
Nutrition in Cystic Fibrosis—Some Notes on the Fat Recommendations


Abstract
1. Introduction
2. Fat Recommendations and Intake
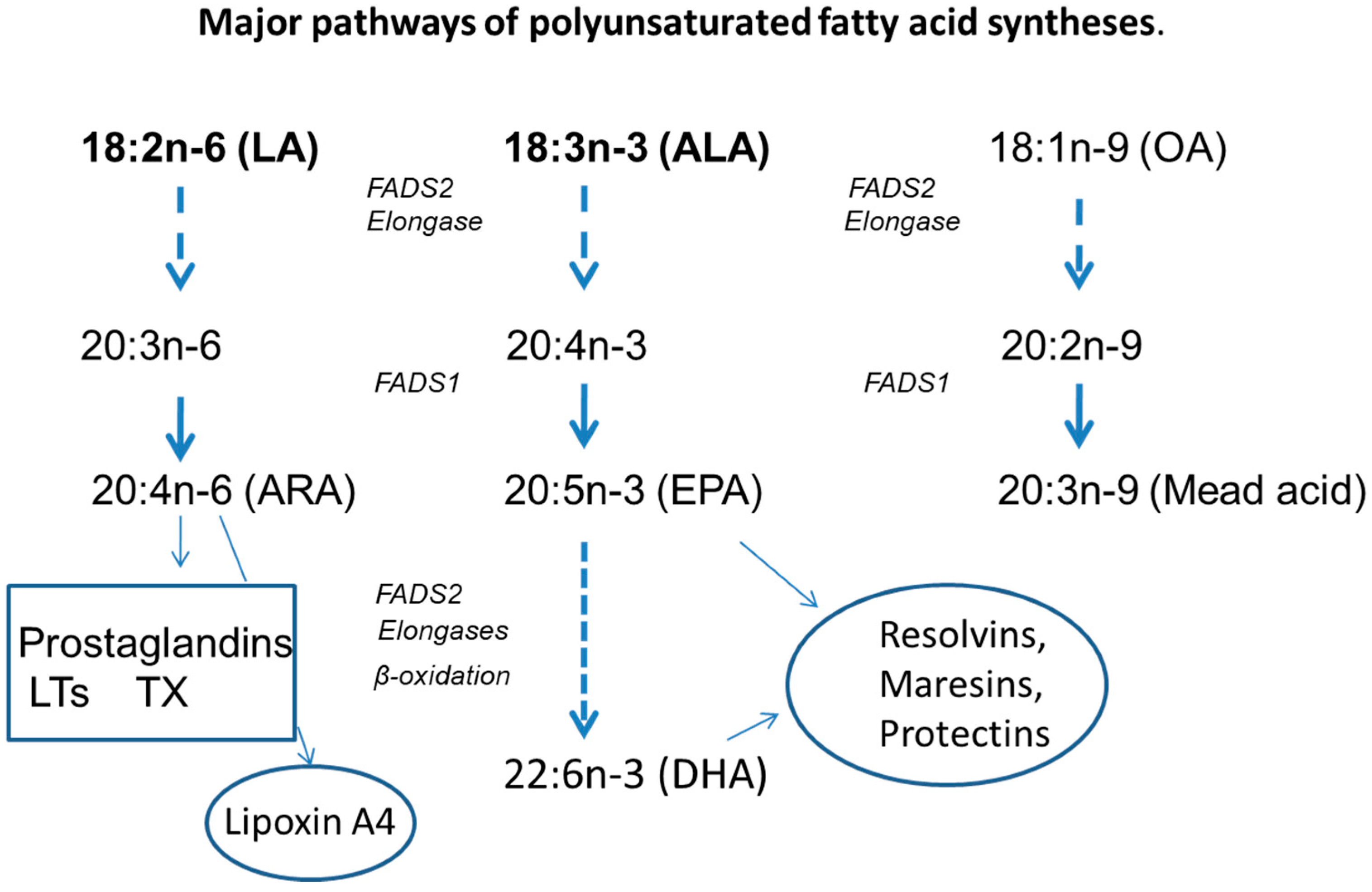
3. Fatty Acid Abnormalities in CF
4. Is Dietary Qualitative Intervention Justified?
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Andersen, D.H. Cystic fibrosis of the pancreas and its relation to celiac disease. A clinical and pathological study. Am. J. Dis. Child 1938, 56, 344–399. [Google Scholar] [CrossRef]
- Fanconi, G.; Uehlinger, E.; Knauer, C. Das Coelioksyndrome bei Angeborener zystischer Pankreas fibromatose und Bronkiektasien. Wien. Med. Wochenschr. 1936, 86, 753–756. [Google Scholar]
- Shteinberg, M.; Haq, I.J.; Polineni, D.; Davies, J.C. Cystic fibrosis. Lancet 2021, 397, 2195–2211. [Google Scholar] [CrossRef]
- Strandvik, B. Care of patients with cystic fibrosis. Treatment, screening and clinical outcome. Ann. Nestlé 2006, 64, 131–140. [Google Scholar]
- Earnest, A.; Salimi, F.; Wainwright, C.E.; Bell, S.C.; Ruseckaite, R.; Ranger, T.; Kotsimbos, T.; Ahern, S. Lung function over the life course of paediatric and adult patients with cystic fibrosis from a large multi-centre registry. Sci. Rep. 2020, 10, 17421. [Google Scholar] [CrossRef]
- North American Cystic Fibrosis Foundation. 2019 Patient Registry Annual Data Report; Cystic Fibrosis Foundation: Bethesda, MD, USA, 2021. [Google Scholar]
- Cystic Fibrosis Canada. 2018 Canadian CF Registry Annual Data Report; Cystic Fibrosis Canada: Toronto, ON, Canada, 2021. [Google Scholar]
- Orenti, A.; Zolin, A.; Jung, A.; van Rens, J. ECFS Patient Registry Annual Report 2019; European Cystic Fibrosis Society: Karup, Denmark, 2021. [Google Scholar]
- Steinkamp, G.; Wiedemann, B. Relationship between nutritional status and lung function in cystic fibrosis: Cross sectional and longitudinal analyses from the German CF quality assurance (CFQA) project. Thorax 2002, 57, 596–601. [Google Scholar] [CrossRef]
- Altman, K.; McDonald, C.M.; Michel, S.H.; Maguiness, K. Nutrition in cystic fibrosis: From the past to the present and into the future. Pediatric Pulmonol. 2019, 54 (Suppl. S3), S56–S73. [Google Scholar] [CrossRef]
- Ashkenazi, M.; Nathan, N.; Sarouk, I.; Aluma, B.E.B.; Dagan, A.; Bezalel, Y.; Keler, S.; Vilozni, D.; Efrati, O. Nutritional Status in Childhood as a Prognostic Factor in Patients with Cystic Fibrosis. Lung 2019, 197, 371–376. [Google Scholar] [CrossRef]
- Van der Haak, N.; King, S.J.; Crowder, T.; Kench, A.; Painter, C.; Saxby, N. Highlights from the nutrition guidelines for cystic fibrosis in Australia and New Zealand. J. Cyst. Fibros. 2020, 19, 16–25. [Google Scholar] [CrossRef]
- Wilschanski, M.; Braegger, C.P.; Colombo, C.; Declercq, D.; Morton, A.; Pancheva, R.; Robberecht, E.; Stern, M.; Strandvik, B.; Wolfe, S.; et al. Highlights of the ESPEN-ESPGHAN-ECFS Guidelines on Nutrition Care for Infants and Children with Cystic Fibrosis. J. Pediatric Gastroenterol. Nutr. 2016, 63, 671–675. [Google Scholar] [CrossRef]
- McDonald, C.M.; Alvarez, J.A.; Bailey, J.; Bowser, E.K.; Farnham, K.; Mangus, M.; Padula, L.; Porco, K.; Rozga, M. Academy of Nutrition and Dietetics: 2020 Cystic Fibrosis Evidence Analysis Center Evidence-Based Nutrition Practice Guideline. J. Acad. Nutr. Diet. 2021, 121, 1591–1636.e3. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, T.; Hempstead, S.E.; Brady, C.; Cannon, C.L.; Clark, K.; Condren, M.E.; Guill, M.F.; Guillerman, R.P.; Leone, C.G.; Maguiness, K.; et al. Clinical Practice Guidelines from the Cystic Fibrosis Foundation for Preschoolers with Cystic Fibrosis. Pediatrics 2016, 137, e20151784. [Google Scholar] [CrossRef] [PubMed]
- Saxby, N.; Painter, C.; Kench, A.; King, S.; Crowder, T.; van der Haak, N. Nutrition Guidelines for Cystic Fibrosis in Australia and New Zealand; Thoracic Society of Australia and New Zealand: Sydney, Australia, 2017. [Google Scholar]
- Turck, D.; Braegger, C.P.; Colombo, C.; Declercq, D.; Morton, A.; Pancheva, R.; Robberecht, E.; Stern, M.; Strandvik, B.; Wolfe, S.; et al. ESPEN-ESPGHAN-ECFS guidelines on nutrition care for infants, children, and adults with cystic fibrosis. Clin. Nutr. 2016, 35, 557–577. [Google Scholar] [CrossRef]
