
Lipid Metabolism and Epigenetics Crosstalk in Prostate Cancer


 , ,
, , 
Abstract
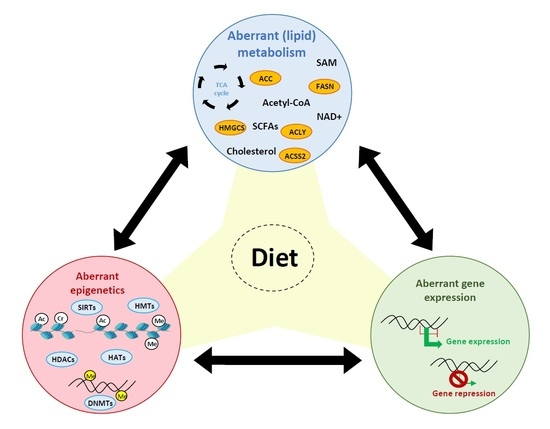
:
1. Introduction
2. Lipid Metabolism Alterations: A Hallmark of PCa
2.1. Alterations of FA Metabolism in PCa
2.1.1. De Novo FA Synthesis
2.1.2. FA Uptake
2.1.3. FA Oxidation (FAO)
2.1.4. FA Storage and Release
2.2. Alterations of Cholesterol Metabolism in PCa
2.3. Alterations of Phospholipid Metabolism in PCa
2.4. Targeting Metabolic Vulnerabilities in PCa
3. The Epigenome of PCa
3.1. Alterations of DNA Methylation in PCa
3.2. Alterations of Histone Modifications in PCa
4. Influence of Diet on PCa, Lipid Metabolism and the Epigenome
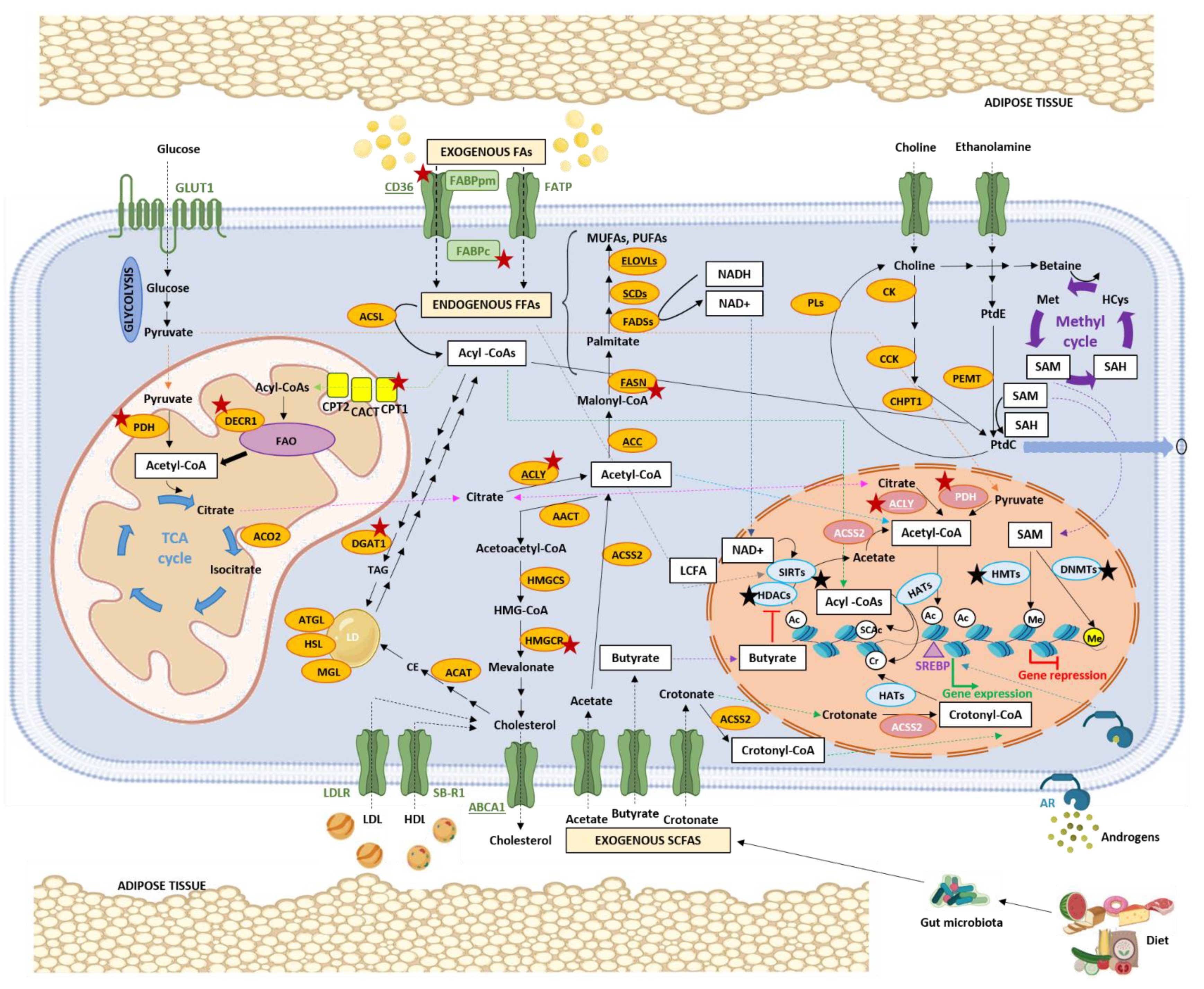
5. Interaction between Lipid Metabolism and Epigenetics in PCa
5.1. Impact of Lipid Metabolism on Epigenetics
5.1.1. FAs, Acetyl-CoA, and Histone Acetylation
5.1.2. SCFAs, Non-Acetyl Acyl-CoAs, and Histone Acylation
5.1.3. SCFAs and Histone Deacetylases
5.1.4. FAs and Sirtuins
5.1.5. Phospholipids, SAM, and Histone Methylation
5.1.6. Nuclear Lipogenic Enzymes
5.2. Impact of Epigenetics on Lipid Metabolism
6. Concluding Remarks and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandhu, S.; Moore, C.M.; Chiong, E.; Beltran, H.; Bristow, R.G.; Williams, S.G. Prostate cancer. Lancet 2021, 398, 1075–1090. [Google Scholar] [CrossRef]
- Gravis, G.; Boher, J.-M.; Chen, Y.-H.; Liu, G.; Fizazi, K.; Carducci, M.A.; Oudard, S.; Joly, F.; Jarrard, D.M.; Soulie, M.; et al. Burden of Metastatic Castrate Naive Prostate Cancer Patients, to Identify Men More Likely to Benefit from Early Docetaxel: Further Analyses of CHAARTED and GETUG-AFU15 Studies. Eur. Urol. 2018, 73, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Francini, E.; Gray, K.P.; Xie, W.; Shaw, G.K.; Valença, L.; Bernard, B.; Albiges, L.; Harshman, L.C.; Kantoff, P.W.; Taplin, M.-E.; et al. Time of metastatic disease presentation and volume of disease are prognostic for metastatic hormone sensitive prostate cancer (mHSPC). Prostate 2018, 78, 889–895. [Google Scholar] [CrossRef]
- Debes, J.D.; Tindall, D.J. The role of androgens and the androgen receptor in prostate cancer. Cancer Lett. 2002, 187, 1–7. [Google Scholar] [CrossRef]
- Davies, A.; Conteduca, V.; Zoubeidi, A.; Beltran, H. Biological Evolution of Castration-resistant Prostate Cancer. Eur. Urol. Focus 2019, 5, 147–154. [Google Scholar] [CrossRef]
- Sartor, O.; de Bono, J.S. Metastatic Prostate Cancer. N. Engl. J. Med. 2018, 378, 645–657. [Google Scholar] [CrossRef]
- de Bono, J.S.; Oudard, S.; Ozguroglu, M.; Hansen, S.; Machiels, J.-P.; Kocak, I.; Gravis, G.; Bodrogi, I.; Mackenzie, M.J.; Shen, L.; et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: A randomised open-label trial. Lancet 2010, 376, 1147–1154. [Google Scholar] [CrossRef]
- de Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B.; Saad, F.; et al. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef]
- Ryan, C.J.; Smith, M.R.; de Bono, J.S.; Molina, A.; Logothetis, C.J.; de Souza, P.; Fizazi, K.; Mainwaring, P.; Piulats, J.M.; Ng, S.; et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N. Engl. J. Med. 2013, 368, 138–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Özgüroğlu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone plus Prednisone in Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2017, 377, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.E.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Bhattacharya, S.; Carles, J.; Chowdhury, S.; et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N. Engl. J. Med. 2014, 371, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, I.D.; Martin, A.J.; Stockler, M.R.; Begbie, S.; Chi, K.N.; Chowdhury, S.; Coskinas, X.; Frydenberg, M.; Hague, W.E.; Horvath, L.G.; et al. Enzalutamide with Standard First-Line Therapy in Metastatic Prostate Cancer. N. Engl. J. Med. 2019, 381, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.-E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, M.; Fizazi, K.; Saad, F.; Rathenborg, P.; Shore, N.; Ferreira, U.; Ivashchenko, P.; Demirhan, E.; Modelska, K.; Phung, D.; et al. Enzalutamide in Men with Nonmetastatic, Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2018, 378, 2465–2474. [Google Scholar] [CrossRef]
- Smith, M.R.; Saad, F.; Chowdhury, S.; Oudard, S.; Hadaschik, B.A.; Graff, J.N.; Olmos, D.; Mainwaring, P.N.; Lee, J.Y.; Uemura, H.; et al. Apalutamide Treatment and Metastasis-free Survival in Prostate Cancer. N. Engl. J. Med. 2018, 378, 1408–1418. [Google Scholar] [CrossRef]
