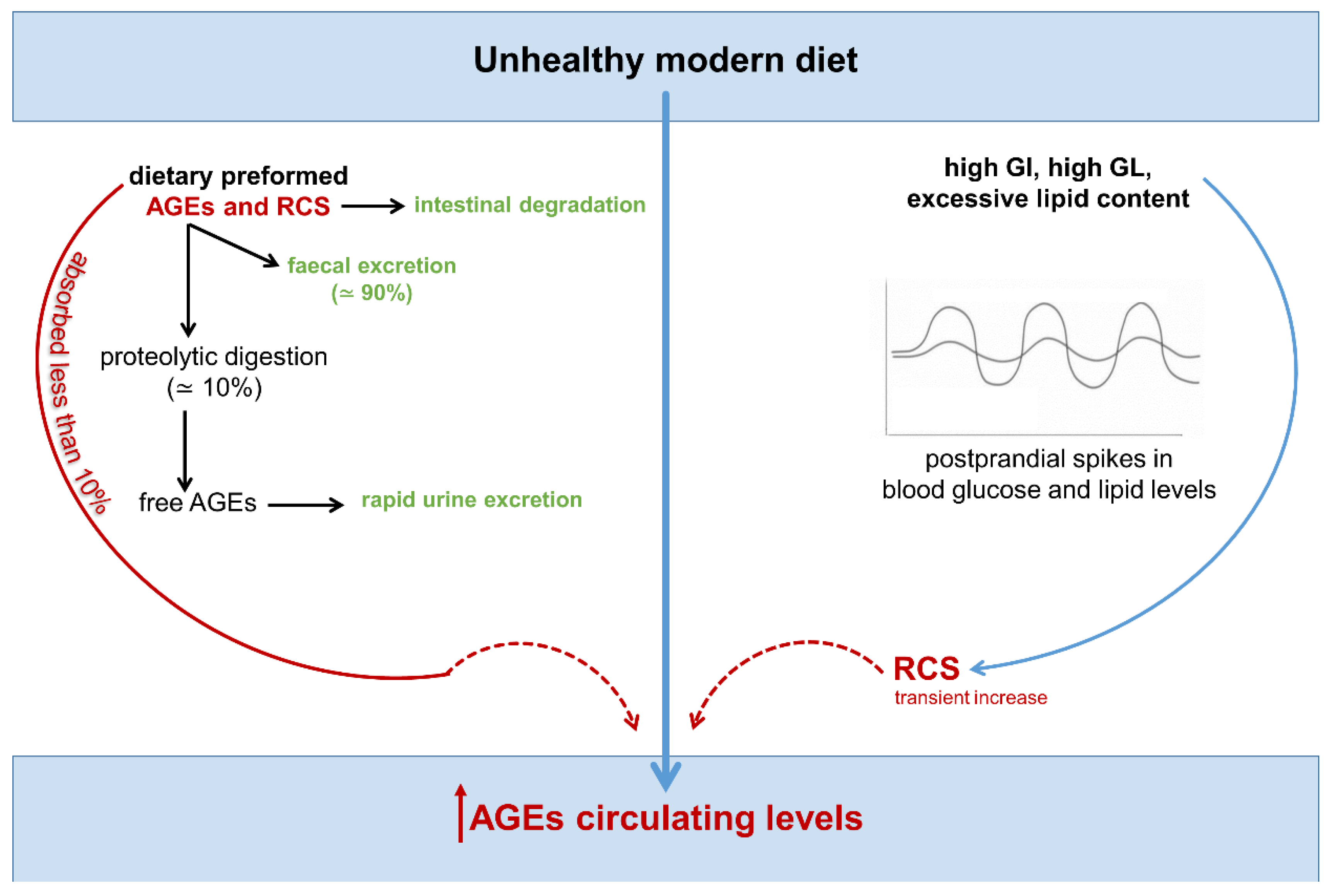
- A modern diet can increase the accumulation of RCS and AGEs through the following mechanisms: 1) the consumption of high glycemic index and lipid-rich foods, which favor greater endogenous production of RCS and AGEs, and 2) intake of highly processed foods rich in preformed RCS/AGEs. This dietary pattern may promote inflammation, atherogenic changes, and tumor growth.
- Exploring therapeutic agents that target carbonyl stress could offer new ways to prevent diet-associated health issues, particularly cardiometabolic diseases and cancer.
Abstract
:1. Introduction
2. Biochemistry of Carbonyl Stress and Targeted Therapeutic Strategies
2.1. Biochemistry
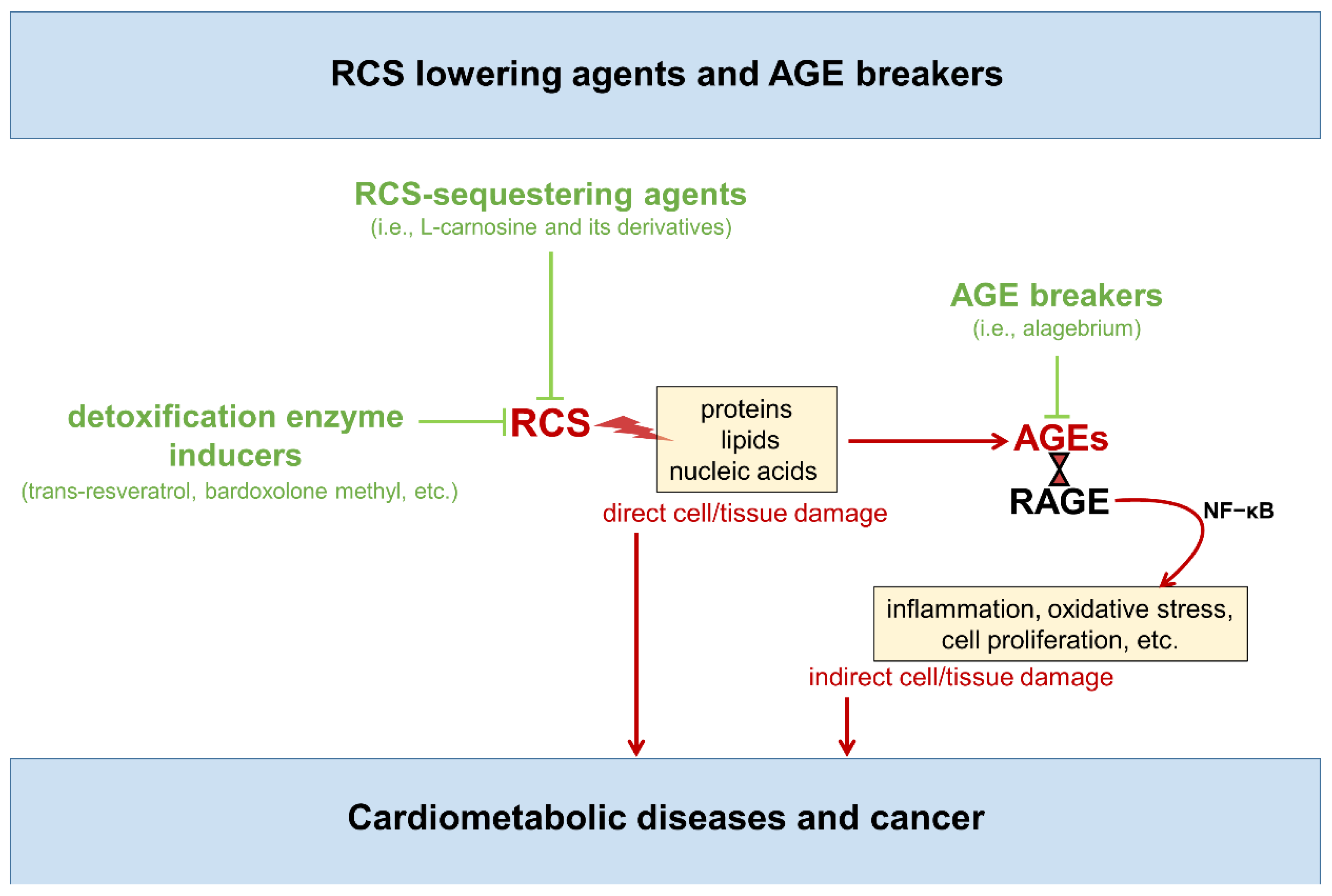
2.2. Targeted Therapies
3. Dietary AGE Intake, Cardiometabolic Disorders, and Cancer
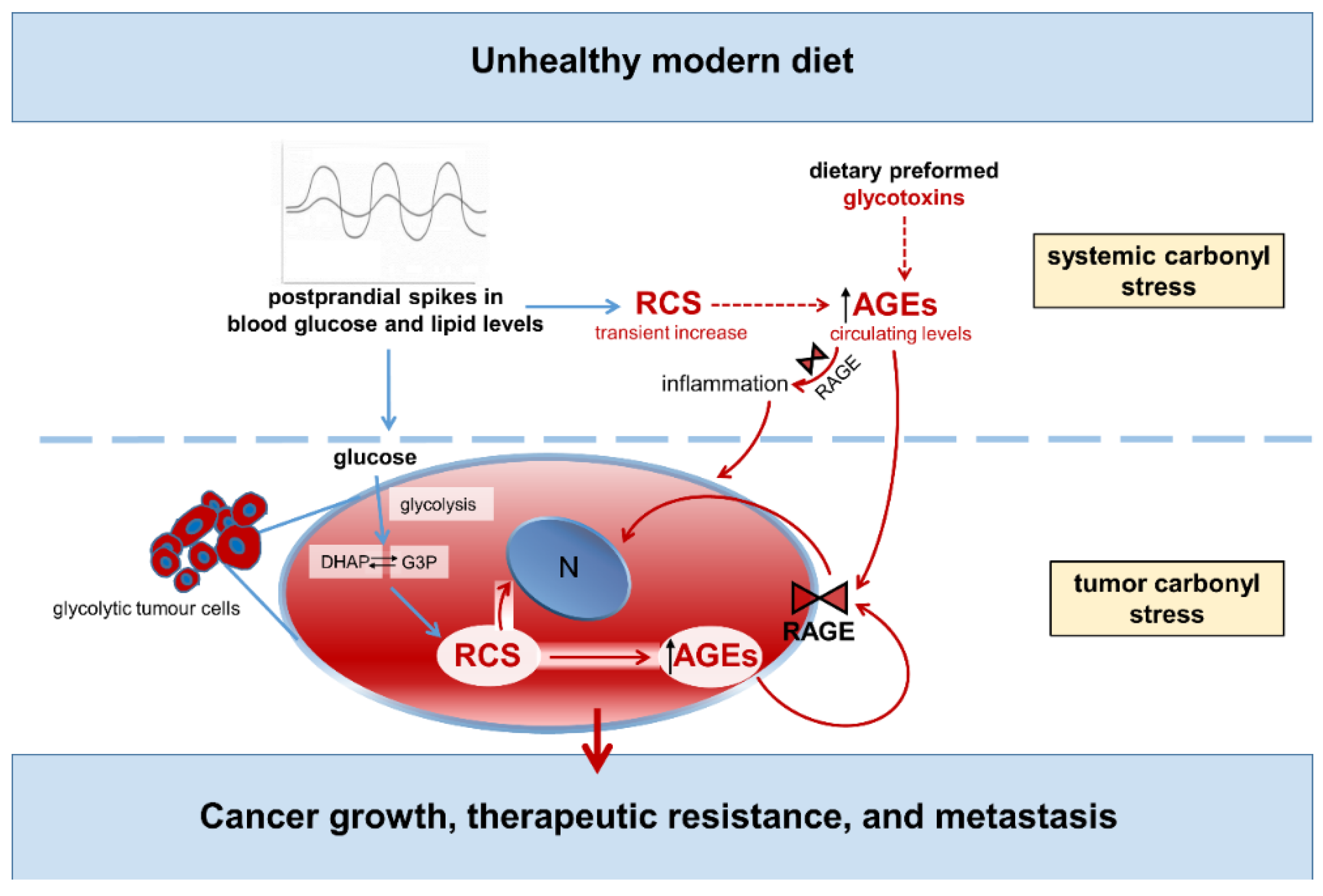
4. Endogenous, Nutrient-Induced Carbonyl Stress: A New Perspective on Diet in Cardiometabolic and Cancer Risk
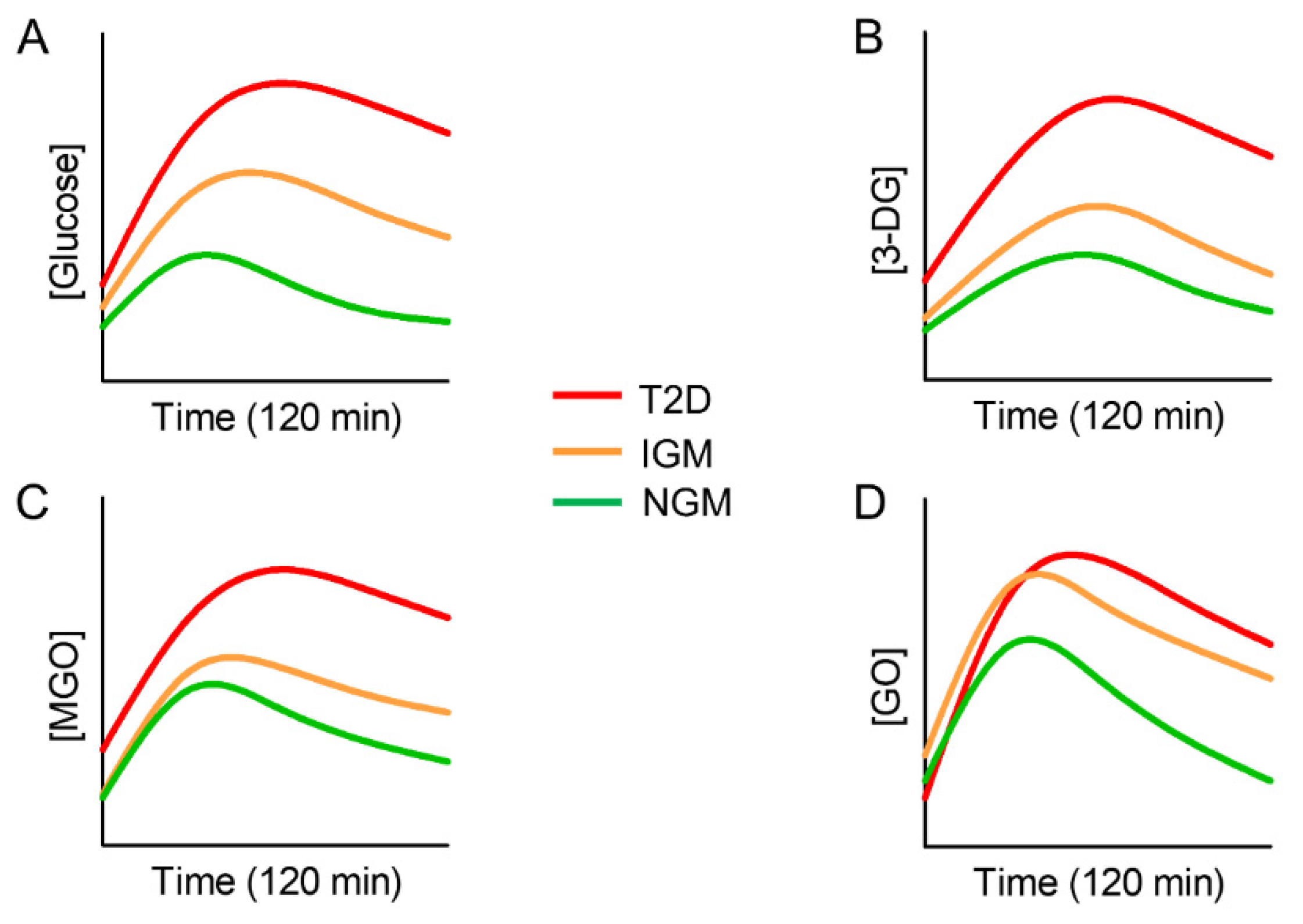
4.1. Endogenous Carbonyl Stress and Cardiometabolic Risk
4.2. Endogenous Carbonyl Stress and Cancer Risk
5. Carbonyl Stress Targeted Therapies to Reduce Cardiometabolic and Cancer Risk
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic Inflammation in the Etiology of Disease across the Life Span. Nat. Med. 2019, 25, 1822. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Proctor, M.J.; McMillan, D.C.; Horgan, P.G.; Fletcher, C.D.; Talwar, D.; Morrison, D.S. Systemic Inflammation Predicts All-Cause Mortality: A Glasgow Inflammation Outcome Study. PLoS ONE 2015, 10, e0116206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bays, H.E. Adiposopathy: Is “Sick Fat” a Cardiovascular Disease? J. Am. Coll. Cardiol. 2011, 57, 2461–2473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasetti Fantauzzi, C.; Iacobini, C.; Menini, S.; Vitale, M.; Sorice, G.P.; Mezza, T.; Cinti, S.; Giaccari, A.; Pugliese, G. Galectin-3 Gene Deletion Results in Defective Adipose Tissue Maturation and Impaired Insulin Sensitivity and Glucose Homeostasis. Sci. Rep. 2020, 10, 20070. [Google Scholar] [CrossRef] [PubMed]
- Arribas-Lorenzo, G.; Morales, F.J. Analysis, Distribution, and Dietary Exposure of Glyoxal and Methylglyoxal in Cookies and Their Relationship with Other Heat-Induced Contaminants. J. Agric. Food Chem. 2010, 58, 2966–2972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettiga, A.; Fiorio, F.; Di Marco, F.; Trevisani, F.; Romani, A.; Porrini, E.; Salonia, A.; Montorsi, F.; Vago, R. The Modern Western Diet Rich in Advanced Glycation End-Products (AGEs): An Overview of Its Impact on Obesity and Early Progression of Renal Pathology. Nutrients 2019, 11, 1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vistoli, G.; de Maddis, D.; Cipak, A.; Zarkovic, N.; Carini, M.; Aldini, G. Advanced Glycoxidation and Lipoxidation End Products (AGEs and ALEs): An Overview of Their Mechanisms of Formation. Free Radic. Res. 2013, 47, 3–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luévano-Contreras, C.; Gómez-Ojeda, A.; Macías-Cervantes, M.H.; Garay-Sevilla, M.E. Dietary Advanced Glycation End Products and Cardiometabolic Risk. Curr. Diabetes Rep. 2017, 17, 63. [Google Scholar] [CrossRef]
- Rask-Madsen, C.; King, G.L. Vascular Complications of Diabetes: Mechanisms of Injury and Protective Factors. Cell Metab. 2013, 17, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baynes, J.W.; Thorpe, S.R. Role of Oxidative Stress in Diabetic Complications: A New Perspective on an Old Paradigm. Diabetes 1999, 48, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mol, M.; Degani, G.; Coppa, C.; Baron, G.; Popolo, L.; Carini, M.; Aldini, G.; Vistoli, G.; Altomare, A. Advanced Lipoxidation End Products (ALEs) as RAGE Binders: Mass Spectrometric and Computational Studies to Explain the Reasons Why. Redox Biol. 2019, 23, 101083. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Schmidt, A.M. Glycation & Insulin Resistance: Novel Mechanisms and Unique Targets? Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aglago, E.K.; Schalkwijk, C.G.; Freisling, H.; Fedirko, V.; Hughes, D.J.; Jiao, L.; Dahm, C.C.; Olsen, A.; Tjønneland, A.; Katzke, V.; et al. Plasma Concentrations of Advanced Glycation End-Products and Colorectal Cancer Risk in the EPIC Study. Carcinogenesis 2021, 42, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Menini, S.; Iacobini, C.; Vitale, M.; Pesce, C.; Pugliese, G. Diabetes and Pancreatic Cancer—A Dangerous Liaison Relying on Carbonyl Stress. Cancers 2021, 13, 313. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced Glycation End Products in Foods and a Practical Guide to Their Reduction in the Diet. J. Am. Diet. Assoc. 2010, 110, 911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, J.; Morrissey, P.A.; Ames, J.M. Nutritional and Toxicological Aspects of the Maillard Browning Reaction in Foods. Crit. Rev. Food Sci. Nutr. 2009, 28, 211–248. [Google Scholar] [CrossRef] [PubMed]
- Lambadiari, V.; Korakas, E.; Tsimihodimos, V. The Impact of Dietary Glycemic Index and Glycemic Load on Postprandial Lipid Kinetics, Dyslipidemia and Cardiovascular Risk. Nutrients 2020, 12, 2204. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, J.H.; Bell, D.S.H. Postprandial Hyperglycemia/Hyperlipidemia (Postprandial Dysmetabolism) Is a Cardiovascular Risk Factor. Am. J. Cardiol. 2007, 100, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; Saito, A.; Kurokawa, K.; Van de Strihou, C.Y. Advanced Glycation and Lipoxidation End Products: Reactive Carbonyl Compounds-related Uraemic Toxicity. Nephrol. Dial. Transplant. 2001, 16, 8–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pamplona, R. Advanced Lipoxidation End-Products. Chem.-Biol. Interact. 2011, 192, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Negre-Salvayre, A.; Coatrieux, C.; Ingueneau, C.; Salvayre, R. Advanced Lipid Peroxidation End Products in Oxidative Damage to Proteins. Potential Role in Diseases and Therapeutic Prospects for the Inhibitors. Br. J. Pharmacol. 2008, 153, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelen, L.; Lund, S.S.; Ferreira, I.; Tarnow, L.; Parving, H.H.; Gram, J.; Winther, K.; Pedersen, O.; Teerlink, T.; Barto, R.; et al. Improved Glycemic Control Induced by Both Metformin and Repaglinide Is Associated with a Reduction in Blood Levels of 3-Deoxyglucosone in Nonobese Patients with Type 2 Diabetes. Eur. J. Endocrinol. 2011, 164, 371–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thornalley, P.J. Endogenous α-Oxoaldehydes and Formation of Protein and Nucleotide Advanced Glycation Endproducts in Tissue Damage. Novartis Found. Symp. 2007, 285, 229–246. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Weickert, M.O.; Qureshi, S.; Kandala, N.B.; Anwar, A.; Waldron, M.; Shafie, A.; Messenger, D.; Fowler, M.; Jenkins, G.; et al. Improved Glycemic Control and Vascular Function in Overweight and Obese Subjects by Glyoxalase 1 Inducer Formulation. Diabetes 2016, 65, 2282–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacobini, C.; Vitale, M.; Pesce, C.; Pugliese, G.; Menini, S. Diabetic Complications and Oxidative Stress: A 20-Year Voyage Back in Time and Back to the Future. Antioxidants 2021, 10, 727. [Google Scholar] [CrossRef] [PubMed]