- Kopp, B.T.; Joseloff, E.; Goetz, D.; Ingram, B.; Heltshe, S.L.; Leung, D.H.; Ramsey, B.W.; McCoy, K.; Borowitz, D. Urinary metabolomics reveals unique metabolic signatures in infants with cystic fibrosis. J. Cyst. Fibros. 2019, 18, 507–515. [Google Scholar] [CrossRef]
- Liessi, N.; Pedemonte, N.; Armirotti, A.; Braccia, C. Proteomics and Metabolomics for Cystic Fibrosis Research. Int. J. Mol. Sci. 2020, 21, 5439. [Google Scholar] [CrossRef]
- Wheelock, C.E.; Strandvik, B. Abnormal n-6 fatty acid metabolism in cystic fibrosis contributes to pulmonary symptoms. Prostaglandins Leukot. Essent. Fat. Acids 2020, 160, 102156. [Google Scholar] [CrossRef]
- Dei Cas, M.; Zulueta, A.; Mingione, A.; Caretti, A.; Ghidoni, R.; Signorelli, P.; Paroni, R. An Innovative Lipidomic Workflow to Investigate the Lipid Profile in a Cystic Fibrosis Cell Line. Cells 2020, 9, 1197. [Google Scholar] [CrossRef]
- Pacetti, D.; Malavolta, M.; Bocci, F.; Boselli, E.; Frega, N.G. High-performance liquid chromatography/electrospray ionization ion-trap tandem mass spectrometric analysis and quantification of phosphatidylcholine molecular species in the serum of cystic fibrosis subjects supplemented with docosahexaenoic acid. Rapid Commun. Mass Spectrom. 2004, 18, 2395–2400. [Google Scholar] [CrossRef]
- Hulbert, A.J.; Turner, N.; Storlien, L.H.; Else, P.L. Dietary fats and membrane function: Implications for metabolism and disease. Biol. Rev. Camb. Philos. Soc. 2005, 80, 155–169. [Google Scholar] [CrossRef]
- Ulven, S.M.; Holven, K.B. Metabolomic and gene expression analysis to study the effects of dietary saturated and polyunsaturated fats. Curr. Opin. Lipidol. 2020, 31, 15–19. [Google Scholar] [CrossRef]
- Gimenez, M.S.; Oliveros, L.B.; Gomez, N.N. Nutritional deficiencies and phospholipid metabolism. Int. J. Mol. Sci. 2011, 12, 2408–2433. [Google Scholar] [CrossRef] [PubMed]
- Corey, M.; McLaughlin, F.J.; Williams, M.; Levison, H. A comparison of survival, growth, and pulmonary function in patients with cystic fibrosis in Boston and Toronto. J. Clin. Epidemiol. 1988, 41, 583–591. [Google Scholar] [CrossRef]
- Lai, H.C.; Corey, M.; FitzSimmons, S.; Kosorok, M.R.; Farrell, P.M. Comparison of growth status of patients with cystic fibrosis between the United States and Canada. Am. J. Clin. Nutr. 1999, 69, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Goss, C.H.; Sykes, J.; Stanojevic, S.; Marshall, B.; Petren, K.; Ostrenga, J.; Fink, A.; Elbert, A.; Quon, B.S.; Stephenson, A.L. Comparison of Nutrition and Lung Function Outcomes in Patients with Cystic Fibrosis Living in Canada and the United States. Am. J. Respir. Crit. Care Med. 2018, 197, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Poulimeneas, D.; Grammatikopoulou, M.G.; Devetzi, P.; Petrocheilou, A.; Kaditis, A.G.; Papamitsou, T.; Doudounakis, S.E.; Vassilakou, T. Adherence to Dietary Recommendations, Nutrient Intake Adequacy and Diet Quality among Pediatric Cystic Fibrosis Patients: Results from the GreeCF Study. Nutrients 2020, 12, 3126. [Google Scholar] [CrossRef]
- Moukarzel, S.; Dyer, R.A.; Innis, S.M. Complex Relation Between Diet and Phospholipid Fatty Acids in Children with Cystic Fibrosis. J. Pediatric Gastroenterol. Nutr. 2017, 64, 598–604. [Google Scholar] [CrossRef]
- Smith, C.; Winn, A.; Seddon, P.; Ranganathan, S. A fat lot of good: Balance and trends in fat intake in children with cystic fibrosis. J. Cyst. Fibros. 2012, 11, 154–157. [Google Scholar] [CrossRef]
- Woestenenk, J.W.; Castelijns, S.J.; van der Ent, C.K.; Houwen, R.H. Dietary intake in children and adolescents with cystic fibrosis. Clin. Nutr. 2014, 33, 528–532. [Google Scholar] [CrossRef]
- Filigno, S.S.; Robson, S.M.; Szczesniak, R.D.; Chamberlin, L.A.; Baker, M.A.; Sullivan, S.M.; Kroner, J.; Powers, S.W. Macronutrient intake in preschoolers with cystic fibrosis and the relationship between macronutrients and growth. J. Cyst. Fibros. 2017, 16, 519–524. [Google Scholar] [CrossRef]
- Calvo-Lerma, J.; Hulst, J.M.; Asseiceira, I.; Claes, I.; Garriga, M.; Colombo, C.; Fornés, V.; Woodcock, S.; Martins, T.; Boon, M.; et al. Nutritional status, nutrient intake and use of enzyme supplements in paediatric patients with Cystic Fibrosis; a European multicentre study with reference to current guidelines. J. Cyst. Fibros. 2017, 16, 510–518. [Google Scholar] [CrossRef]