- Chi, K.N.; Agarwal, N.; Bjartell, A.; Chung, B.H.; Pereira de Santana Gomes, A.J.; Given, R.; Juárez Soto, Á.; Merseburger, A.S.; Özgüroğlu, M.; Uemura, H.; et al. Apalutamide for Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2019, 381, 13–24. [Google Scholar] [CrossRef]
- Fizazi, K.; Shore, N.; Tammela, T.L.; Ulys, A.; Vjaters, E.; Polyakov, S.; Jievaltas, M.; Luz, M.; Alekseev, B.; Kuss, I.; et al. Darolutamide in Nonmetastatic, Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2019, 380, 1235–1246. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Ci, X.; Choi, S.Y.C.; Crea, F.; Lin, D.; Wang, Y. Molecular events in neuroendocrine prostate cancer development. Nat. Rev. Urol. 2021, 18, 581–596. [Google Scholar] [CrossRef]
- Perry, A.S.; Watson, R.W.G.; Lawler, M.; Hollywood, D. The epigenome as a therapeutic target in prostate cancer. Nat. Rev. Urol. 2010, 7, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.S.K.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Dent, S.Y.R. Chromatin modifiers and remodellers: Regulators of cellular differentiation. Nat. Rev. Genet. 2014, 15, 93–106. [Google Scholar] [CrossRef] [Green Version]
- Berdasco, M.; Esteller, M. Aberrant epigenetic landscape in cancer: How cellular identity goes awry. Dev. Cell 2010, 19, 698–711. [Google Scholar] [CrossRef] [Green Version]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome-biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Aguilera, O.; Fernández, A.F.; Muñoz, A.; Fraga, M.F. Epigenetics and environment: A complex relationship. J. Appl. Physiol. 2010, 109, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Labbé, D.P.; Zadra, G.; Ebot, E.M.; Mucci, L.A.; Kantoff, P.W.; Loda, M.; Brown, M. Role of diet in prostate cancer: The epigenetic link. Oncogene 2015, 34, 4683–4691. [Google Scholar] [CrossRef] [Green Version]
- Narita, S.; Tsuchiya, N.; Saito, M.; Inoue, T.; Kumazawa, T.; Yuasa, T.; Nakamura, A.; Habuchi, T. Candidate genes involved in enhanced growth of human prostate cancer under high fat feeding identified by microarray analysis. Prostate 2008, 68, 321–335. [Google Scholar] [CrossRef]
- Llaverias, G.; Danilo, C.; Wang, Y.; Witkiewicz, A.K.; Daumer, K.; Lisanti, M.P.; Frank, P.G. A Western-type diet accelerates tumor progression in an autochthonous mouse model of prostate cancer. Am. J. Pathol. 2010, 177, 3180–3191. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.; Venkateswaran, V.; Fleshner, N.E.; Klotz, L.H.; Cox, M.E. The impact of diet and micronutrient supplements on the expression of neuroendocrine markers in murine Lady transgenic prostate. Prostate 2008, 68, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Ferro, M.; Terracciano, D.; Buonerba, C.; Lucarelli, G.; Bottero, D.; Perdonà, S.; Autorino, R.; Serino, A.; Cantiello, F.; Damiano, R.; et al. The emerging role of obesity, diet and lipid metabolism in prostate cancer. Futur. Oncol. 2017, 13, 285–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buschemeyer, W.C.; Freedland, S.J. Obesity and prostate cancer: Epidemiology and clinical implications. Eur. Urol. 2007, 52, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, J.V.; Heemers, H.; van de Sande, T.; de Schrijver, E.; Brusselmans, K.; Heyns, W.; Verhoeven, G. Androgens, lipogenesis and prostate cancer. J. Steroid Biochem. Mol. Biol. 2004, 92, 273–279. [Google Scholar] [CrossRef]
- Scaglia, N.; Frontini-López, Y.R.; Zadra, G. Prostate Cancer Progression: As a Matter of Fats. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Muoio, D.M. Metabolic inflexibility: When mitochondrial indecision leads to metabolic gridlock. Cell 2014, 159, 1253–1262. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.A.; Zubair, H.; Anand, S.; Srivastava, S.K.; Singh, S.; Singh, A.P. Dysregulation of metabolic enzymes in tumor and stromal cells: Role in oncogenesis and therapeutic opportunities. Cancer Lett. 2020, 473, 176–185. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, D.A.; McGuire, S.E. Tumour metabolism and its unique properties in prostate adenocarcinoma. Nat. Rev. Urol. 2020, 17, 214–231. [Google Scholar] [CrossRef]
- Costello, L.C.; Franklin, R.B. Prostate epithelial cells utilize glucose and aspartate as the carbon sources for net citrate production. Prostate 1989, 15, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Uo, T.; Sprenger, C.C.; Plymate, S.R. Androgen Receptor Signaling and Metabolic and Cellular Plasticity During Progression to Castration Resistant Prostate Cancer. Front. Oncol. 2020, 10, 580617. [Google Scholar] [CrossRef]