- Traverso, N.; Menini, S.; Odetti, P.; Pronzato, M.A.; Cottalasso, D.; Marinari, U.M. Lipoperoxidation in Hepatic Subcellular Compartments of Diabetic Rats. Free Radic. Biol. Med. 1999, 26, 538–547. [Google Scholar] [CrossRef]
- Niwa, T.; Takeda, N.; Yoshizumi, H.; Tatematsu, A.; Ohara, M.; Tomiyama, S.; Niimura, K. Presence of 3-Deoxyglucosone, a Potent Protein Crosslinking Intermediate of Maillard Reaction, in Diabetic Serum. Biochem. Biophys. Res. Commun. 1993, 196, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.X.; Requena, J.R.; Jenkins, A.J.; Lyons, T.J.; Baynes, J.W.; Thorpe, S.R. The Advanced Glycation End Product, N∊-(Carboxymethyl)Lysine, Is a Product of Both Lipid Peroxidation and Glycoxidation Reactions. J. Biol. Chem. 1996, 271, 9982–9986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucala, R.; Makita, Z.; Koschinsky, T.; Cerami, A.; Vlassara, H. Lipid Advanced Glycosylation: Pathway for Lipid Oxidation in Vivo. Proc. Natl. Acad. Sci. USA 1993, 90, 6434. [Google Scholar] [CrossRef] [Green Version]
- Requena, J.R.; Ahmed, M.U.; Fountain, C.W.; Degenhardt, T.P.; Reddy, S.; Perez, C.; Lyons, T.J.; Jenkins, A.J.; Baynes, J.W.; Thorpe, S.R. Carboxymethylethanolamine, a Biomarker of Phospholipid Modification during the Maillard Reaction in Vivo. J. Biol. Chem. 1997, 272, 17473–17479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistry, N.; Podmore, I.; Cooke, M.; Butler, P.; Griffiths, H.; Herbert, K.; Lunec, J. Novel Monoclonal Antibody Recognition of Oxidative DNA Damage Adduct, Deoxycytidine-Glyoxal. Lab. Investig. 2003, 83, 241–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, S.A.; Thornalley, P.J. The Formation of Methylglyoxal from Triose Phosphates. Eur. J. Biochem. 1993, 212, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Allaman, I.; Bélanger, M.; Magistretti, P.J. Methylglyoxal, the Dark Side of Glycolysis. Front. Neurosci. 2015, 9, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinohara, M.; Thornalley, P.J.; Giardino, I.; Beisswenger, P.; Thorpe, S.R.; Onorato, J.; Brownlee, M. Overexpression of Glyoxalase-I in Bovine Endothelial Cells Inhibits Intracellular Advanced Glycation Endproduct Formation and Prevents Hyperglycemia-Induced Increases in Macromolecular Endocytosis. J. Clin. Investig. 1998, 101, 1142. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. Modification of the Glyoxalase System in Human Red Blood Cells by Glucose in Vitro. Biochem. J. 1988, 254, 751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlassara, H. Recent Progress in Advanced Glycation End Products and Diabetic Complications. Diabetes 1997, 46, S19–S25. [Google Scholar] [CrossRef]
- Watanabe, M.; Toyomura, T.; Wake, H.; Liu, K.; Teshigawara, K.; Takahashi, H.; Nishibori, M.; Mori, S. Differential Contribution of Possible Pattern-Recognition Receptors to Advanced Glycation End Product–Induced Cellular Responses in Macrophage-like RAW264.7 Cells. Biotechnol. Appl. Biochem. 2020, 67, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N. Advanced Glycation Endproducts—Role in Pathology of Diabetic Complications. Diabetes Res. Clin. Pract. 2005, 67, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Kellow, N.J.; Savige, G.S. Dietary Advanced Glycation End-Product Restriction for the Attenuation of Insulin Resistance, Oxidative Stress and Endothelial Dysfunction: A Systematic Review. Eur. J. Clin. Nutr. 2013, 67, 239–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, S.; Akhter, F.; Shahab, U.; Rafi, Z.; Khan, M.S.; Nabi, R.; Khan, M.S.; Ahmad, K.; Ashraf, J.M.; Moinuddin. Do All Roads Lead to the Rome? The Glycation Perspective! Semin. Cancer Biol. 2018, 49, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Khan, H.; Siddiqui, Z.; Khan, M.Y.; Rehman, S.; Shahab, U.; Godovikova, T.; Silnikov, V.; Moinuddin. AGEs, RAGEs and s-RAGE, Friend or Foe for Cancer. Semin. Cancer Biol. 2018, 49, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Stolzenberg-Solomon, R.; Zimmerman, T.P.; Duan, Z.; Chen, L.; Kahle, L.; Risch, A.; Subar, A.F.; Cross, A.J.; Hollenbeck, A.; et al. Dietary Consumption of Advanced Glycation End Products and Pancreatic Cancer in the Prospective NIH-AARP Diet and Health Study. Am. J. Clin. Nutr. 2015, 101, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, L.L.; Park, S.; Park, Y.; Colditz, G.A.; Anbardar, N.; Turner, D.P. Dietary Advanced Glycation End Products and Risk of Postmenopausal Breast Cancer in the NIH-AARP Diet and Health Study. Cancer 2020, 126, 2648. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.; Jacobs, K.; Haucke, E.; Santos, A.N.; Grune, T.; Simm, A. Role of Advanced Glycation End Products in Cellular Signaling. Redox Biol. 2014, 2, 411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manigrasso, M.B.; Juranek, J.; Ramasamy, R.; Schmidt, A.M. Unlocking the Biology of RAGE in Diabetic Microvascular Complications. Trends Endocrinol. Metab. TEM 2014, 25, 15–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soman, S.; Raju, R.; Sandhya, V.K.; Advani, J.; Khan, A.A.; Harsha, H.C.; Prasad, T.S.K.; Sudhakaran, P.R.; Pandey, A.; Adishesha, P.K. A Multicellular Signal Transduction Network of AGE/RAGE Signaling. J. Cell Commun. Signal. 2013, 7, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pesce, C.; Menini, S.; Pricci, F.; Favre, A.; Leto, G.; Dimario, U.; Pugliese, G. Glomerular Cell Replication and Cell Loss through Apoptosis in Experimental Diabetes Mellitus. Nephron 2002, 90, 484–488. [Google Scholar] [CrossRef]
- Aldini, G.; Facino, R.M.; Beretta, G.; Carini, M. Carnosine and Related Dipeptides as Quenchers of Reactive Carbonyl Species: From Structural Studies to Therapeutic Perspectives. BioFactors 2005, 24, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Regazzoni, L.; de Courten, B.; Garzon, D.; Altomare, A.; Marinello, C.; Jakubova, M.; Vallova, S.; Krumpolec, P.; Carini, M.; Ukropec, J.; et al. A Carnosine Intervention Study in Overweight Human Volunteers: Bioavailability and Reactive Carbonyl Species Sequestering Effect. Sci. Rep. 2016, 6, 27224. [Google Scholar] [CrossRef]