- Stallings, V.A.; Tindall, A.M.; Mascarenhas, M.R.; Maqbool, A.; Schall, J.I. Improved residual fat malabsorption and growth in children with cystic fibrosis treated with a novel oral structured lipid supplement: A randomized controlled trial. PLoS ONE 2020, 15, e0232685. [Google Scholar] [CrossRef]
- Kindstedt-Arfwidson, K.; Strandvik, B. Food intake in patients with cystic fibrosis on an ordinary diet. Scand. J. Gastroenterol. Suppl. 1988, 143, 160–162. [Google Scholar] [CrossRef]
- Van Egmond, A.W.; Kosorok, M.R.; Koscik, R.; Laxova, A.; Farrell, P.M. Effect of linoleic acid intake on growth of infants with cystic fibrosis. Am. J. Clin. Nutr. 1996, 63, 746–752. [Google Scholar] [CrossRef]
- Walkowiak, J.; Przyslawski, J. Five-year prospective analysis of dietary intake and clinical status in malnourished cystic fibrosis patients. J. Hum. Nutr. Diet 2003, 16, 225–231. [Google Scholar] [CrossRef]
- Strandvik, B.; Lundquist-Persson, C.; Sabel, K.-G. Early behavior and development are influenced by the n-6 and n-3 status in prematures. Oleagineux Corps Gras Lipides 2011, 18, 297–300. [Google Scholar] [CrossRef][Green Version]
- Blasbalg, T.L.; Hibbeln, J.R.; Ramsden, C.E.; Majchrzak, S.F.; Rawlings, R.R. Changes in consumption of omega-3 and omega-6 fatty acids in the United States during the 20th century. Am. J. Clin. Nutr. 2011, 93, 950–962. [Google Scholar] [CrossRef]
- Heird, W.C. The role of polyunsaturated fatty acids in term and preterm infants and breastfeeding mothers. Pediatric Clin. N. Am. 2001, 48, 173–188. [Google Scholar] [CrossRef]
- Otto, S.J.; Houwelingen, A.C.; Antal, M.; Manninen, A.; Godfrey, K.; López-Jaramillo, P.; Hornstra, G. Maternal and neonatal essential fatty acid status in phospholipids: An international comparative study. Eur. J. Clin. Nutr. 1997, 51, 232–242. [Google Scholar] [CrossRef]
- Strandvik, B.; Gronowitz, E.; Enlund, F.; Martinsson, T.; Wahlström, J. Essential fatty acid deficiency in relation to genotype in patients with cystic fibrosis. J. Pediatric 2001, 139, 650–655. [Google Scholar] [CrossRef]
- Walkowiak, J.; Lisowska, A.; Blaszczynski, M.; Przyslawski, J.; Walczak, M. Polyunsaturated fatty acids in cystic fibrosis are related to nutrition and clinical expression of the disease. J. Pediatric Gastroenterol. Nutr. 2007, 45, 488–489. [Google Scholar] [CrossRef]
- Sinaasappel, M.; Stern, M.; Littlewood, J.; Wolfe, S.; Steinkamp, G.; Heijerman, H.G.; Robberecht, E.; Döring, G. Nutrition in patients with cystic fibrosis: A European Consensus. J. Cyst. Fibros. 2002, 1, 51–75. [Google Scholar] [CrossRef]
- Kuo, P.T.; Huang, N.N.; Bassett, D.R. The fatty acid composition of the serum chylomicrons and adipose tissue of children with cystic fibrosis of the pancreas. J. Pediatric 1962, 60, 394–403. [Google Scholar] [CrossRef]
- Colombo, C.; Nobili, R.M.; Alicandro, G. Challenges with optimizing nutrition in cystic fibrosis. Expert Rev. Respir. Med. 2019, 13, 533–544. [Google Scholar] [CrossRef]
- Hankard, R.; Munck, A.; Navarro, J. Nutrition and growth in cystic fibrosis. Horm. Res. 2002, 58 (Suppl. S1), 16–20. [Google Scholar] [CrossRef]
- Munck, A. Nutritional considerations in patients with cystic fibrosis. Expert Rev. Respir. Med. 2010, 4, 47–56. [Google Scholar] [CrossRef]
- Borowitz, D.; Baker, R.D.; Stallings, V. Consensus report on nutrition for pediatric patients with cystic fibrosis. J. Pediatric Gastroenterol. Nutr. 2002, 35, 246–259. [Google Scholar] [CrossRef]
- Strandvik, B. Fatty acid metabolism in cystic fibrosis. Prostaglandins Leukot. Essent. Fat. Acids 2010, 83, 121–129. [Google Scholar] [CrossRef]
- Gibson, R.A.; Teubner, J.K.; Haines, K.; Cooper, D.M.; Davidson, G.P. Relationships between pulmonary function and plasma fatty acid levels in cystic fibrosis patients. J. Pediatric Gastroenterol. Nutr. 1986, 5, 408–415. [Google Scholar] [CrossRef]
- Rogiers, V.; Vercruysse, A.; Dab, I.; Crokaert, R.; Vis, H.L. Fatty acid pattern of platelet phospholipids in cystic fibrosis. Eur. J. Pediatric 1984, 142, 305–306. [Google Scholar] [CrossRef]
- Freedman, S.D.; Blanco, P.G.; Zaman, M.M.; Shea, J.C.; Ollero, M.; Hopper, I.K.; Weed, D.A.; Gelrud, A.; Regan, M.M.; Laposata, M.; et al. Association of cystic fibrosis with abnormalities in fatty acid metabolism. N. Engl. J. Med. 2004, 350, 560–569. [Google Scholar] [CrossRef]