- Richter, J.A.; Rodríguez, M.; Rioja, J.; Peñuelas, I.; Martí-Climent, J.; Garrastachu, P.; Quincoces, G.; Zudaire, J.; García-Velloso, M.J. Dual tracer 11C-choline and FDG-PET in the diagnosis of biochemical prostate cancer relapse after radical treatment. Mol. imaging Biol. 2010, 12, 210–217. [Google Scholar] [CrossRef]
- Jadvar, H.; Desai, B.; Ji, L.; Conti, P.S.; Dorff, T.B.; Groshen, S.G.; Gross, M.E.; Pinski, J.K.; Quinn, D.I. Prospective evaluation of 18F-NaF and 18F-FDG PET/CT in detection of occult metastatic disease in biochemical recurrence of prostate cancer. Clin. Nucl. Med. 2012, 37, 637–643. [Google Scholar] [CrossRef] [Green Version]
- Costello, L.C.; Franklin, R.B. A comprehensive review of the role of zinc in normal prostate function and metabolism; and its implications in prostate cancer. Arch. Biochem. Biophys. 2016, 611, 100–112. [Google Scholar] [CrossRef] [Green Version]
- Bader, D.A.; Hartig, S.M.; Putluri, V.; Foley, C.; Hamilton, M.P.; Smith, E.A.; Saha, P.K.; Panigrahi, A.; Walker, C.; Zong, L.; et al. Mitochondrial pyruvate import is a metabolic vulnerability in androgen receptor-driven prostate cancer. Nat. Metab. 2019, 1, 70–85. [Google Scholar] [CrossRef]
- Chetta, P.; Zadra, G. Metabolic reprogramming as an emerging mechanism of resistance to endocrine therapies in prostate cancer. Cancer Drug Resist. 2021. [Google Scholar] [CrossRef]
- Papachristodoulou, A.; Rodriguez-Calero, A.; Panja, S.; Margolskee, E.; Virk, R.K.; Milner, T.A.; Martina, L.P.; Kim, J.Y.; Di Bernardo, M.; Williams, A.B.; et al. NKX3.1 Localization to Mitochondria Suppresses Prostate Cancer Initiation. Cancer Discov. 2021, 11, 2316–2333. [Google Scholar] [CrossRef] [PubMed]
- Heemers, H.V.; Verhoeven, G.; Swinnen, J.V. Androgen Activation of the Sterol Regulatory Element-Binding Protein Pathway: Current Insights. Mol. Endocrinol. 2006, 20, 2265–2277. [Google Scholar] [CrossRef]
- Li, X.; Wu, J.B.; Chung, L.W.K.; Huang, W.-C. Anti-cancer efficacy of SREBP inhibitor, alone or in combination with docetaxel, in prostate cancer harboring p53 mutations. Oncotarget 2015, 6, 41018–41032. [Google Scholar] [CrossRef] [PubMed]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.-L.; Schulze, A. SREBP Activity Is Regulated by mTORC1 and Contributes to Akt-Dependent Cell Growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Zhang, J.; Sampieri, K.; Clohessy, J.G.; Mendez, L.; Gonzalez-Billalabeitia, E.; Liu, X.-S.; Lee, Y.-R.; Fung, J.; Katon, J.M.; et al. An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat. Genet. 2018, 50, 206–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Röhrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef]
- Hamada, S.; Horiguchi, A.; Kuroda, K.; Ito, K.; Asano, T.; Miyai, K.; Iwaya, K. Increased fatty acid synthase expression in prostate biopsy cores predicts higher Gleason score in radical prostatectomy specimen. BMC Clin. Pathol. 2014, 14, 3. [Google Scholar] [CrossRef] [Green Version]
- Fritz, V.; Benfodda, Z.; Rodier, G.; Henriquet, C.; Iborra, F.; Avancès, C.; Allory, Y.; de la Taille, A.; Culine, S.; Blancou, H.; et al. Abrogation of De novo Lipogenesis by Stearoyl-CoA Desaturase 1 Inhibition Interferes with Oncogenic Signaling and Blocks Prostate Cancer Progression in Mice. Mol. Cancer Ther. 2010, 9, 1740–1754. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Makino, A.; Hullin-Matsuda, F.; Kobayashi, T.; Furihata, M.; Chung, S.; Ashida, S.; Miki, T.; Fujioka, T.; Shuin, T.; et al. Novel Lipogenic Enzyme ELOVL7 Is Involved in Prostate Cancer Growth through Saturated Long-Chain Fatty Acid Metabolism. Cancer Res. 2009, 69, 8133–8140. [Google Scholar] [CrossRef] [Green Version]
- Zadra, G.; Ribeiro, C.F.; Chetta, P.; Ho, Y.; Cacciatore, S.; Gao, X.; Syamala, S.; Bango, C.; Photopoulos, C.; Huang, Y.; et al. Inhibition of de novo lipogenesis targets androgen receptor signaling in castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 631–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.; Carriveau, W.J.; Li, J.; Campbell, S.L.; Kopinski, P.K.; Lim, H.-W.; Daurio, N.; Trefely, S.; Won, K.-J.; Wallace, D.C.; et al. Targeting ACLY sensitizes castration-resistant prostate cancer cells to AR antagonism by impinging on an ACLY-AMPK-AR feedback mechanism. Oncotarget 2016, 7, 43713–43730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushita, M.; Fujita, K.; Nonomura, N. Influence of Diet and Nutrition on Prostate Cancer. Int. J. Mol. Sci. 2020, 21, 1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tousignant, K.D.; Rockstroh, A.; Taherian Fard, A.; Lehman, M.L.; Wang, C.; McPherson, S.J.; Philp, L.K.; Bartonicek, N.; Dinger, M.E.; Nelson, C.C.; et al. Lipid Uptake Is an Androgen-Enhanced Lipid Supply Pathway Associated with Prostate Cancer Disease Progression and Bone Metastasis. Mol. Cancer Res. 2019, 17, 1166–1179. [Google Scholar] [CrossRef] [Green Version]