- Baye, E.; Ukropec, J.; de Courten, M.P.J.; Mousa, A.; Kurdiova, T.; Johnson, J.; Wilson, K.; Plebanski, M.; Aldini, G.; Ukropcova, B.; et al. Carnosine Supplementation Improves Serum Resistin Concentrations in Overweight or Obese Otherwise Healthy Adults: A Pilot Randomized Trial. Nutrients 2018, 10, 1258. [Google Scholar] [CrossRef] [Green Version]
- Anderson, E.J.; Vistoli, G.; Katunga, L.A.; Funai, K.; Regazzoni, L.; Monroe, T.B.; Gilardoni, E.; Cannizzaro, L.; Colzani, M.; de Maddis, D.; et al. A Carnosine Analog Mitigates Metabolic Disorders of Obesity by Reducing Carbonyl Stress. J. Clin. Investig. 2018, 128, 5280. [Google Scholar] [CrossRef]
- Menini, S.; Iacobini, C.; Ricci, C.; Blasetti Fantauzzi, C.; Pugliese, G. Protection from Diabetes-Induced Atherosclerosis and Renal Disease by d-Carnosine-Octylester: Effects of Early vs Late Inhibition of Advanced Glycation End-Products in Apoe-Null Mice. Diabetologia 2015, 58, 845–853. [Google Scholar] [CrossRef] [Green Version]
- Menini, S.; Iacobini, C.; Ricci, C.; Scipioni, A.; Blasetti Fantauzzi, C.; Giaccari, A.; Salomone, E.; Canevotti, R.; Lapolla, A.; Orioli, M.; et al. D-Carnosine Octylester Attenuates Atherosclerosis and Renal Disease in ApoE Null Mice Fed a Western Diet through Reduction of Carbonyl Stress and Inflammation. Br. J. Pharmacol. 2012, 166, 1344. [Google Scholar] [CrossRef] [Green Version]
- Iacobini, C.; Menini, S.; Blasetti Fantauzzi, C.; Pesce, C.M.; Giaccari, A.; Salomone, E.; Lapolla, A.; Orioli, M.; Aldini, G.; Pugliese, G. FL-926-16, a Novel Bioavailable Carnosinase-Resistant Carnosine Derivative, Prevents Onset and Stops Progression of Diabetic Nephropathy in Db/Db Mice. Br. J. Pharmacol. 2018, 175, 53–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menini, S.; Iacobini, C.; Blasetti Fantauzzi, C.; Pugliese, G. L-Carnosine and Its Derivatives as New Therapeutic Agents for the Prevention and Treatment of Vascular Complications of Diabetes. Curr. Med. Chem. 2019, 27, 1744–1763. [Google Scholar] [CrossRef] [PubMed]
- Bakris, G.L.; Bank, A.J.; Kass, D.A.; Neutel, J.M.; Preston, R.A.; Oparil, S. Advanced Glycation End-Product Cross-Link BreakersA Novel Approach to Cardiovascular Pathologies Related to the Aging Process. Am. J. Hypertens. 2004, 17, 23S–30S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, V.P.; Beyaz, A. Inhibitors of the Maillard Reaction and AGE Breakers as Therapeutics for Multiple Diseases. Drug Discov. Today 2006, 11, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.F.; Ramasamy, R.; Naka, Y.; Schmidt, A.M. Glycation, Inflammation, and RAGE. Circ. Res. 2003, 93, 1159–1169. [Google Scholar] [CrossRef] [Green Version]
- Coughlan, M.T.; Forbes, J.M.; Cooper, M.E. Role of the AGE Crosslink Breaker, Alagebrium, as a Renoprotective Agent in Diabetes. Kidney Int. 2007, 72, S54–S60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, E.M. Reactive Carbonyls and Oxidative Stress: Potential for Therapeutic Intervention. Pharmacol. Ther. 2007, 115, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Aldini, G.; Dalle-Donne, I.; Facino, R.M.; Milzani, A.; Carini, M. Intervention Strategies to Inhibit Protein Carbonylation by Lipoxidation-Derived Reactive Carbonyls. Med. Res. Rev. 2007, 27, 817–868. [Google Scholar] [CrossRef] [PubMed]
- Bolton, W.K.; Cattran, D.C.; Williams, M.E.; Adler, S.G.; Appel, G.B.; Cartwright, K.; Foiles, P.G.; Freedman, B.I.; Raskin, P.; Ratner, R.E.; et al. Randomized Trial of an Inhibitor of Formation of Advanced Glycation End Products in Diabetic Nephropathy. Am. J. Nephrol. 2004, 24, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Raval, A.D.; Thakker, D.; Rangoonwala, A.N.; Gor, D.; Walia, R. Vitamin B and Its Derivatives for Diabetic Kidney Disease. Cochrane Database Syst. Rev. 2015, 1, CD009403. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.E.; Bolton, W.K.; Khalifah, R.G.; Degenhardt, T.P.; Schotzinger, R.J.; McGill, J.B. Effects of Pyridoxamine in Combined Phase 2 Studies of Patients with Type 1 and Type 2 Diabetes and Overt Nephropathy. Am. J. Nephrol. 2007, 27, 605–614. [Google Scholar] [CrossRef]
- Lewis, E.J.; Greene, T.; Spitalewiz, S.; Blumenthal, S.; Berl, T.; Hunsicker, L.G.; Pohl, M.A.; Rohde, R.D.; Raz, I.; Yerushalmy, Y.; et al. Pyridorin in Type 2 Diabetic Nephropathy. J. Am. Soc. Nephrol. JASN 2012, 23, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drozak, J.; Veiga-da-Cunha, M.; Vertommen, D.; Stroobant, V.; Van Schaftingen, E. Molecular Identification of Carnosine Synthase as ATP-Grasp Domain-Containing Protein 1 (ATPGD1). J. Biol. Chem. 2010, 285, 9346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, V.; Klessens, C.Q.F.; Baelde, H.J.; Singler, B.; Veraar, K.A.M.; Zutinic, A.; Drozak, J.; Zschocke, J.; Schmitt, C.P.; de Heer, E. Intrinsic Carnosine Metabolism in the Human Kidney. Amino Acids 2015, 47, 2541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen, B.; Hohenadel, D.; Brinkkoetter, P.; Peters, V.; Rind, N.; Fischer, C.; Rychlik, I.; Cerna, M.; Romzova, M.; de Heer, E.; et al. Carnosine as a Protective Factor in Diabetic Nephropathy. Diabetes 2005, 54, 2320–2327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldini, G.; Orioli, M.; Rossoni, G.; Savi, F.; Braidotti, P.; Vistoli, G.; Yeum, K.J.; Negrisoli, G.; Carini, M. The Carbonyl Scavenger Carnosine Ameliorates Dyslipidaemia and Renal Function in Zucker Obese Rats. J. Cell. Mol. Med. 2011, 15, 1339. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, T.; Schilperoort, M.; Zhang, S.; Braun, J.D.; Qiu, J.; Rodriguez, A.; Pastene, D.O.; Krämer, B.K.; Köppel, H.; Baelde, H.; et al. Carnosine Attenuates the Development of Both Type 2 Diabetes and Diabetic Nephropathy in BTBR Ob/Ob Mice. Sci. Rep. 2017, 7, 44492. [Google Scholar] [CrossRef]
- Nokin, M.J.; Durieux, F.; Peixoto, P.; Chiavarina, B.; Peulen, O.; Blomme, A.; Turtoi, A.; Costanza, B.; Smargiasso, N.; Baiwir, D.; et al. Methylglyoxal, a Glycolysis Side-Product, Induces Hsp90 Glycation and YAP-Mediated Tumor Growth and Metastasis. eLife 2016, 5, e19375. [Google Scholar] [CrossRef]