- Mischler, E.H.; Parrell, S.W.; Farrell, P.M.; Raynor, W.J.; Lemen, R.J. Correction of linoleic acid deficiency in cystic fibrosis. Pediatric Res. 1986, 20, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Moonen, C.T.; Dimand, R.J.; Cox, K.L. The noninvasive determination of linoleic acid content of human adipose tissue by natural abundance carbon-13 nuclear magnetic resonance. Magn. Reason. Med. 1988, 6, 140–157. [Google Scholar] [CrossRef] [PubMed]
- Underwood, B.A.; Denning, C.R.; Navab, M. Polyunsaturated fatty acids and tocopherol levels in patients with cystic fibrosis. Ann. N. Y. Acad. Sci. 1972, 203, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Chase, H.P.; Cotton, E.K.; Elliott, R.B. Intravenous linoleic acid supplementation in children with cystic fibrosis. Pediatrics 1979, 64, 207–213. [Google Scholar] [CrossRef]
- Elliott, R.B. A therapeutic trial of fatty acid supplementation in cystic fibrosis. Pediatrics 1976, 57, 474–479. [Google Scholar] [CrossRef]
- Hjelte, L.; Nilsson, K.; Moen, I.E.; Lindblad, A.; Mared, L.; Pressler, T.; Fluge, G. Linoleic acid but not EPA and DHA correlates to prognostic markers in Scandinavian CF patients. J. Cyst. Fibros. 2008, 7, S93. [Google Scholar]
- Lloyd-Still, J.D.; Bibus, D.M.; Powers, C.A.; Johnson, S.B.; Holman, R.T. Essential fatty acid deficiency and predisposition to lung disease in cystic fibrosis. Acta Paediatr. 1996, 85, 1426–1432. [Google Scholar] [CrossRef]
- Maqbool, A.; Schall, J.I.; Garcia-Espana, J.F.; Zemel, B.S.; Strandvik, B.; Stallings, V.A. Serum linoleic acid status as a clinical indicator of essential fatty acid status in children with cystic fibrosis. J. Pediatric Gastroenterol. Nutr. 2008, 47, 635–644. [Google Scholar] [CrossRef]
- Risé, P.; Volpi, S.; Colombo, C.; Padoan, R.F.; D’Orazio, C.; Ghezzi, S.; Melotti, P.; Bennato, V.; Agostoni, C.; Assael, B.M.; et al. Whole blood fatty acid analysis with micromethod in cystic fibrosis and pulmonary disease. J. Cyst. Fibros. 2010, 9, 228–233. [Google Scholar] [CrossRef]
- Lai, H.C.; Kosorok, M.R.; Laxova, A.; Davis, L.A.; FitzSimmon, S.C.; Farrell, P.M. Nutritional status of patients with cystic fibrosis with meconium ileus: A comparison with patients without meconium ileus and diagnosed early through neonatal screening. Pediatrics 2000, 105, 53–61. [Google Scholar] [CrossRef]
- Shoff, S.M.; Ahn, H.Y.; Davis, L.; Lai, H. Temporal associations among energy intake, plasma linoleic acid, and growth improvement in response to treatment initiation after diagnosis of cystic fibrosis. Pediatrics 2006, 117, 391–400. [Google Scholar] [CrossRef]
- Sanders, D.B.; Zhang, Z.; Farrell, P.M.; Lai, H.J. Early life growth patterns persist for 12 years and impact pulmonary outcomes in cystic fibrosis. J. Cyst. Fibros. 2018, 17, 528–535. [Google Scholar] [CrossRef]
- Strandvik, B.; Berg, U.; Kallner, A.; Kusoffsky, E. Effect on renal function of essential fatty acid supplementation in cystic fibrosis. J. Pediatric 1989, 115, 242–250. [Google Scholar] [CrossRef]
- Strandvik, B.; Hultcrantz, R. Liver function and morphology during long-term fatty acid supplementation in cystic fibrosis. Liver 1994, 14, 32–36. [Google Scholar] [CrossRef]
- Aldámiz-Echevarría, L.; Prieto, J.A.; Andrade, F.; Elorz, J.; Sojo, A.; Lage, S.; Sanjurjo, P.; Vázquez, C.; Rodríguez-Soriano, J. Persistence of essential fatty acid deficiency in cystic fibrosis despite nutritional therapy. Pediatric Res. 2009, 66, 585–589. [Google Scholar] [CrossRef]
- Lindblad, A.; Glaumann, H.; Strandvik, B. Natural history of liver disease in cystic fibrosis. Hepatology 1999, 30, 1151–1158. [Google Scholar] [CrossRef]
- Colombo, C.; Alicandro, G.; Oliver, M.; Lewindon, P.J.; Ramm, G.A.; Ooi, C.Y.; Alghisi, F.; Kashirskaya, N.; Kondratyeva, E.; Corti, F.; et al. Ursodeoxycholic acid and liver disease associated with cystic fibrosis: A multicenter cohort study. J. Cyst. Fibros. 2021, in press. [CrossRef]
- Harayama, T.; Riezman, H. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. [Google Scholar] [CrossRef]
- Strandvik, B.; Svensson, E.; Seyberth, H.W. Prostanoid biosynthesis in patients with cystic fibrosis. Prostaglandins Leukot. Essent. Fat. Acids 1996, 55, 419–425. [Google Scholar] [CrossRef]