- Febbraio, M.; Hajjar, D.P.; Silverstein, R.L. CD36: A class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J. Clin. Investig. 2001, 108, 785–791. [Google Scholar] [CrossRef]
- Pascual, G.; Domínguez, D.; Elosúa-Bayes, M.; Beckedorff, F.; Laudanna, C.; Bigas, C.; Douillet, D.; Greco, C.; Symeonidi, A.; Hernández, I.; et al. Dietary palmitic acid promotes a prometastatic memory via Schwann cells. Nature 2021, 599, 485–490. [Google Scholar] [CrossRef]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martín, M.; Castellanos, A.; Attolini, C.S.-O.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef]
- Pan, J.; Fan, Z.; Wang, Z.; Dai, Q.; Xiang, Z.; Yuan, F.; Yan, M.; Zhu, Z.; Liu, B.; Li, C. CD36 mediates palmitate acid-induced metastasis of gastric cancer via AKT/GSK-3β/β-catenin pathway. J. Exp. Clin. Cancer Res. 2019, 38, 52. [Google Scholar] [CrossRef] [Green Version]
- Watt, M.J.; Clark, A.K.; Selth, L.A.; Haynes, V.R.; Lister, N.; Rebello, R.; Porter, L.H.; Niranjan, B.; Whitby, S.T.; Lo, J.; et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Stremmel, W.; Strohmeyer, G.; Borchard, F.; Kochwa, S.; Berk, P.D. Isolation and partial characterization of a fatty acid binding protein in rat liver plasma membranes. Proc. Natl. Acad. Sci. USA 1985, 82, 4–8. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S.; Bernlohr, D.A. Metabolic functions of FABPs—mechanisms and therapeutic implications. Nat. Rev. Endocrinol. 2015, 11, 592–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, R.; Hammamieh, R.; Neill, R.; Melhem, M.; Jett, M. Expression pattern of fatty acid-binding proteins in human normal and cancer prostate cells and tissues. Clin. Cancer Res. 2001, 7, 1706–1715. [Google Scholar] [PubMed]
- O’Reilly, M.W.; House, P.J.; Tomlinson, J.W. Understanding androgen action in adipose tissue. J. Steroid Biochem. Mol. Biol. 2014, 143, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Pinthus, J.H.; Lu, J.-P.; Bidaisee, L.A.; Lin, H.; Bryskine, I.; Gupta, R.S.; Singh, G. Androgen-dependent regulation of medium and long chain fatty acids uptake in prostate cancer. Prostate 2007, 67, 1330–1338. [Google Scholar] [CrossRef]
- Wajchenberg, B.L. Subcutaneous and visceral adipose tissue: Their relation to the metabolic syndrome. Endocr. Rev. 2000, 21, 697–738. [Google Scholar] [CrossRef]
- Nguyen, P.L.; Alibhai, S.M.H.; Basaria, S.; D’Amico, A.V.; Kantoff, P.W.; Keating, N.L.; Penson, D.F.; Rosario, D.J.; Tombal, B.; Smith, M.R. Adverse effects of androgen deprivation therapy and strategies to mitigate them. Eur. Urol. 2015, 67, 825–836. [Google Scholar] [CrossRef]
- Forootan, F.S.; Forootan, S.S.; Malki, M.I.; Chen, D.; Li, G.; Lin, K.; Rudland, P.S.; Foster, C.S.; Ke, Y. The expression of C-FABP and PPARγ and their prognostic significance in prostate cancer. Int. J. Oncol. 2014, 44, 265–275. [Google Scholar] [CrossRef]
- Adamson, J.; Morgan, E.A.; Beesley, C.; Mei, Y.; Foster, C.S.; Fujii, H.; Rudland, P.S.; Smith, P.H.; Ke, Y. High-level expression of cutaneous fatty acid-binding protein in prostatic carcinomas and its effect on tumorigenicity. Oncogene 2003, 22, 2739–2749. [Google Scholar] [CrossRef] [Green Version]
- Carbonetti, G.; Converso, C.; Clement, T.; Wang, C.; Trotman, L.C.; Ojima, I.; Kaczocha, M. Docetaxel/cabazitaxel and fatty acid binding protein 5 inhibitors produce synergistic inhibition of prostate cancer growth. Prostate 2020, 80, 88–98. [Google Scholar] [CrossRef]