- Menini, S.; Iacobini, C.; de Latouliere, L.; Manni, I.; Vitale, M.; Pilozzi, E.; Pesce, C.; Cappello, P.; Novelli, F.; Piaggio, G.; et al. Diabetes Promotes Invasive Pancreatic Cancer by Increasing Systemic and Tumour Carbonyl Stress in KrasG12D/+ Mice. J. Exp. Clin. Cancer Res. CR 2020, 39, 152. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xia, N.; Förstermann, U. Cardiovascular Effects and Molecular Targets of Resveratrol. Nitric Oxide 2012, 26, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Yang, Y.X.; Zhe, H.; He, Z.X.; Zhou, S.F. Bardoxolone Methyl (CDDO-Me) as a Therapeutic Agent: An Update on Its Pharmacokinetic and Pharmacodynamic Properties. Drug Des. Dev. Ther. 2014, 8, 2075. [Google Scholar] [CrossRef] [Green Version]
- Ooi, B.K.; Chan, K.G.; Goh, B.H.; Yap, W.H. The Role of Natural Products in Targeting Cardiovascular Diseases via Nrf2 Pathway: Novel Molecular Mechanisms and Therapeutic Approaches. Front. Pharmacol. 2018, 9, 1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnefont-Rousselot, D. Resveratrol and Cardiovascular Diseases. Nutrients 2016, 8, 250. [Google Scholar] [CrossRef] [PubMed]
- Tellone, E.; Galtieri, A.; Russo, A.; Giardina, B.; Ficarra, S. Resveratrol: A Focus on Several Neurodegenerative Diseases. Oxidative Med. Cell. Longev. 2015, 2015, 392169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calkins, M.J.; Johnson, D.A.; Townsend, J.A.; Vargas, M.R.; Dowell, J.A.; Williamson, T.P.; Kraft, A.D.; Lee, J.M.; Li, J.; Johnson, J.A. The Nrf2/ARE Pathway as a Potential Therapeutic Target in Neurodegenerative Disease. Antioxid. Redox Signal. 2009, 11, 497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, K.J.; Baur, J.A.; Lewis, K.N.; Peshkin, L.; Price, N.L.; Labinskyy, N.; Swindell, W.R.; Kamara, D.; Minor, R.K.; Perez, E.; et al. Resveratrol Delays Age-Related Deterioration and Mimics Transcriptional Aspects of Dietary Restriction without Extending Lifespan. Cell Metab. 2008, 8, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robledinos-Antón, N.; Fernández-Ginés, R.; Manda, G.; Cuadrado, A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxidative Med. Cell. Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef] [PubMed]
- Chaplin, A.; Carpéné, C.; Mercader, J. Resveratrol, Metabolic Syndrome, and Gut Microbiota. Nutrients 2018, 10, 1651. [Google Scholar] [CrossRef] [Green Version]
- Baye, E.; Ukropec, J.; de Courten, M.P.J.; Kurdiova, T.; Krumpolec, P.; Fernández-Real, J.M.; Aldini, G.; Ukropcova, B.; de Courten, B. Carnosine Supplementation Reduces Plasma Soluble Transferrin Receptor in Healthy Overweight or Obese Individuals: A Pilot Randomised Trial. Amino Acids 2018, 51, 73–81. [Google Scholar] [CrossRef]
- Baye, E.; Ukropec, J.; de Courten, M.P.; Vallova, S.; Krumpolec, P.; Kurdiova, T.; Aldini, G.; Ukropcova, B.; de Courten, B. Effect of Carnosine Supplementation on the Plasma Lipidome in Overweight and Obese Adults: A Pilot Randomised Controlled Trial. Sci. Rep. 2017, 7, 17458. [Google Scholar] [CrossRef] [PubMed]
- Elbarbary, N.S.; Ismail, E.A.R.; El-Naggar, A.R.; Hamouda, M.H.; El-Hamamsy, M. The Effect of 12 Weeks Carnosine Supplementation on Renal Functional Integrity and Oxidative Stress in Pediatric Patients with Diabetic Nephropathy: A Randomized Placebo-Controlled Trial. Pediatric Diabetes 2018, 19, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Menon, K.; Marquina, C.; Hoj, P.; Liew, D.; Mousa, A.; de Courten, B. Carnosine and Histidine-Containing Dipeptides Improve Dyslipidemia: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Nutr. Rev. 2020, 78, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Baye, E.; Menon, K.; de Courten, M.P.; Earnest, A.; Cameron, J.; de Courten, B. Does Supplementation with Carnosine Improve Cardiometabolic Health and Cognitive Function in Patients with Pre-Diabetes and Type 2 Diabetes? Study Protocol for a Randomised, Double-Blind, Placebo-Controlled Trial. BMJ Open 2017, 7, e017691. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative Stress and Diabetic Complications. Circ. Res. 2010, 107, 1058. [Google Scholar] [CrossRef] [Green Version]
- Maillard, L.C. Action des acides amines sur les sucres: Formation des melanoidines par voie methodique. Comptes Rendus Hebd. Séances l’Académie Sci. 1912, 154, 66–68. [Google Scholar]
- Poulsen, M.W.; Hedegaard, R.V.; Andersen, J.M.; de Courten, B.; Bügel, S.; Nielsen, J.; Skibsted, L.H.; Dragsted, L.O. Advanced Glycation Endproducts in Food and Their Effects on Health. Food Chem. Toxicol. 2013, 60, 10–37. [Google Scholar] [CrossRef]
- Scheijen, J.L.J.M.; Clevers, E.; Engelen, L.; Dagnelie, P.C.; Brouns, F.; Stehouwer, C.D.A.; Schalkwijk, C.G. Analysis of Advanced Glycation Endproducts in Selected Food Items by Ultra-Performance Liquid Chromatography Tandem Mass Spectrometry: Presentation of a Dietary AGE Database. Food Chem. 2016, 190, 1145–1150. [Google Scholar] [CrossRef] [PubMed]
- Scheijen, J.L.J.M.; Hanssen, N.M.J.; Van Greevenbroek, M.M.; Van der Kallen, C.J.; Feskens, E.J.M.; Stehouwer, C.D.A.; Schalkwijk, C.G. Dietary Intake of Advanced Glycation Endproducts Is Associated with Higher Levels of Advanced Glycation Endproducts in Plasma and Urine: The CODAM Study. Clin. Nutr. 2018, 37, 919–925. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Cai, W.; Peppa, M.; Goodman, S.; Ferrucci, L.; Striker, G.; Vlassara, H. Circulating Glycotoxins and Dietary Advanced Glycation Endproducts: Two Links to Inflammatory Response, Oxidative Stress, and Aging. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2007, 62, 427. [Google Scholar] [CrossRef] [Green Version]
- Uribarri, J.; Cai, W.; Woodward, M.; Tripp, E.; Goldberg, L.; Pyzik, R.; Yee, K.; Tansman, L.; Chen, X.; Mani, V.; et al. Elevated Serum Advanced Glycation Endproducts in Obese Indicate Risk for the Metabolic Syndrome: A Link between Healthy and Unhealthy Obesity? J. Clin. Endocrinol. Metab. 2015, 100, 1957. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Cai, W.; Pyzik, R.; Goodman, S.; Chen, X.; Zhu, L.; Ramdas, M.; Striker, G.E.; Vlassara, H. Suppression of Native Defense Mechanisms, SIRT1 and PPARγ, by Dietary Glycoxidants Precedes Disease in Adult Humans; Relevance to Lifestyle-Engendered Chronic Diseases. Amino Acids 2014, 46, 301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, P.; Huang, C.; Hsu, C.; Yin, M.; Guo, Y. Association of Dietary AGEs with Circulating AGEs, Glycated LDL, IL-1α and MCP-1 Levels in Type 2 Diabetic Patients. Eur. J. Nutr. 2010, 49, 429–434. [Google Scholar] [CrossRef]
- Angoorani, P.; Ejtahed, H.S.; Mirmiran, P.; Mirzaei, S.; Azizi, F. Dietary Consumption of Advanced Glycation End Products and Risk of Metabolic Syndrome. Int. J. Food Sci. Nutr. 2016, 67, 170–176. [Google Scholar] [CrossRef]
- Vlassara, H.; Cai, W.; Crandall, J.; Goldberg, T.; Oberstein, R.; Dardaine, V.; Peppa, M.; Rayfield, E.J. Inflammatory Mediators Are Induced by Dietary Glycotoxins, a Major Risk Factor for Diabetic Angiopathy. Proc. Natl. Acad. Sci. USA 2002, 99, 15596. [Google Scholar] [CrossRef] [Green Version]