- Dodge, J.A.; Custance, J.M.; Goodchild, M.C.; Laing, S.C.; Vaughan, M. Paradoxical effects of essential fatty acid supplementation on lipid profiles and sweat electrolytes in cystic fibrosis. Br. J. Nutr. 1990, 63, 259–271. [Google Scholar] [CrossRef]
- Van, D.; Beerthuis, R.K.; Nugteren, D.H.; Vonkeman, H. The biosynthesis of prostaglandins. Biochim. Biophys. Acta 1964, 90, 204–207. [Google Scholar] [CrossRef]
- Brenner, R.R.; Peluffo, R.O.; Nervi, A.M.; De Thomas, M.E. Competitive effect of alpha- and gamma-lionlenyl-CoA in linoleyl-CoA desaturation to gamma-linolenyl-CoA. Biochim. Biophys. Acta 1969, 176, 420–422. [Google Scholar] [CrossRef]
- Peluffo, R.O.; Nervi, A.M.; Brenner, R.R. Linoleic acid desaturation activity of liver microsomes of essential fatty acid deficient and sufficient rats. Biochim. Biophys. Acta 1976, 441, 25–31. [Google Scholar] [CrossRef]
- Seegmiller, A.C. Abnormal unsaturated fatty acid metabolism in cystic fibrosis: Biochemical mechanisms and clinical implications. Int. J. Mol. Sci. 2014, 15, 16083–16099. [Google Scholar] [CrossRef]
- Strandvik, B.; Brönnegård, M.; Gilljam, H.; Carlstedt-Duke, J. Relation between defective regulation of arachidonic acid release and symptoms in cystic fibrosis. Scand. J. Gastroenterol. Suppl. 1988, 143, 1–4. [Google Scholar] [CrossRef]
- Carlstedt-Duke, J.; Brönnegård, M.; Strandvik, B. Pathological regulation of arachidonic acid release in cystic fibrosis: The putative basic defect. Proc. Natl. Acad. Sci. USA 1986, 83, 9202–9206. [Google Scholar] [CrossRef]
- Levistre, R.; Lemnaouar, M.; Rybkine, T.; Béréziat, G.; Masliah, J. Increase of bradykinin-stimulated arachidonic acid release in a delta F508 cystic fibrosis epithelial cell line. Biochim. Biophys. Acta 1993, 1181, 233–239. [Google Scholar] [CrossRef]
- Berguerand, M.; Klapisz, E.; Thomas, G.; Humbert, L.; Jouniaux, A.M.; Olivier, J.L.; Béréziat, G.; Masliah, J. Differential stimulation of cytosolic phospholipase A2 by bradykinin in human cystic fibrosis cell lines. Am. J. Respir. Cell Mol. Biol. 1997, 17, 481–490. [Google Scholar] [CrossRef]
- Bhura-Bandali, F.N.; Suh, M.; Man, S.F.; Clandinin, M.T. The deltaF508 mutation in the cystic fibrosis transmembrane conductance regulator alters control of essential fatty acid utilization in epithelial cells. J. Nutr. 2000, 130, 2870–2875. [Google Scholar] [CrossRef]
- Miele, L.; Cordella-Miele, E.; Xing, M.; Frizzell, R.; Mukherjee, A.B. Cystic fibrosis gene mutation (deltaF508) is associated with an intrinsic abnormality in Ca2+-induced arachidonic acid release by epithelial cells. DNA Cell Biol. 1997, 16, 749–759. [Google Scholar] [CrossRef]
- Ulane, M.M.; Butler, J.D.; Peri, A.; Miele, L.; Ulane, R.E.; Hubbard, V.S. Cystic fibrosis and phosphatidylcholine biosynthesis. Clin. Chim. Acta 1994, 230, 109–116. [Google Scholar] [CrossRef]
- Strandvik, B. Long Chain Fatty Acid Metabolism and Essential Fatty Acid Deficiency with Special Emphasis on Cystic Fibrosis; Bracco, U., Deckelbaum, R., Eds.; Raven Press: New York, NY, USA, 1992; Volume 28, pp. 159–167. [Google Scholar]
- Strandvik, B. Relation between essential fatty acid metabolism and gastrointestinal symptoms in cystic fibrosis. Acta Paediatr. Scand. Suppl. 1989, 363, 58–63. [Google Scholar] [CrossRef]
- Van Biervliet, S.; Van Biervliet, J.P.; Robberecht, E.; Christophe, A. Fatty acid composition of serum phospholipids in cystic fibrosis (CF) patients with or without CF related liver disease. Clin. Chem. Lab. Med. 2010, 48, 1751–1755. [Google Scholar] [CrossRef]
- Drzymała-Czyż, S.; Szczepanik, M.; Krzyżanowska, P.; Duś-Żuchowska, M.; Pogorzelski, A.; Sapiejka, E.; Juszczak, P.; Lisowska, A.; Koletzko, B.; Walkowiak, J. Serum Phospholipid Fatty Acid Composition in Cystic Fibrosis Patients with and without Liver Cirrhosis. Ann. Nutr. Metab. 2017, 71, 91–98. [Google Scholar] [CrossRef]
- Beharry, S.; Ackerley, C.; Corey, M.; Kent, G.; Heng, Y.M.; Christensen, H.; Luk, C.; Yantiss, R.K.; Nasser, I.A.; Zaman, M.; et al. Long-term docosahexaenoic acid therapy in a congenic murine model of cystic fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G839–G848. [Google Scholar] [CrossRef]