- Liu, Y. Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis. 2006, 9, 230–234. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Temkin, S.M.; Hawkridge, A.M.; Guo, C.; Wang, W.; Wang, X.-Y.; Fang, X. Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer Lett. 2018, 435, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Shao, Y.; Zhao, X.; Hong, C.S.; Wang, F.; Lu, X.; Li, J.; Ye, G.; Yan, M.; Zhuang, Z.; et al. Integration of Metabolomics and Transcriptomics Reveals Major Metabolic Pathways and Potential Biomarker Involved in Prostate Cancer. Mol. Cell. Proteom. 2016, 15, 154–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlaepfer, I.R.; Rider, L.; Rodrigues, L.U.; Gijón, M.A.; Pac, C.T.; Romero, L.; Cimic, A.; Sirintrapun, S.J.; Glodé, L.M.; Eckel, R.H.; et al. Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Mol. Cancer Ther. 2014, 13, 2361–2371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nassar, Z.D.; Mah, C.Y.; Dehairs, J.; Burvenich, I.J.; Irani, S.; Centenera, M.M.; Helm, M.; Shrestha, R.K.; Moldovan, M.; Don, A.S.; et al. Human DECR1 is an androgen-repressed survival factor that regulates PUFA oxidation to protect prostate tumor cells from ferroptosis. Elife 2020, 9, e54166. [Google Scholar] [CrossRef]
- Walther, T.C.; Farese, R.V. Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem. 2012, 81, 687–714. [Google Scholar] [CrossRef] [Green Version]
- Bersuker, K.; Olzmann, J.A. Establishing the lipid droplet proteome: Mechanisms of lipid droplet protein targeting and degradation. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 2017, 1862, 1166–1177. [Google Scholar] [CrossRef]
- Petan, T.; Jarc, E.; Jusović, M. Lipid Droplets in Cancer: Guardians of Fat in a Stressful World. Molecules 2018, 23, 1941. [Google Scholar] [CrossRef] [Green Version]
- Swinnen, J.V.; Verhoeven, G. Androgens and the control of lipid metabolism in human prostate cancer cells. J. Steroid Biochem. Mol. Biol. 1998, 65, 191–198. [Google Scholar] [CrossRef]
- Nardi, F.; Franco, O.E.; Fitchev, P.; Morales, A.; Vickman, R.E.; Hayward, S.W.; Crawford, S.E. DGAT1 Inhibitor Suppresses Prostate Tumor Growth and Migration by Regulating Intracellular Lipids and Non-Centrosomal MTOC Protein GM130. Sci. Rep. 2019, 9, 3035. [Google Scholar] [CrossRef]
- Smirnova, E.; Goldberg, E.B.; Makarova, K.S.; Lin, L.; Brown, W.J.; Jackson, C.L. ATGL has a key role in lipid droplet/adiposome degradation in mammalian cells. EMBO Rep. 2006, 7, 106–113. [Google Scholar] [CrossRef]
- Mitra, R.; Le, T.T.; Gorjala, P.; Goodman, O.B. Positive regulation of prostate cancer cell growth by lipid droplet forming and processing enzymes DGAT1 and ABHD5. BMC Cancer 2017, 17, 631. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zhou, G.; Aras, S.; He, Z.; Lucas, S.; Podgorski, I.; Skar, W.; Granneman, J.G.; Wang, J. Loss of ABHD5 promotes the aggressiveness of prostate cancer cells. Sci. Rep. 2017, 7, 13021. [Google Scholar] [CrossRef] [Green Version]
- Anderson, L.A.; McTernan, P.G.; Harte, A.L.; Barnett, A.H.; Kumar, S. The regulation of HSL and LPL expression by DHT and flutamide in human subcutaneous adipose tissue. Diabetes. Obes. Metab. 2002, 4, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Chen, S.; Ng, P.; Bubley, G.J.; Nelson, P.S.; Mostaghel, E.A.; Marck, B.; Matsumoto, A.M.; Simon, N.I.; Wang, H.; et al. Intratumoral De Novo Steroid Synthesis Activates Androgen Receptor in Castration-Resistant Prostate Cancer and Is Upregulated by Treatment with CYP17A1 Inhibitors. Cancer Res. 2011, 71, 6503–6513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomura, D.K.; Long, J.Z.; Niessen, S.; Hoover, H.S.; Ng, S.-W.; Cravatt, B.F. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 2010, 140, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Nomura, D.K.; Lombardi, D.P.; Chang, J.W.; Niessen, S.; Ward, A.M.; Long, J.Z.; Hoover, H.H.; Cravatt, B.F. Monoacylglycerol Lipase Exerts Dual Control over Endocannabinoid and Fatty Acid Pathways to Support Prostate Cancer. Chem. Biol. 2011, 18, 846–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostaghel, E.A. Steroid hormone synthetic pathways in prostate cancer. Transl. Androl. Urol. 2013, 2, 212–227. [Google Scholar] [CrossRef]
- Huang, B.; Song, B.; Xu, C. Cholesterol metabolism in cancer: Mechanisms and therapeutic opportunities. Nat. Metab. 2020, 2, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Pelton, K.; Freeman, M.R.; Solomon, K.R. Cholesterol and prostate cancer. Curr. Opin. Pharmacol. 2012, 12, 751–759. [Google Scholar] [CrossRef] [Green Version]