- Uribarri, J.; Peppa, M.; Cai, W.; Goldberg, T.; Lu, M.; He, C.; Vlassara, H. Restriction of Dietary Glycotoxins Reduces Excessive Advanced Glycation End Products in Renal Failure Patients. J. Am. Soc. Nephrol. 2003, 14, 728–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlassara, H.; Cai, W.; Goodman, S.; Pyzik, R.; Yong, A.; Chen, X.; Zhu, L.; Neade, T.; Beeri, M.; Silverman, J.M.; et al. Protection against Loss of Innate Defenses in Adulthood by Low Advanced Glycation End Products (AGE) Intake: Role of the Antiinflammatory AGE Receptor-1. J. Clin. Endocrinol. Metab. 2009, 94, 4483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlassara, H.; Cai, W.; Tripp, E.; Pyzik, R.; Yee, K.; Goldberg, L.; Tansman, L.; Chen, X.; Mani, V.; Fayad, Z.A.; et al. Oral AGE Restriction Ameliorates Insulin Resistance in Obese Individuals with the Metabolic Syndrome: A Randomised Controlled Trial. Diabetologia 2016, 59, 2181. [Google Scholar] [CrossRef] [PubMed]
- Schröter, D.; Höhn, A. Role of Advanced Glycation End Products in Carcinogenesis and Their Therapeutic Implications. Curr. Pharm. Des. 2019, 24, 261. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.H.; Detty, S.Q.; Agrawal, D.K. Clinical Implications of High-Mobility Group Box-1 (HMGB1) and the Receptor for Advanced Glycation End-Products (RAGE) in Cutaneous Malignancy: A Systematic Review. Anticancer Res. 2017, 37, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Sims, G.P.; Rowe, D.C.; Rietdijk, S.T.; Herbst, R.; Coyle, A.J. HMGB1 and RAGE in Inflammation and Cancer. Annu. Rev. Immunol. 2010, 28, 367–388. [Google Scholar] [CrossRef]
- Menini, S.; Iacobini, C.; de Latouliere, L.; Manni, I.; Ionta, V.; Blasetti Fantauzzi, C.; Pesce, C.; Cappello, P.; Novelli, F.; Piaggio, G.; et al. The Advanced Glycation End-Product Nϵ-Carboxymethyllysine Promotes Progression of Pancreatic Cancer: Implications for Diabetes-Associated Risk and Its Prevention. J. Pathol. 2020, 245, 197–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riehl, A.; Németh, J.; Angel, P.; Hess, J. The Receptor RAGE: Bridging Inflammation and Cancer. Cell Commun. Signal. CCS 2009, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Gebhardt, C.; Riehl, A.; Durchdewald, M.; Németh, J.; Fürstenberger, G.; Müller-Decker, K.; Enk, A.; Arnold, B.; Bierhaus, A.; Nawroth, P.P.; et al. RAGE Signaling Sustains Inflammation and Promotes Tumor Development. J. Exp. Med. 2008, 205, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taguchi, A.; Blood, D.C.; del Toro, G.; Canet, A.; Lee, D.C.; Qu, W.; Tanji, N.; Lu, Y.; Lalla, E.; Fu, C.; et al. Blockade of RAGE–Amphoterin Signalling Suppresses Tumour Growth and Metastases. Nature 2000, 405, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Tang, D.; Schapiro, N.E.; Livesey, K.M.; Farkas, A.; Loughran, P.; Bierhaus, A.; Lotze, M.T.; Zeh, H.J. The Receptor for Advanced Glycation End-Products (RAGE) Sustains Autophagy and Limits Apoptosis, Promoting Pancreatic Tumor Cell Survival. Cell Death Differ. 2010, 17, 666. [Google Scholar] [CrossRef] [Green Version]
- Birlouez-Aragon, I.; Saavedra, G.; Tessier, F.J.; Galinier, A.; Ait-Ameur, L.; Lacoste, F.; Niamba, C.N.; Alt, N.; Somoza, V.; Lecerf, J.M. A Diet Based on High-Heat-Treated Foods Promotes Risk Factors for Diabetes Mellitus and Cardiovascular Diseases. Am. J. Clin. Nutr. 2010, 91, 1220–1226. [Google Scholar] [CrossRef] [Green Version]
- Uribarri, J.; Cai, W.; Ramdas, M.; Goodman, S.; Pyzik, R.; Chen, X.; Zhu, L.; Striker, G.E.; Vlassara, H. Restriction of Advanced Glycation End Products Improves Insulin Resistance in Human Type 2 Diabetes: Potential Role of AGER1 and SIRT1. Diabetes Care 2011, 34, 1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado-Andrade, C. Carboxymethyl-Lysine: Thirty Years of Investigation in the Field of AGE Formation. Food Funct. 2016, 7, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Roncero-Ramos, I.; Delgado-Andrade, C.; Tessier, F.J.; Niquet-Léridon, C.; Strauch, C.; Monnier, V.M.; Navarro, M.P. Metabolic Transit of Nε-Carboxymethyl-Lysine after Consumption of AGEs from Bread Crust. Food Funct. 2013, 4, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Koschinsky, T.; He, C.J.; Mitsuhashi, T.; Bucala, R.; Liu, C.; Buenting, C.; Heitmann, K.; Vlassara, H. Orally Absorbed Reactive Glycation Products (Glycotoxins): An Environmental Risk Factor in Diabetic Nephropathy. Proc. Natl. Acad. Sci. USA 1997, 94, 6474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degen, J.; Vogel, M.; Richter, D.; Hellwig, M.; Henle, T. Metabolic Transit of Dietary Methylglyoxal. J. Agric. Food Chem. 2013, 61, 10253–10260. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Sabol, J.; Mitsuhashi, T.; Vlassara, H. Dietary glycotoxins: Inhibition of reactive products by aminoguanidine facilitates renal clearance and reduces tissue sequestration. Diabetes 1999, 48, 1308–1315. [Google Scholar] [CrossRef] [PubMed]
- Hellwig, M.; Matthes, R.; Peto, A.; Lobner, J.; Henle, T. N-epsilon-fructosyllysine and N-epsilon-carboxymethyllysine, but not lysinoalanine, are available for absorption after simulated gastrointestinal digestion. Amino Acids 2014, 46, 289e99. [Google Scholar] [CrossRef] [PubMed]
- Monnier, L.; Mas, E.; Ginet, C.; Michel, F.; Villon, L.; Cristol, J.P.; Colette, C. Activation of Oxidative Stress by Acute Glucose Fluctuations Compared With Sustained Chronic Hyperglycemia in Patients With Type 2 Diabetes. JAMA 2006, 295, 1681–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coussens, L.M.; Werb, Z. Inflammation and Cancer. Nature 2002, 420, 860. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167. [Google Scholar] [CrossRef] [PubMed]