- Freedman, S.D.; Katz, M.H.; Parker, E.M.; Laposata, M.; Urman, M.Y.; Alvarez, J.G. A membrane lipid imbalance plays a role in the phenotypic expression of cystic fibrosis in cftr(−/−) mice. Proc. Natl. Acad. Sci. USA 1999, 96, 13995–14000. [Google Scholar] [CrossRef]
- Watson, H.; Stackhouse, C. Omega-3 fatty acid supplementation for cystic fibrosis. Cochrane Database Syst. Rev. 2020, 4, Cd002201. [Google Scholar] [CrossRef]
- Wassall, S.R.; Leng, X.; Canner, S.W.; Pennington, E.R.; Kinnun, J.J.; Cavazos, A.T.; Dadoo, S.; Johnson, D.; Heberle, F.A.; Katsaras, J.; et al. Docosahexaenoic acid regulates the formation of lipid rafts: A unified view from experiment and simulation. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1985–1993. [Google Scholar] [CrossRef]
- Chiang, N.; Serhan, C.N. Specialized pro-resolving mediator network: An update on production and actions. Essays Biochem. 2020, 64, 443–462. [Google Scholar] [CrossRef]
- Philippe, R.; Urbach, V. Specialized Pro-Resolving Lipid Mediators in Cystic Fibrosis. Int. J. Mol. Sci. 2018, 19, 2865. [Google Scholar] [CrossRef]
- Recchiuti, A.; Isopi, E.; Romano, M.; Mattoscio, D. Roles of Specialized Pro-Resolving Lipid Mediators in Autophagy and Inflammation. Int. J. Mol. Sci. 2020, 21, 6637. [Google Scholar] [CrossRef]
- Mingione, A.; Ottaviano, E.; Barcella, M.; Merelli, I.; Rosso, L.; Armeni, T.; Cirilli, N.; Ghidoni, R.; Borghi, E.; Signorelli, P. Cystic Fibrosis Defective Response to Infection Involves Autophagy and Lipid Metabolism. Cells 2020, 9, 1845. [Google Scholar] [CrossRef] [PubMed]
- Isopi, E.; Mattoscio, D.; Codagnone, M.; Mari, V.C.; Lamolinara, A.; Patruno, S.; D’Aurora, M.; Cianci, E.; Nespoli, A.; Franchi, S.; et al. Resolvin D1 Reduces Lung Infection and Inflammation Activating Resolution in Cystic Fibrosis. Front. Immunol. 2020, 11, 581. [Google Scholar] [CrossRef] [PubMed]
- López-Neyra, A.; Suárez, L.; Muñoz, M.; de Blas, A.; Ruiz de Valbuena, M.; Garriga, M.; Calvo, J.; Ribes, C.; Girón Moreno, R.; Máiz, L.; et al. Long-term docosahexaenoic acid (DHA) supplementation in cystic fibrosis patients: A randomized, multi-center, double-blind, placebo-controlled trial. Prostaglandins Leukot. Essent. Fat. Acids 2020, 162, 102186. [Google Scholar] [CrossRef] [PubMed]
- Urbach, V.; Higgins, G.; Buchanan, P.; Ringholz, F. The role of Lipoxin A4 in Cystic Fibrosis Lung Disease. Comput. Struct. Biotechnol. J. 2013, 6, e201303018. [Google Scholar] [CrossRef][Green Version]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef]
- Van Biervliet, S.; Vanbillemont, G.; Van Biervliet, J.P.; Declercq, D.; Robberecht, E.; Christophe, A. Relation between fatty acid composition and clinical status or genotype in cystic fibrosis patients. Ann. Nutr. Metab. 2007, 51, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Welsh, M.J.; Smith, A.E. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 1993, 73, 1251–1254. [Google Scholar] [CrossRef]
- De Boeck, K.; Amaral, M.D. Progress in therapies for cystic fibrosis. Lancet Respir. Med. 2016, 4, 662–674. [Google Scholar] [CrossRef]
- Zardini Buzatto, A.; Abdel Jabar, M.; Nizami, I.; Dasouki, M.; Li, L.; Abdel Rahman, A.M. Lipidome Alterations Induced by Cystic Fibrosis, CFTR Mutation, and Lung Function. J. Proteome Res. 2021, 20, 549–564. [Google Scholar] [CrossRef]
- Guerrera, I.C.; Astarita, G.; Jais, J.P.; Sands, D.; Nowakowska, A.; Colas, J.; Sermet-Gaudelus, I.; Schuerenberg, M.; Piomelli, D.; Edelman, A.; et al. A novel lipidomic strategy reveals plasma phospholipid signatures associated with respiratory disease severity in cystic fibrosis patients. PLoS ONE 2009, 4, e7735. [Google Scholar] [CrossRef] [PubMed]
- Korotkova, M.; Strandvik, B. Essential fatty acid deficiency affects the fatty acid composition of the rat small intestinal and colonic mucosa differently. Biochim. Biophys. Acta 2000, 1487, 319–325. [Google Scholar] [CrossRef]
- Witters, P.; Dupont, L.; Vermeulen, F.; Proesmans, M.; Cassiman, D.; Wallemacq, P.; De Boeck, K. Lung transplantation in cystic fibrosis normalizes essential fatty acid profiles. J. Cyst. Fibros. 2013, 12, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Hanssens, L.; Duchateau, J.; Namane, S.A.; Malfroot, A.; Knoop, C.; Casimir, G. Influence of lung transplantation on the essential fatty acid profile in cystic fibrosis. Prostaglandins Leukot. Essent. Fat. Acids 2020, 158, 102060. [Google Scholar] [CrossRef]