- Caro-Maldonado, A.; Camacho, L.; Zabala-Letona, A.; Torrano, V.; Fernández-Ruiz, S.; Zamacola-Bascaran, K.; Arreal, L.; Valcárcel-Jiménez, L.; Martín-Martín, N.; Flores, J.M.; et al. Low-dose statin treatment increases prostate cancer aggressiveness. Oncotarget 2018, 9, 1494–1504. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, J.L.; Brown, M.S. The LDL receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stopsack, K.H.; Gerke, T.A.; Andrén, O.; Andersson, S.-O.; Giovannucci, E.L.; Mucci, L.A.; Rider, J.R. Cholesterol uptake and regulation in high-grade and lethal prostate cancers. Carcinogenesis 2017, 38, 806–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schörghofer, D.; Kinslechner, K.; Preitschopf, A.; Schütz, B.; Röhrl, C.; Hengstschläger, M.; Stangl, H.; Mikula, M. The HDL receptor SR-BI is associated with human prostate cancer progression and plays a possible role in establishing androgen independence. Reprod. Biol. Endocrinol. 2015, 13, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, S.; Li, J.; Lee, S.-Y.; Lee, H.J.; Shao, T.; Song, B.; Cheng, L.; Masterson, T.A.; Liu, X.; Ratliff, T.L.; et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 2014, 19, 393–406. [Google Scholar] [CrossRef] [Green Version]
- Marín-Aguilera, M.; Pereira, M.V.; Jiménez, N.; Reig, Ò.; Cuartero, A.; Victoria, I.; Aversa, C.; Ferrer-Mileo, L.; Prat, A.; Mellado, B. Glutamine and Cholesterol Plasma Levels and Clinical Outcomes of Patients with Metastatic Castration-Resistant Prostate Cancer Treated with Taxanes. Cancers 2021, 13, 4960. [Google Scholar] [CrossRef]
- Tarling, E.J.; de Aguiar Vallim, T.Q.; Edwards, P.A. Role of ABC transporters in lipid transport and human disease. Trends Endocrinol. Metab. 2013, 24, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Moon, S.-H.; Huang, C.-H.; Houlihan, S.L.; Regunath, K.; Freed-Pastor, W.A.; Morris, J.P.; Tschaharganeh, D.F.; Kastenhuber, E.R.; Barsotti, A.M.; Culp-Hill, R.; et al. p53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell 2019, 176, 564–580.e19. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Tontonoz, P. Phospholipid Remodeling in Physiology and Disease. Annu. Rev. Physiol. 2019, 81, 165–188. [Google Scholar] [CrossRef]
- Swinnen, J.V.; Van Veldhoven, P.P.; Timmermans, L.; De Schrijver, E.; Brusselmans, K.; Vanderhoydonc, F.; Van de Sande, T.; Heemers, H.; Heyns, W.; Verhoeven, G. Fatty acid synthase drives the synthesis of phospholipids partitioning into detergent-resistant membrane microdomains. Biochem. Biophys. Res. Commun. 2003, 302, 898–903. [Google Scholar] [CrossRef]
- Glunde, K.; Bhujwalla, Z.M.; Ronen, S.M. Choline metabolism in malignant transformation. Nat. Rev. Cancer 2011, 11, 835–848. [Google Scholar] [CrossRef] [Green Version]
- Awwad, H.M.; Geisel, J.; Obeid, R. The role of choline in prostate cancer. Clin. Biochem. 2012, 45, 1548–1553. [Google Scholar] [CrossRef] [PubMed]
- Fuccio, C.; Schiavina, R.; Castellucci, P.; Rubello, D.; Martorana, G.; Celli, M.; Malizia, C.; Profitos, M.B.; Marzola, M.C.; Pettinato, V.; et al. Androgen deprivation therapy influences the uptake of 11C-choline in patients with recurrent prostate cancer: The preliminary results of a sequential PET/CT study. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1985–1989. [Google Scholar] [CrossRef] [PubMed]
- von Eyben, F.E.; Kairemo, K. Meta-analysis of (11)C-choline and (18)F-choline PET/CT for management of patients with prostate cancer. Nucl. Med. Commun. 2014, 35, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, E.P.; Weiss, S.B. The function of cytidine coenzymes in the biosynthesis of phospholipides. J. Biol. Chem. 1956, 222, 193–214. [Google Scholar] [CrossRef]
- Ramírez de Molina, A.; Rodríguez-González, A.; Gutiérrez, R.; Martínez-Piñeiro, L.; Sánchez, J.; Bonilla, F.; Rosell, R.; Lacal, J. Overexpression of choline kinase is a frequent feature in human tumor-derived cell lines and in lung, prostate, and colorectal human cancers. Biochem. Biophys. Res. Commun. 2002, 296, 580–583. [Google Scholar] [CrossRef]
- Hemdan, T.; Turker, P.; Malmström, P.-U.; Segersten, U. Choline-phosphate cytidylyltransferase-α as a possible predictor of survival and response to cisplatin neoadjuvant chemotherapy in urothelial cancer of the bladder. Scand. J. Urol. 2018, 52, 200–205. [Google Scholar] [CrossRef]
- Jia, M.; Andreassen, T.; Jensen, L.; Bathen, T.F.; Sinha, I.; Gao, H.; Zhao, C.; Haldosen, L.-A.; Cao, Y.; Girnita, L.; et al. Estrogen Receptor α Promotes Breast Cancer by Reprogramming Choline Metabolism. Cancer Res. 2016, 76, 5634–5646. [Google Scholar] [CrossRef] [Green Version]
- Wen, S.; He, Y.; Wang, L.; Zhang, J.; Quan, C.; Niu, Y.; Huang, H. Aberrant activation of super enhancer and choline metabolism drive antiandrogen therapy resistance in prostate cancer. Oncogene 2020, 39, 6556–6571. [Google Scholar] [CrossRef]