- Fiolet, T.; Srour, B.; Sellem, L.; Kesse-Guyot, E.; Allès, B.; Méjean, C.; Deschasaux, M.; Fassier, P.; Latino-Martel, P.; Beslay, M.; et al. Consumption of Ultra-Processed Foods and Cancer Risk: Results from NutriNet-Santé Prospective Cohort. BMJ 2018, 360, k322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Keefe, J.H.; Gheewala, N.M.; O’Keefe, J.O. Dietary Strategies for Improving Post-Prandial Glucose, Lipids, Inflammation, and Cardiovascular Health. J. Am. Coll. Cardiol. 2008, 51, 249–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Title, L.M.; Cummings, P.M.; Giddens, K.; Nassar, B.A. Oral Glucose Loading Acutely Attenuates Endothelium-Dependent Vasodilation in Healthy Adults without Diabetes: An Effect Prevented by Vitamins C and E. J. Am. Coll. Cardiol. 2000, 36, 2185–2191. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, M.; Holst, J.J.; Björck, I.M. Metabolic Effects of Amino Acid Mixtures and Whey Protein in Healthy Subjects: Studies Using Glucose-Equivalent Drinks. Am. J. Clin. Nutr. 2007, 85, 996–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coutinho, M.; Gerstein, H.C.; Wang, Y.; Yusuf, S. The Relationship between Glucose and Incident Cardiovascular Events. A Metaregression Analysis of Published Data from 20 Studies of 95,783 Individuals Followed for 12.4 Years. Diabetes Care 1999, 22, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M.; Hirsch, I.B. Glycemic Variability: A Hemoglobin A1c–Independent Risk Factor for Diabetic Complications. JAMA 2006, 295, 1707–1708. [Google Scholar] [CrossRef]
- Ceriello, A.; Assaloni, R.; da Ros, R.; Maier, A.; Piconi, L.; Quagliaro, L.; Esposito, K.; Giugliano, D. Effect of Atorvastatin and Irbesartan, Alone and in Combination, on Postprandial Endothelial Dysfunction, Oxidative Stress, and Inflammation in Type 2 Diabetic Patients. Circulation 2005, 111, 2518–2524. [Google Scholar] [CrossRef]
- Maessen, D.E.; Hanssen, N.M.; Scheijen, J.L.; Van der Kallen, C.J.; Van Greevenbroek, M.M.; Stehouwer, C.D.; Schalkwijk, C.G. Post–Glucose Load Plasma α-Dicarbonyl Concentrations Are Increased in Individuals with Impaired Glucose Metabolism and Type 2 Diabetes: The CODAM Study. Diabetes Care 2015, 38, 913–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schalkwijk, C.G.; Stehouwer, C.D.A. Methylglyoxal, a Highly Reactive Dicarbonyl Compound, in Diabetes, Its Vascular Complications, and Other Age-Related Diseases. Physiol. Rev. 2019, 100, 407–461. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H.; Uribarri, J. Advanced Glycation End Products (AGE) and Diabetes: Cause, Effect, or Both? Curr. Diabetes Rep. 2014, 14, 453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. We Need to Talk about the Warburg Effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef]
- Chiavarina, B.; Nokin, M.J.; Durieux, F.; Bianchi, E.; Turtoi, A.; Peulen, O.; Peixoto, P.; Irigaray, P.; Uchida, K.; Belpomme, D.; et al. Triple Negative Tumors Accumulate Significantly Less Methylglyoxal Specific Adducts than Other Human Breast Cancer Subtypes. Oncotarget 2014, 5, 5472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nokin, M.J.; Durieux, F.; Bellier, J.; Peulen, O.; Uchida, K.; Spiegel, D.A.; Cochrane, J.R.; Hutton, C.A.; Castronovo, V.; Bellahcène, A. Hormetic Potential of Methylglyoxal, a Side-Product of Glycolysis, in Switching Tumours from Growth to Death. Sci. Rep. 2017, 7, 11722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentile, F.; Arcaro, A.; Pizzimenti, S.; Daga, M.; Cetrangolo, G.P.; Dianzani, C.; Lepore, A.; Graf, M.; Ames, P.R.J.; Barrera, G. DNA Damage by Lipid Peroxidation Products: Implications in Cancer, Inflammation and Autoimmunity. AIMS Genet. 2017, 4, 103. [Google Scholar] [CrossRef]
- Zarkovic, N.; Ilic, Z.; Jurin, M.; Schaur, R.J.; Puhl, H.; Esterbauer, H. Stimulation of HeLa Cell Growth by Physiological Concentrations of 4-Hydroxynonenal. Cell Biochem. Funct. 1993, 11, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Nokin, M.J.; Bellier, J.; Durieux, F.; Peulen, O.; Rademaker, G.; Gabriel, M.; Monseur, C.; Charloteaux, B.; Verbeke, L.; Van Laere, S.; et al. Methylglyoxal, a Glycolysis Metabolite, Triggers Metastasis through MEK/ERK/SMAD1 Pathway Activation in Breast Cancer. Breast Cancer Res. BCR 2019, 21, 11. [Google Scholar] [CrossRef] [PubMed]
- Bellier, J.; Nokin, M.J.; Caprasse, M.; Tiamiou, A.; Blomme, A.; Scheijen, J.L.; Koopmansch, B.; MacKay, G.M.; Chiavarina, B.; Costanza, B.; et al. Methylglyoxal Scavengers Resensitize KRAS-Mutated Colorectal Tumors to Cetuximab. Cell Rep. 2020, 30, 1400–1416.e6. [Google Scholar] [CrossRef] [PubMed]
- Wondrak, G.T.; Jacobson, M.K.; Jacobson, E.L. Antimelanoma Activity of Apoptogenic Carbonyl Scavengers. J. Pharmacol. Exp. Ther. 2006, 316, 805–814. [Google Scholar] [CrossRef] [Green Version]
- Van Heijst, J.W.J.; Niessen, H.W.M.; Musters, R.J.; Van Hinsbergh, V.W.M.; Hoekman, K.; Schalkwijk, C.G. Argpyrimidine-Modified Heat Shock Protein 27 in Human Non-Small Cell Lung Cancer: A Possible Mechanism for Evasion of Apoptosis. Cancer Lett. 2006, 241, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Oya-Ito Tomoko, T.; Naito, Y.; Takagi, T.; Handa, O.; Matsui, H.; Yamada, M.; Shima, K.; Yoshikawa, T. Heat-Shock Protein 27 (Hsp27) as a Target of Methylglyoxal in Gastrointestinal Cancer. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2011, 1812, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Nandakumar, N.; Shi, Y.; Manzano, M.; Smith, A.; Graham, G.; Gupta, S.; Vietsch, E.E.; Laughlin, S.Z.; Wadhwa, M.; et al. Downstream of Mutant KRAS, the Transcription Regulator YAP Is Essential for Neoplastic Progression to Pancreatic Ductal Adenocarcinoma. Sci. Signal. 2014, 7, ra42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, D. The Hippo Signaling Pathway in Development and Cancer. Dev. Cell 2010, 19, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Eynde, M.D.G.; Kusters, Y.H.A.M.; Houben, A.J.H.M.; Scheijen, J.L.J.M.; Van Duynhoven, J.; Fazelzadeh, P.; Joris, P.J.; Plat, J.; Mensink, R.P.; Hanssen, N.M.J.; et al. Diet-Induced Weight Loss Reduces Postprandial Dicarbonyl Stress in Abdominally Obese Men: Secondary Analysis of a Randomized Controlled Trial. Clin. Nutr. 2021, 40, 2654–2662. [Google Scholar] [CrossRef] [PubMed]
- Maessen, D.E.; Hanssen, N.M.; Lips, M.A.; Scheijen, J.L.; Willems Van Dijk, K.; Pijl, H.; Stehouwer, C.D.; Schalkwijk, C.G. Energy Restriction and Roux-En-Y Gastric Bypass Reduce Postprandial α-Dicarbonyl Stress in Obese Women with Type 2 Diabetes. Diabetologia 2016, 59, 2013–2017. [Google Scholar] [CrossRef]
- de Courten, B.; Jakubova, M.; de Courten, M.P.; Kukurova, I.J.; Vallova, S.; Krumpolec, P.; Valkovic, L.; Kurdiova, T.; Garzon, D.; Barbaresi, S.; et al. Effects of Carnosine Supplementation on Glucose Metabolism: Pilot Clinical Trial. Obesity 2016, 24, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Al-Sawalha, N.A.; Alshogran, O.Y.; Awawdeh, M.S.; Almomani, B.A. The Effects of L-Carnosine on Development of Metabolic Syndrome in Rats. Life Sci. 2019, 237, 116905. [Google Scholar] [CrossRef]
- Menon, K.; Marquina, C.; Liew, D.; Mousa, A.; de Courten, B. Histidine-containing dipeptides reduce central obesity and improve glycaemic outcomes: A systematic review and meta-analysis of randomized controlled trials. Obes. Rev. 2020, 21, e12975. [Google Scholar] [CrossRef] [PubMed]