- Parsons, H.G.; O’Loughlin, E.V.; Forbes, D.; Cooper, D.; Gall, D.G. Supplemental calories improve essential fatty acid deficiency in cystic fibrosis patients. Pediatric Res. 1988, 24, 353–356. [Google Scholar] [CrossRef]
- Rogiers, V.; Dab, I.; Crokaert, R.; Vis, H.L. Long chain non-esterified fatty acid pattern in plasma of cystic fibrosis patients and their parents. Pediatric Res. 1980, 14, 1088–1091. [Google Scholar] [CrossRef]
- Harindhanavudhi, T.; Wang, Q.; Dunitz, J.; Moran, A.; Moheet, A. Prevalence and factors associated with overweight and obesity in adults with cystic fibrosis: A single-center analysis. J. Cyst. Fibros. 2020, 19, 139–145. [Google Scholar] [CrossRef]
- Hanssens, L.S.; Duchateau, J.; Casimir, G.J. CFTR Protein: Not Just a Chloride Channel? Cells 2021, 10, 2844. [Google Scholar] [CrossRef]
- Nowak, J.K.; Szczepanik, M.; Wojsyk-Banaszak, I.; Mądry, E.; Wykrętowicz, A.; Krzyżanowska-Jankowska, P.; Drzymała-Czyż, S.; Nowicka, A.; Pogorzelski, A.; Sapiejka, E.; et al. Cystic fibrosis dyslipidaemia: A cross-sectional study. J. Cyst. Fibros. 2019, 18, 566–571. [Google Scholar] [CrossRef]
- Shaikhkhalil, A.K.; Freeman, A.J.; Sathe, M. Variations in Nutrition Practices in Cystic Fibrosis: A Survey of the DIGEST Program. Nutr. Clin. Pract. 2021, 36, 1247–1251. [Google Scholar] [CrossRef]
- Strandvik, B. Is the ENaC Dysregulation in CF an Effect of Protein-Lipid Interaction in the Membranes? Int. J. Mol. Sci. 2021, 22, 2739. [Google Scholar] [CrossRef] [PubMed]
- Rosenlund, M.L.; Selekman, J.A.; Kim, H.K.; Kritchevsky, D. Dietary essential fatty acids in cystic fibrosis. Pediatrics 1977, 59, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Sigström, L.; Strandvik, B. Erythrocyte sodium-potassium transport in cystic fibrosis. Pediatric Res. 1992, 31, 425–427. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Still, J.D.; Powers, C.A.; Hoffman, D.R.; Boyd-Trull, K.; Lester, L.A.; Benisek, D.C.; Arterburn, L.M. Bioavailability and safety of a high dose of docosahexaenoic acid triacylglycerol of algal origin in cystic fibrosis patients: A randomized, controlled study. Nutrition 2006, 22, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Alicandro, G.; Faelli, N.; Gagliardini, R.; Santini, B.; Magazzù, G.; Biffi, A.; Risé, P.; Galli, C.; Tirelli, A.S.; Loi, S.; et al. A randomized placebo-controlled study on high-dose oral algal docosahexaenoic acid supplementation in children with cystic fibrosis. Prostaglandins Leukot. Essent. Fat. Acids 2013, 88, 163–169. [Google Scholar] [CrossRef]
- Van Biervliet, S.; Devos, M.; Delhaye, T.; Van Biervliet, J.P.; Robberecht, E.; Christophe, A. Oral DHA supplementation in DeltaF508 homozygous cystic fibrosis patients. Prostaglandins Leukot. Essent. Fat. Acids 2008, 78, 109–115. [Google Scholar] [CrossRef]
- Simon, M.; Dalle Molle, R.; Silva, F.M.; Rodrigues, T.W.; Feldmann, M.; Forte, G.C.; Marostica, P.J.C. Antioxidant Micronutrients and Essential Fatty Acids Supplementation on Cystic Fibrosis Outcomes: A Systematic Review. J. Acad. Nutr. Diet. 2020, 120, 1016–1033. [Google Scholar] [CrossRef]
- Bass, R.; Brownell, J.N.; Stallings, V.A. The Impact of Highly Effective CFTR Modulators on Growth and Nutrition Status. Nutrients 2021, 13, 2907. [Google Scholar] [CrossRef]
- Harris, J.K.; Wagner, B.D.; Zemanick, E.T.; Robertson, C.E.; Stevens, M.J.; Heltshe, S.L.; Rowe, S.M.; Sagel, S.D. Changes in Airway Microbiome and Inflammation with Ivacaftor Treatment in Patients with Cystic Fibrosis and the G551D Mutation. Ann. Am. Thorac. Soc. 2020, 17, 212–220. [Google Scholar] [CrossRef]
- Veltman, M.; De Sanctis, J.B.; Stolarczyk, M.; Klymiuk, N.; Bähr, A.; Brouwer, R.W.; Oole, E.; Shah, J.; Ozdian, T.; Liao, J.; et al. CFTR Correctors and Antioxidants Partially Normalize Lipid Imbalance but not Abnormal Basal Inflammatory Cytokine Profile in CF Bronchial Epithelial Cells. Front. Physiol. 2021, 12, 619442. [Google Scholar] [CrossRef]
- Vertex. Pharmaceuticals; Incorporated. Prescribing Information. TRIKAFTA (Elexaccaftor/Tezacaftor/Ivacaftor; Ivacaftor Tablets. Table 4: Pharmacokinetic Parameters of TRIKAFTA Components. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/212273s000lbl.pdf (accessed on 10 February 2022).