- Raittinen, P.V.H.; Syvälä, H.; Tammela, T.L.J.; Häkkinen, M.R.; Ilmonen, P.; Auriola, S.; Murtola, T.J. Atorvastatin induces adrenal androgen downshift in men with prostate cancer: A post Hoc analysis of a pilot adaptive Randomised clinical trial. EBioMedicine 2021, 68, 103432. [Google Scholar] [CrossRef]
- Jeong, I.G.; Lim, B.; Yun, S.-C.; Lim, J.H.; Hong, J.H.; Kim, C.-S. Adjuvant Low-dose Statin Use after Radical Prostatectomy: The PRO-STAT Randomized Clinical Trial. Clin. Cancer Res. 2021, 27, 5004–5011. [Google Scholar] [CrossRef]
- Longo, J.; Hamilton, R.J.; Masoomian, M.; Khurram, N.; Branchard, E.; Mullen, P.J.; Elbaz, M.; Hersey, K.; Chadwick, D.; Ghai, S.; et al. A pilot window-of-opportunity study of preoperative fluvastatin in localized prostate cancer. Prostate Cancer Prostatic Dis. 2020, 23, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Falchook, G.; Infante, J.; Arkenau, H.-T.; Patel, M.R.; Dean, E.; Borazanci, E.; Brenner, A.; Cook, N.; Lopez, J.; Pant, S.; et al. First-in-human study of the safety, pharmacokinetics, and pharmacodynamics of first-in-class fatty acid synthase inhibitor TVB-2640 alone and with a taxane in advanced tumors. EClinicalMedicine 2021, 34, 100797. [Google Scholar] [CrossRef] [PubMed]
- Ct, W.; Morris, J.R. Genes, genetics, and epigenetics: A correspondence. Science 2001, 293, 1103–1105. [Google Scholar] [CrossRef] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Holoch, D.; Moazed, D. RNA-mediated epigenetic regulation of gene expression. Nat. Rev. Genet. 2015, 16, 71–84. [Google Scholar] [CrossRef]
- Längst, G.; Manelyte, L. Chromatin Remodelers: From Function to Dysfunction. Genes 2015, 6, 299–324. [Google Scholar] [CrossRef]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Koldobskiy, M.A.; Göndör, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef]
- Kukkonen, K.; Taavitsainen, S.; Huhtala, L.; Uusi-Makela, J.; Granberg, K.J.; Nykter, M.; Urbanucci, A. Chromatin and Epigenetic Dysregulation of Prostate Cancer Development, Progression, and Therapeutic Response. Cancers 2021, 13, 3325. [Google Scholar] [CrossRef]
- Yegnasubramanian, S.; De Marzo, A.M.; Nelson, W.G. Prostate Cancer Epigenetics: From Basic Mechanisms to Clinical Implications. Cold Spring Harb. Perspect. Med. 2019, 9, a030445. [Google Scholar] [CrossRef]
- Nelson, W.G.; De Marzo, A.M.; Yegnasubramanian, S. Minireview: Epigenetic Alterations in Human Prostate Cancers. Endocrinology 2009, 150, 3991–4002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, A.; Zoubeidi, A.; Selth, L.A. The epigenetic and transcriptional landscape of neuroendocrine prostate cancer. Endocr. Relat. Cancer 2020, 27, R35–R50. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zheng, D.; Zhou, T.; Song, H.; Hulsurkar, M.; Su, N.; Liu, Y.; Wang, Z.; Shao, L.; Ittmann, M.; et al. Androgen deprivation promotes neuroendocrine differentiation and angiogenesis through CREB-EZH2-TSP1 pathway in prostate cancers. Nat. Commun. 2018, 9, 4080. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Al-Muftah, M.A.; Al-Kowari, M.K.; Abuaqel, S.W.J.; Al-Rumaihi, K.; Al-Bozom, I.; Li, P.; Chouchane, L. Targeting Wnt/EZH2/microRNA-708 signaling pathway inhibits neuroendocrine differentiation in prostate cancer. Cell Death Discov. 2019, 5, 139. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [Green Version]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Baubec, T.; Schubeler, D. Genomic patterns and context specific interpretation of DNA methylation. Curr. Opin. Genet. Dev. 2014, 25, 85–92. [Google Scholar] [CrossRef]
- Locke, W.J.; Guanzon, D.; Ma, C.; Liew, Y.J.; Duesing, K.R.; Fung, K.Y.C.; Ross, J.P. DNA Methylation Cancer Biomarkers: Translation to the Clinic. Front. Genet. 2019, 10, 1150. [Google Scholar] [CrossRef]