- Menon, K.; de Courten, B.; Magliano, D.J.; Ademi, Z.; Liew, D.; Zomer, E. The Cost-Effectiveness of Supplemental Carnosine in Type 2 Diabetes. Nutrients 2022, 14, 215. [Google Scholar] [CrossRef] [PubMed]
- Haus, J.M.; Thyfault, J.P. Therapeutic Potential of Carbonyl-Scavenging Carnosine Derivative in Metabolic Disorders. J. Clin. Investig. 2018, 128, 5198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-Mass Index and Incidence of Cancer: A Systematic Review and Meta-Analysis of Prospective Observational Studies. Lancet 2008, 371, 569–578. [Google Scholar] [CrossRef]
- Vlassara, H. Advanced Glycation in Health and Disease: Role of the Modern Environment. Ann. N. Y. Acad. Sci. 2005, 1043, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, H.; Mashima, T.; Yamamoto, K.; Tsuruo, T. Modulation of Heat-Shock Protein 27 (Hsp27) Anti-Apoptotic Activity by Methylglyoxal Modification. J. Biol. Chem. 2002, 277, 45770–45775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlassara, H.; Uribarri, J.; Cai, W.; Goodman, S.; Pyzik, R.; Post, J.; Grosjean, F.; Woodward, M.; Striker, G.E. Effects of Sevelamer on HbA1c, Inflammation, and Advanced Glycation End Products in Diabetic Kidney Disease. Clin. J. Am. Soc. Nephrol. CJASN 2012, 7, 934. [Google Scholar] [CrossRef]
- Ferramosca, E.; Burke, S.; Chasan-Taber, S.; Ratti, C.; Chertow, G.M.; Raggi, P. Potential Antiatherogenic and Anti-Inflammatory Properties of Sevelamer in Maintenance Hemodialysis Patients. Am. Heart J. 2005, 149, 820–825. [Google Scholar] [CrossRef]
- Ketteler, M.; Rix, M.; Fan, S.; Pritchard, N.; Oestergaard, O.; Chasan-Taber, S.; Heaton, J.; Duggal, A.; Kalra, P.A. Efficacy and Tolerability of Sevelamer Carbonate in Hyperphosphatemic Patients Who Have Chronic Kidney Disease and Are Not on Dialysis. Clin. J. Am. Soc. Nephrol. CJASN 2008, 3, 1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Titan, S.M.; Zatz, R.; Graciolli, F.G.; dos Reis, L.M.; Barros, R.T.; Jorgetti, V.; Moysés, R.M.A. FGF-23 as a Predictor of Renal Outcome in Diabetic Nephropathy. Clin. J. Am. Soc. Nephrol. CJASN 2011, 6, 241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakuta, T.; Tanaka, R.; Hyodo, T.; Suzuki, H.; Kanai, G.; Nagaoka, M.; Takahashi, H.; Hirawa, N.; Oogushi, Y.; Miyata, T.; et al. Effect of Sevelamer and Calcium-Based Phosphate Binders on Coronary Artery Calcification and Accumulation of Circulating Advanced Glycation End Products in Hemodialysis Patients. Am. J. Kidney Dis. 2011, 57, 422–431. [Google Scholar] [CrossRef]
- Russo, D.; Miranda, I.; Ruocco, C.; Battaglia, Y.; Buonanno, E.; Manzi, S.; Russo, L.; Scafarto, A.; Andreucci, V.E. The Progression of Coronary Artery Calcification in Predialysis Patients on Calcium Carbonate or Sevelamer. Kidney Int. 2007, 72, 1255–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shantouf, R.; Budoff, M.J.; Ahmadi, N.; Tiano, J.; Flores, F.; Kalantar-Zadeh, K. Effects of Sevelamer and Calcium-Based Phosphate Binders on Lipid and Inflammatory Markers in Hemodialysis Patients. Am. J. Nephrol. 2008, 28, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yubero-Serrano, E.M.; Woodward, M.; Poretsky, L.; Vlassara, H.; Striker, G.E. Effects of Sevelamer Carbonate on Advanced Glycation End Products and Antioxidant/Pro-Oxidant Status in Patients with Diabetic Kidney Disease. Clin. J. Am. Soc. Nephrol. CJASN 2015, 10, 759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggiero, B.; Trillini, M.; Tartaglione, L.; Rotondi, S.; Perticucci, E.; Tripepi, R.; Aparicio, C.; Lecchi, V.; Perna, A.; Peraro, F.; et al. Effects of Sevelamer Carbonate in Patients with CKD and Proteinuria: The ANSWER Randomized Trial. Am. J. Kidney Dis. 2019, 74, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Biruete, A.; Hill Gallant, K.M.; Lindemann, S.R.; Wiese, G.N.; Chen, N.X.; Moe, S.M. Phosphate Binders and Non-Phosphate Effects in the Gastrointestinal Tract. J. Ren. Nutr. Off. J. Counc. Ren. Nutr. Natl. Kidney Found. 2020, 30, 4–10. [Google Scholar] [CrossRef]
- Pearson-Stuttard, J.; Bennett, J.; Cheng, Y.J.; Vamos, E.P.; Cross, A.J.; Ezzati, M.; Gregg, E.W. Trends in Predominant Causes of Death in Individuals with and without Diabetes in England from 2001 to 2018: An Epidemiological Analysis of Linked Primary Care Records. Lancet Diabetes Endocrinol. 2021, 9, 165–173. [Google Scholar] [CrossRef]
- Song, M. Cancer overtakes vascular disease as leading cause of excess death associated with diabetes. Lancet Diabetes Endocrinol. 2021, 9, 131–133. [Google Scholar] [CrossRef]
- Mozaffarian, D. Dietary and Policy Priorities for Cardiovascular Disease, Diabetes, and Obesity—A Comprehensive Review. Circulation 2016, 133, 187–225. [Google Scholar] [CrossRef] [PubMed]
- Perez-Martinez, P.; Garcia-Quintana, J.M.; Yubero-Serrano, E.M.; Tasset-Cuevas, I.; Tunez, I.; Garcia-Rios, A.; Delgado-Lista, J.; Marin, C.; Perez-Jimenez, F.; Roche, H.M.; et al. Postprandial Oxidative Stress Is Modified by Dietary Fat: Evidence from a Human Intervention Study. Clin. Sci. 2010, 119, 251–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
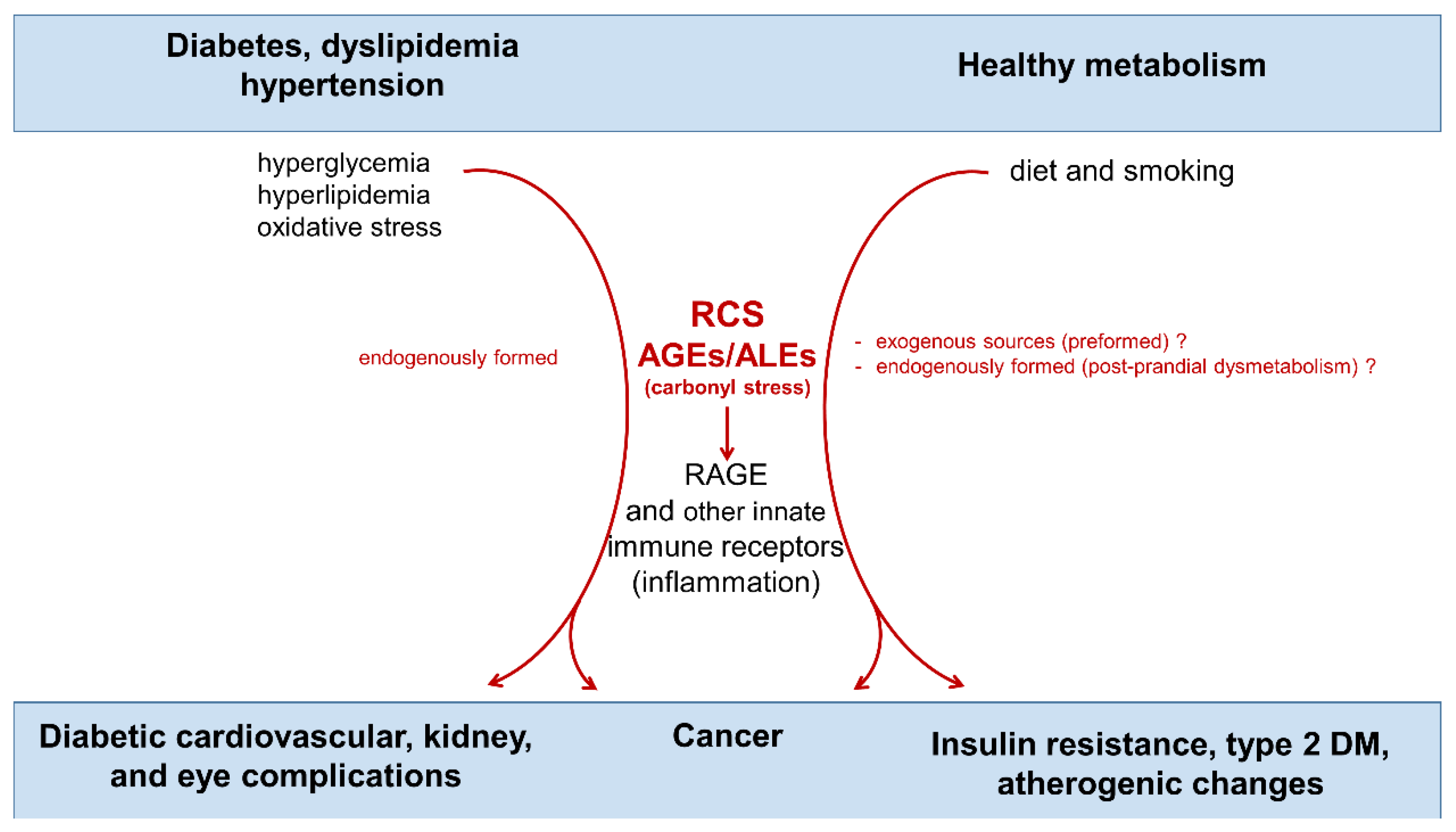






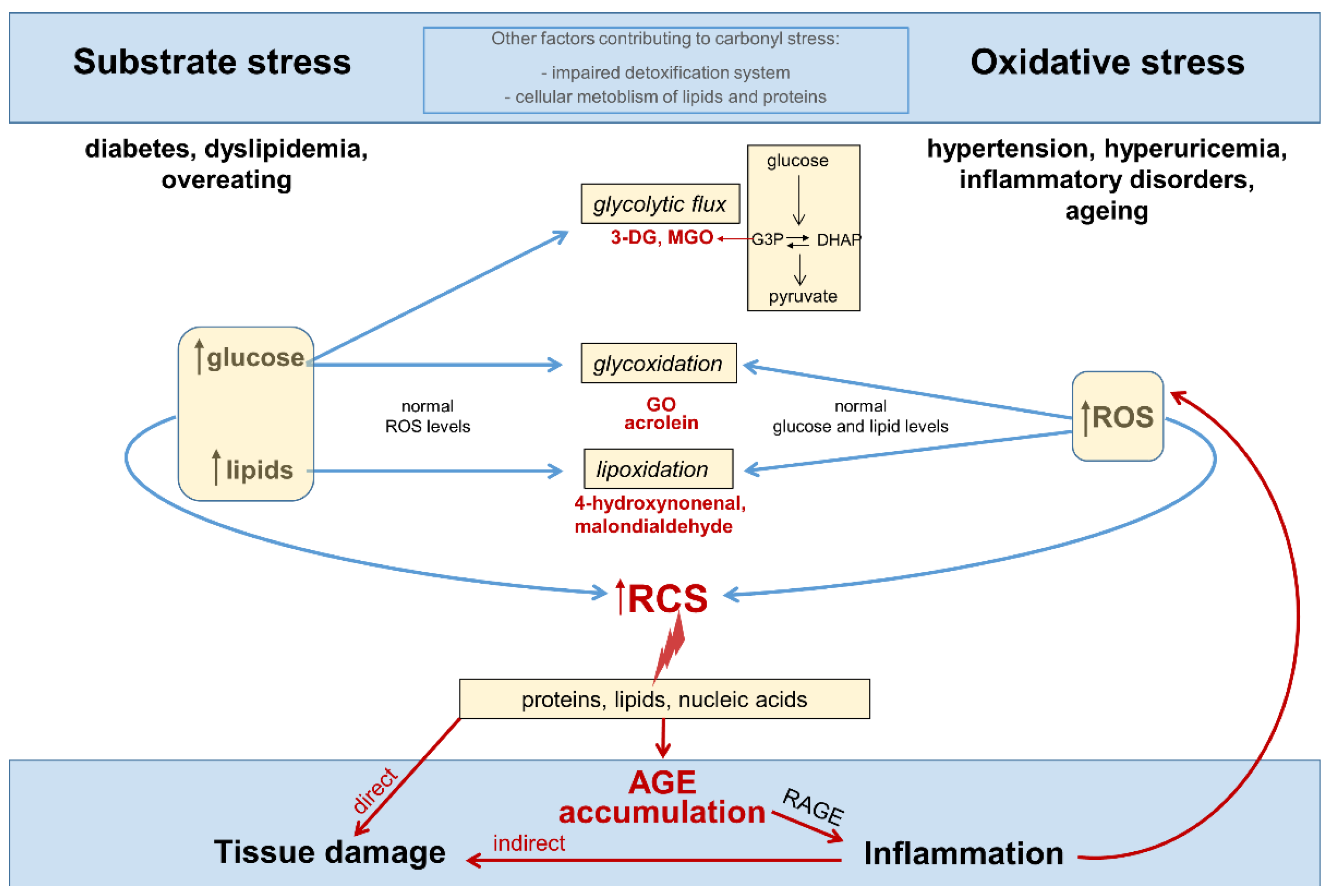
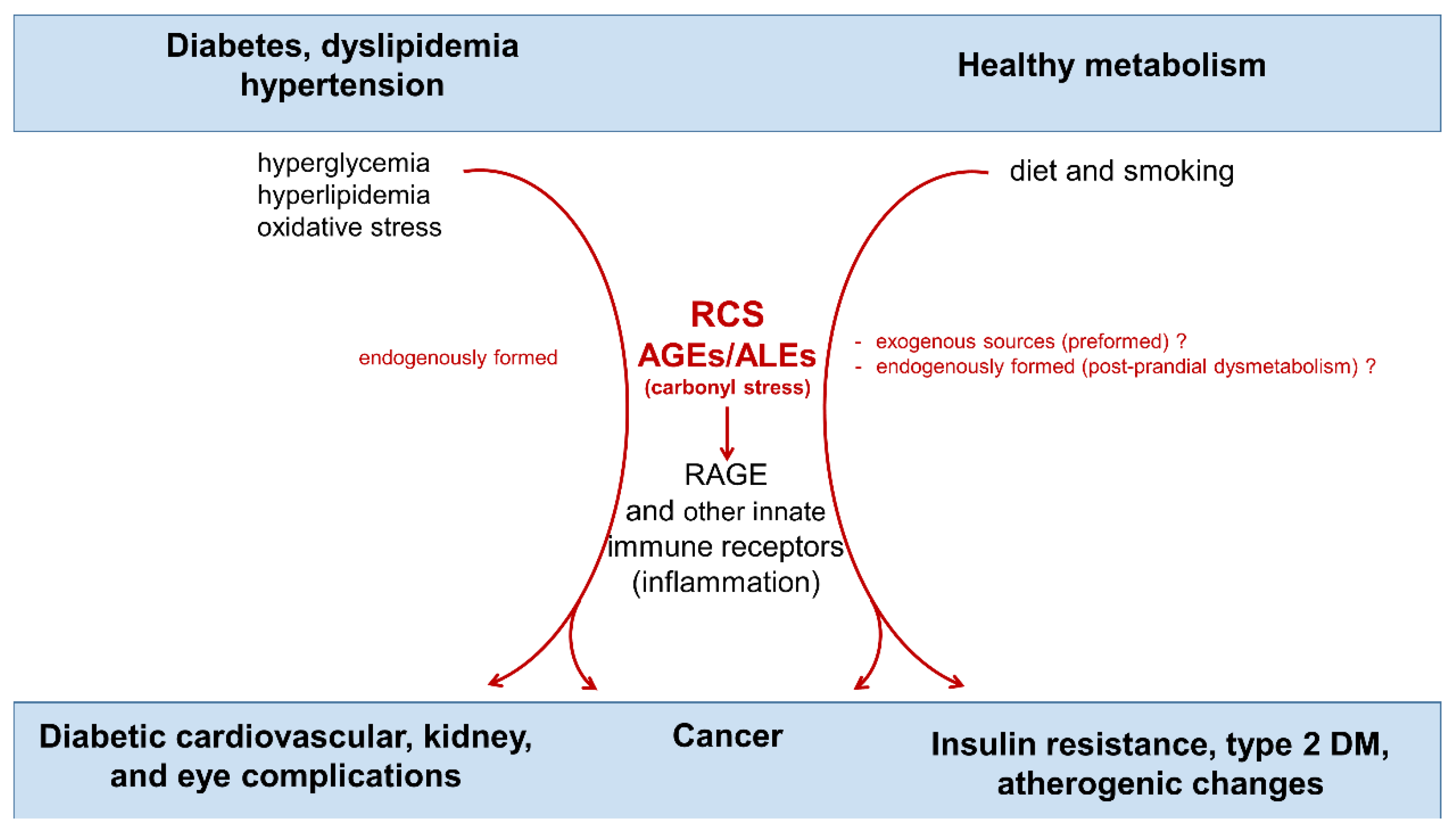{kind=link}
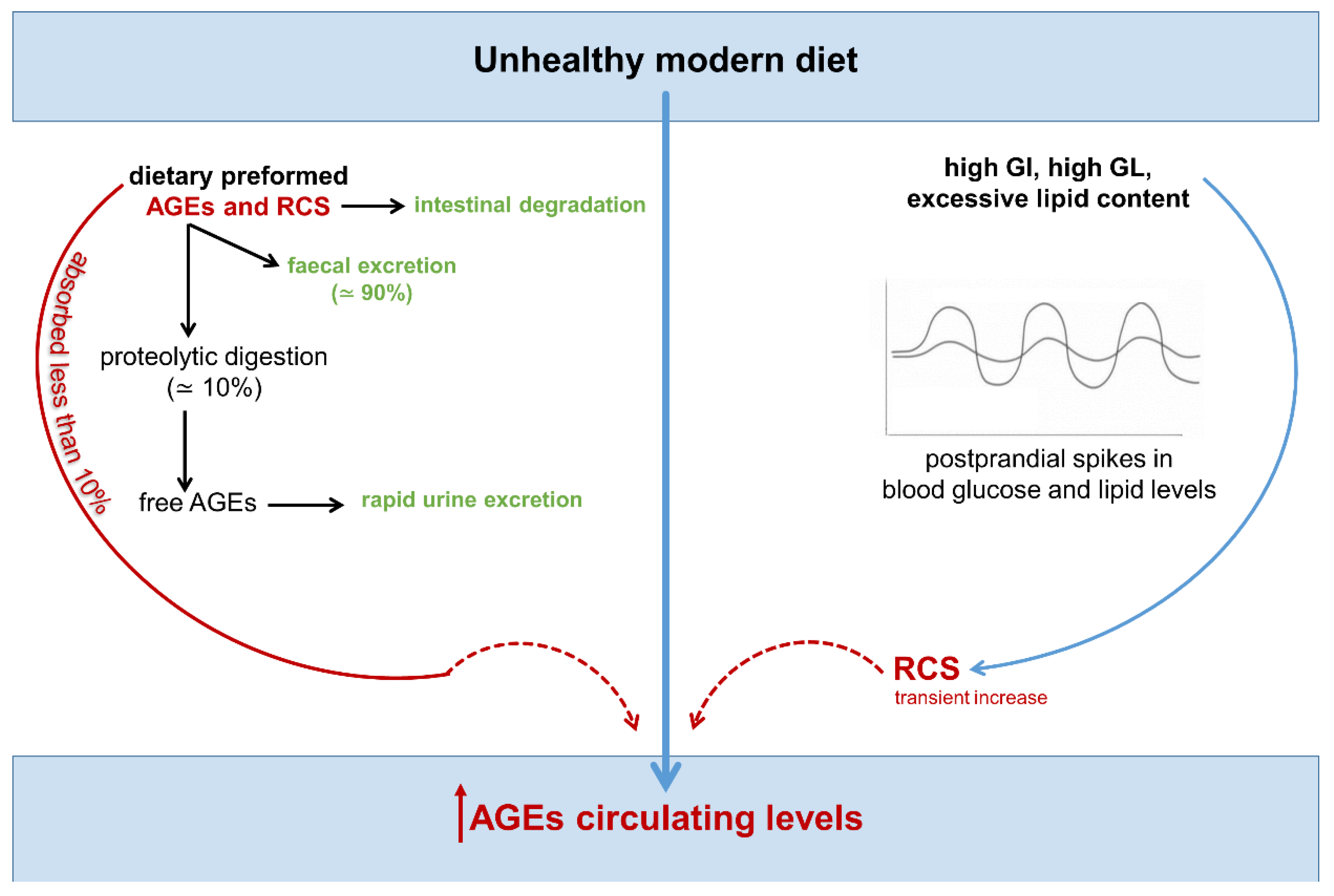
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Food Item | AGE kU/100 g | Serving Size (g) | AGE kU/Serving |
|---|---|---|---|
| Bread/cereals/Breakfast foods/snacks | |||
| Biscuit | 1470 | 30 | 441 |
| Bread, white, slice | 83 | 30 | 25 |
| Bread, white, slice, toasted | 107 | 30 | 32 |
| Bread, whole wheat, slice | 103 | 30 | 31 |
| Bread, whole wheat, slice, toasted | 137 | 30 | 41 |
| Chips, potato | 2883 | 30 | 865 |
| Corn Flakes | 233 | 30 | 70 |
| Cracker, wheat | 857 | 30 | 257 |
| Croissant, butter | 1113 | 30 | 334 |
| French toast | 850 | 30 | 255 |
| Muffin, bran | 340 | 30 | 102 |
| Oatmeal, dry | 13 | 30 | 4 |
| Pancake, homemade | 973 | 30 | 292 |
| Pie, apple | 637 | 191 | |
| Popcorn | 133 | 30 | 40 |
| Rice Krispies | 2000 | 30 | 600 |
| Waffle, toasted | 2870 | 30 | 861 |
| Popcorn | 133 | 30 | 40 |
| Grains/legumes/soy derivatives | |||
| Beans, red kidney, raw | 116 | 100 | 116 |
| Beans, red kidney, cooked 1h | 298 | 100 | 298 |
| Pasta, cooked | 112 | 100 | 112 |
| Milk, soy | 31 | 250 (mL) | 77 |
| Rice, cooked | 9 | 100 | 9 |
| Soy burger | 130 | 30 | 39 |
| Tofu, raw | 788 | 90 | 709 |
| Tofu, broiled | 4107 | 90 | 3696 |
| Starchy vegetables | |||
| Corn, canned | 20 | 100 | 20 |
| Potato, white, boiled | 17 | 100 | 17 |
| Potato, white, roasted | 218 | 100 | 218 |