- Laselva, O.; Guerra, L.; Castellani, S.; Favia, M.; Di Gioia, S.; Conese, M. Small-molecule drugs for cystic fibrosis: Where are we now? Pulm. Pharmacol. Ther. 2021, 72, 102098. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.R.; Gilljam, M.; Olesen, H.V.; Høiby, N.; Karpati, F.; Johansson, E.; Krantz, C.; Skov, M.; Pressler, T.; Lindblad, A. Maintaining normal lung function in children with cystic fibrosis is possible with aggressive treatment regardless of Pseudomonas aeruginosa infections. Acta Paediatr. 2021, 110, 2607–2609. [Google Scholar] [CrossRef] [PubMed]
- Garić, D.; Dumut, D.C.; Shah, J.; De Sanctis, J.B.; Radzioch, D. The role of essential fatty acids in cystic fibrosis and normalizing effect of fenretinide. Cell. Mol. Life Sci. 2020, 77, 4255–4267. [Google Scholar] [CrossRef] [PubMed]
- Vandebrouck, C.; Ferreira, T. Glued in lipids: Lipointoxication in cystic fibrosis. EBioMedicine 2020, 61, 103038. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.G.; Seegmiller, A. The effects of ivacaftor on CF fatty acid metabolism: An analysis from the GOAL study. J. Cyst. Fibros. 2017, 16, 132–138. [Google Scholar] [CrossRef]
- Strandvik, B.; Walkowiak, J.; Drzymala-Czyz, S.; Colombo, C.; Alicandro, A.; Bakkeheim, E.; Badolato, R.; Hansen, C. A double-blind randomised multi-centre European study of linoleic acid supplementation for one year in patients with cystic fibrosis. J. Cyst. Fibros. 2021, 20, S56. [Google Scholar] [CrossRef]


{kind=link}
| Fat | USA | Canada | UK | Belgium | Netherlands | Italy | Spain | Sweden |
|---|---|---|---|---|---|---|---|---|
| Soybean oil | 61.8 | 1.79 | 13.5 | 11.6 | 11.6 | 10.6 | 14.1 | 0.16 |
| Sunflower oil | 0.52 | 0.94 | 7.07 | 9.54 | 1.69 | 31.6 | 11.7 | 6.99 |
| Corn oil | 6.67 | 2.79 | 1.25 | 4.82 | 1.83 | 1.61 | 2.88 | 0.16 |
| Olive oil | 2.67 | 3.00 | 2.67 | 3.42 | 2.30 | 30.9 | 27.2 | 2.48 |
| Ratio n-6 richoils/olive oil | 25.8 | 1.84 | 8.17 | 7.60 | 6.73 | 1.42 | 1.05 | 2.95 |
| Cream + milk | 24.2 | 19.6 | 21.2 | 33.6 | 33.2 | 17.0 | 19.2 | 28.6 |
| Butter | 5.41 | 8.27 | 6.43 | ND | 13.31 | 5.5 | 2.78 | 11.3 |
| Animal fat, raw | 5.84 | 12.9 | 5.66 | 26.7 | 10.8 | 2.47 | 8.45 | 5.87 |
| Fish | 1.17 | 1.12 | 2.29 | 1.85 | 1.51 | 2.94 | 2.14 | 1.50 |
| Ratio n-6 rich oils/ dairy + animal fat | 2.30 | 0.17 | 0.81 | 0.43 | 0.35 | 2.25 | 1.04 | 0.21 |
| Ratio n-6 rich oils/ dairy + butter + animal fat | 1.95 | 0.14 | 0.66 | ND | 0.27 | 1.75 | 0.94 | 0.16 |
| Total fat, (based on presented fat categories) | 106 | 47.4 | 57.4 | 88.1+ | 74.3 | 71.7 | 61.3 | 54.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strandvik, B. Nutrition in Cystic Fibrosis—Some Notes on the Fat Recommendations. Nutrients 2022, 14, 853. https://doi.org/10.3390/nu14040853
Strandvik B. Nutrition in Cystic Fibrosis—Some Notes on the Fat Recommendations. Nutrients. 2022; 14(4):853. https://doi.org/10.3390/nu14040853
Chicago/Turabian StyleStrandvik, Birgitta. 2022. "Nutrition in Cystic Fibrosis—Some Notes on the Fat Recommendations" Nutrients 14, no. 4: 853. https://doi.org/10.3390/nu14040853
APA StyleStrandvik, B. (2022). Nutrition in Cystic Fibrosis—Some Notes on the Fat Recommendations. Nutrients, 14(4), 853. https://doi.org/10.3390/nu14040853