| Potato, white, french fries | 1522 | 100 | 1522 |
| Fruits and vegetables (raw, unless specified otherwise) | |||
| Apple | 13 | 100 | 13 |
| Apple, baked | 45 | 100 | 45 |
| Banana | 9 | 100 | 9 |
| Carrots, canned | 10 | 100 | 10 |
| Onion | 36 | 100 | 36 |
| Tomato | 23 | 100 | 23 |
| Vegetables, grilled (broccoli, carrots, celery) | 226 | 100 | 226 |
| Meat and fish/seafood | |||
| Beef, raw | 707 | 90 | 636 |
| Beef, roast | 6071 | 90 | 5464 |
| Beef, steak, broiled | 7479 | 90 | 6731 |
| Chicken, breast, raw | 769 | 90 | 692 |
| Chicken, breast, boiled in water | 1210 | 90 | 1089 |
| Chicken, breast, roasted | 4768 | 90 | 4291 |
| Chicken, breast, breaded, fried | 9691 | 90 | 8965 |
| Lamb, leg, raw | 826 | 90 | 743 |
| Lamb, leg, broiled | 2431 | 90 | 2188 |
| Pork, bacon, fried | 91,577 | 13 | 11,905 |
| Pork, ham, smoked | 2349 | 90 | 2114 |
| Pork, ribs, roasted | 4430 | 90 | 3987 |
| Pork, sausage, Italian, raw | 1861 | 90 | 1675 |
| Pork, liverwurst | 633 | 90 | 570 |
| Pork, sausage, Italian, BBQ | 4839 | 90 | 4355 |
| Salmon, raw | 528 | 90 | 475 |
| Salmon, smoked | 572 | 90 | 515 |
| Salmon, broiled | 4334 | 90 | 3901 |
| Shrimp, raw | 1003 | 90 | 903 |
| Shrimp, fried | 4328 | 90 | 3895 |
| Tuna, canned with oil | 1740 | 90 | 1566 |
| Tuna, broiled | 5150 | 90 | 4635 |
| Turkey, breast, roasted | 4669 | 90 | 4202 |
| Milk, milk products, and cheese | |||
| Cheese, American, white, processed | 8677 | 30 | 2603 |
| Cheese, brie | 5597 | 30 | 503 |
| Cheese, cheddar | 5523 | 30 | 1657 |
| Cheese, feta, Greek, | 8423 | 30 | 2527 |
| Cheese, mozzarella | 1677 | 30 | 503 |
| Cheese, parmesan, grated | 16,900 | 15 | 2535 |
| Cheese, Swiss, processed | 4470 | 30 | 1341 |
| Milk, whole | 5 | 250 (mL) | 12 |
| Pudding, chocolate | 17 | 120 | 20 |
| Yogurt, vanilla | 3 | 250 | 8 |
| Eggs | |||
| Egg, poached | 90 | 30 | 27 |
| Egg, scrambled, pan, butter | 337 | 30 | 101 |
| Egg, omelet, pan, butter | 507 | 30 | 152 |
| Egg, fried | 2749 | 45 | 1237 |
| Beverages | AGE kU/mL | Serving size (mL) | AGE kU/Serving |
| Beer | 1.20 | 250 | 3 |
| Coca Cola | 2.80 | 250 | 7 |
| Coffee | 1.60 | 250 | 4 |
| Fruit juice, orange | 6 | 250 | 14 |
| Tea | 1.20 | 250 | 3 |
| Wine | 11.20 | 250 | 28 |
| Study | Intervention | Population/Animal Model | Main Outcomes/Purpose |
|---|---|---|---|
| Clinical | |||
| Van den Eynde et al. [143] | calorie restriction | 52 abdominally obese men, 25 lean men (18–65 years) | weight loss associated with reduced postprandial iAUC of MGO, GO, and 3-DG in abdominally obese individuals |
| Maessen et al. [144] | calorie restriction or RYGB | obese women without (n = 27) or with (n = 27) T2D, 12 lean women | weight loss associated with reduced postprandial α-dicarbonyl levels in diabetic women |
| Regazzoni et al. [50] | L-carnosine supplementation (2 g/day for 12 weeks) | 29 overweight to obese individuals, 8 females and 21 males | increased urinary excretion of carnosine-acrolein adducts (acrolein detoxification), |
| de Courten et al. [145] | L-carnosine supplementation (2 g/day for 12 weeks), compared to placebo | 30 overweight to obese individuals, 15 per treatment arm | reduced fasting insulin and insulin resistance, and normalization of 2-h glucose and insulin after 75-g glucose load |
| Baye et al. [84] | L-carnosine supplementation (2 g/day for 12 weeks), compared to placebo | 24 overweight to obese individuals (13 in L-carnosine, 11 in placebo group) | plasma lipidome changes associated with improved insulin sensitivity and secretion, and low serum carnosinase 1 activity |
| Baye et al. [83] | L-carnosine supplementation (2 g/day for 12 weeks), compared to placebo | 26 overweight to obese individuals (14 in L-carnosine, 11 in placebo group) | iron metabolism changes associated with low serum carnosinase 1 activity and increased urinary carnosine concentration |
| Elbarbary et al. [85] | L-carnosine supplementation (1 g/day for 12 weeks), compared to placebo | 90 patients with diabetic nephropathy | improvement of glycemic control, oxidative stress, and renal function |
| Baye et al. [87] (Study protocol for an RCT) | L-carnosine supplementation (2 g/day for 12 weeks), compared to placebo | 50 participants with pre-diabetes and T2D randomly assigned to the intervention or control group | to analyze changes in metabolic, cardiovascular, and cognitive parameters |
| Preclinical | |||
| Anderson et al. [52] | Carnosinol supplementation (10 to 45 mg/kg/day for 6 to 12 weeks), compared to placebo | GPx4+/− and WT mice fed a high-fat/high-sucrose diet, and rats fed a 60% high fat diet, compared to chow fed mice and rats | improved glycemic control and muscle insulin sensitivity in mouse models of severe carbonyl stress and diet-induced obesity |
| Aldini et al. [70] | L-carnosine and D-carnosine supplementation (30 mg/kg/day for 24 weeks), compared to placebo | Zucker obese rat | improved obese-related disorders (dyslipidemia, hypertension, and renal injury) |
| Albrecht et al. [71] | L-carnosine supplementation (45 mg/kg/day for 18 weeks), compared to placebo | BTBR ob/ob mice (T2D model) | elevated carnosine and carnosine-carbonyl adducts associated with improved glucose metabolism, albuminuria, and glomerular pathology |
| Al-Sawalha et al. [146] | L-carnosine supplementation (45 mg/kg/day for 16 weeks), compared to placebo | Wistar rats fed a high-fat high-carbohydrate diet (metabolic syndrome model) | reduced blood pressure and glucose, normalized total cholesterol and low-density lipoprotein levels |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iacobini, C.; Vitale, M.; Haxhi, J.; Pesce, C.; Pugliese, G.; Menini, S. Food-Related Carbonyl Stress in Cardiometabolic and Cancer Risk Linked to Unhealthy Modern Diet. Nutrients 2022, 14, 1061. https://doi.org/10.3390/nu14051061
Iacobini C, Vitale M, Haxhi J, Pesce C, Pugliese G, Menini S. Food-Related Carbonyl Stress in Cardiometabolic and Cancer Risk Linked to Unhealthy Modern Diet. Nutrients. 2022; 14(5):1061. https://doi.org/10.3390/nu14051061
Chicago/Turabian StyleIacobini, Carla, Martina Vitale, Jonida Haxhi, Carlo Pesce, Giuseppe Pugliese, and Stefano Menini. 2022. "Food-Related Carbonyl Stress in Cardiometabolic and Cancer Risk Linked to Unhealthy Modern Diet" Nutrients 14, no. 5: 1061. https://doi.org/10.3390/nu14051061
APA StyleIacobini, C., Vitale, M., Haxhi, J., Pesce, C., Pugliese, G., & Menini, S. (2022). Food-Related Carbonyl Stress in Cardiometabolic and Cancer Risk Linked to Unhealthy Modern Diet. Nutrients, 14(5), 1061. https://doi.org/10.3390/nu14051061