- Bhasin, J.M.; Lee, B.H.; Matkin, L.; Taylor, M.G.; Hu, B.; Xu, Y.; Magi-Galluzzi, C.; Klein, E.A.; Ting, A.H. Methylome-wide Sequencing Detects DNA Hypermethylation Distinguishing Indolent from Aggressive Prostate Cancer. Cell Rep. 2015, 13, 2135–2146. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Dhanasekaran, S.M.; Prensner, J.R.; Cao, X.; Robinson, D.; Kalyana-Sundaram, S.; Huang, C.; Shankar, S.; Jing, X.; Iyer, M.; et al. Deep sequencing reveals distinct patterns of DNA methylation in prostate cancer. Genome Res. 2011, 21, 1028–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.G.; Chen, W.S.; Li, H.; Foye, A.; Zhang, M.; Sjöström, M.; Aggarwal, R.; Playdle, D.; Liao, A.; Alumkal, J.J.; et al. The DNA methylation landscape of advanced prostate cancer. Nat. Genet. 2020, 52, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D.; Annala, M.; Aprikian, A.; Armenia, J.; Arora, A.; et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graça, I.; Pereira-Silva, E.; Henrique, R.; Packham, G.; Crabb, S.J.; Jerónimo, C. Epigenetic modulators as therapeutic targets in prostate cancer. Clin. Epigenetics 2016, 8, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jerónimo, C.; Bastian, P.J.; Bjartell, A.; Carbone, G.M.; Catto, J.W.F.; Clark, S.J.; Henrique, R.; Nelson, W.G.; Shariat, S.F. Epigenetics in Prostate Cancer: Biologic and Clinical Relevance. Eur. Urol. 2011, 60, 753–766. [Google Scholar] [CrossRef]
- McKee, T.C.; Tricoli, J.V. Epigenetics of Prostate Cancer. In Cancer Epigenetics; Verma, M., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2015; Volume 1238, pp. 217–234. ISBN 978-1-4939-1803-4. [Google Scholar]
- Sugiura, M.; Sato, H.; Kanesaka, M.; Imamura, Y.; Sakamoto, S.; Ichikawa, T.; Kaneda, A. Epigenetic modifications in prostate cancer. Int. J. Urol. 2021, 28, 140–149. [Google Scholar] [CrossRef]
- Nakayama, T.; Watanabe, M.; Suzuki, H.; Toyota, M.; Sekita, N.; Hirokawa, Y.; Mizokami, A.; Ito, H.; Yatani, R.; Shiraishi, T. Epigenetic Regulation of Androgen Receptor Gene Expression in Human Prostate Cancers. Lab. Investig. 2000, 80, 1789–1796. [Google Scholar] [CrossRef] [Green Version]
- Buj, R.; Mallona, I.; Díez-Villanueva, A.; Barrera, V.; Mauricio, D.; Puig-Domingo, M.; Reverter, J.L.; Matias-Guiu, X.; Azuara, D.; Ramírez, J.L.; et al. Quantification of Unmethylated Alu (QUAlu): A tool to assess global hypomethylation in routine clinical samples. Oncotarget 2016, 7, 10536–10546. [Google Scholar] [CrossRef] [Green Version]
- Schulz, W.A.; Elo, J.P.; Florl, A.R.; Pennanen, S.; Santourlidis, S.; Engers, R.; Buchardt, M.; Seifert, H.-H.; Visakorpi, T. Genomewide DNA hypomethylation is associated with alterations on chromosome 8 in prostate carcinoma. Genes Chromosom. Cancer 2002, 35, 58–65. [Google Scholar] [CrossRef]
- Zelic, R.; Fiano, V.; Grasso, C.; Zugna, D.; Pettersson, A.; Gillio-Tos, A.; Merletti, F.; Richiardi, L. Global DNA hypomethylation in prostate cancer development and progression: A systematic review. Prostate Cancer Prostatic Dis. 2015, 18, 1–12. [Google Scholar] [CrossRef]
- Bedford, M.T.; van Helden, P.D. Hypomethylation of DNA in pathological conditions of the human prostate. Cancer Res. 1987, 47, 5274–5276. [Google Scholar] [PubMed]
- Stewart, D.J.; Issa, J.-P.; Kurzrock, R.; Nunez, M.I.; Jelinek, J.; Hong, D.; Oki, Y.; Guo, Z.; Gupta, S.; Wistuba, I.I. Decitabine Effect on Tumor Global DNA Methylation and Other Parameters in a Phase I Trial in Refractory Solid Tumors and Lymphomas. Clin. Cancer Res. 2009, 15, 3881–3888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, A.S.; Foley, R.; Woodson, K.; Lawler, M. The emerging roles of DNA methylation in the clinical management of prostate cancer. Endocr. Relat. Cancer 2006, 13, 357–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulukuri, S.M.K.; Rao, J.S. Activation of p53/p21Waf1/Cip1 pathway by 5-aza-2’-deoxycytidine inhibits cell proliferation, induces pro-apoptotic genes and mitogen-activated protein kinases in human prostate cancer cells. Int. J. Oncol. 2005, 26, 863–871. [Google Scholar] [CrossRef]
- Festuccia, C.; Gravina, G.L.; D’Alessandro, A.M.; Millimaggi, D.; Di Rocco, C.; Dolo, V.; Ricevuto, E.; Vicentini, C.; Bologna, M. Downmodulation of dimethyl transferase activity enhances tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in prostate cancer cells. Int. J. Oncol. 2008, 33, 381–388. [Google Scholar] [CrossRef] [Green Version]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Chervona, Y.; Costa, M. Histone modifications and cancer: Biomarkers of prognosis? Am. J. Cancer Res. 2012, 2, 589–597. [Google Scholar]
- Bianco-Miotto, T.; Chiam, K.; Buchanan, G.; Jindal, S.; Day, T.K.; Thomas, M.; Pickering, M.A.; O’Loughlin, M.A.; Ryan, N.K.; Raymond, W.A.; et al. Global Levels of Specific Histone Modifications and an Epigenetic Gene Signature Predict Prostate Cancer Progression and Development. Cancer Epidemiol. Biomarkers Prev. 2010, 19, 2611–2622. [Google Scholar] [CrossRef] [Green Version]
- Ellinger, J.; Kahl, P.; von der Gathen, J.; Rogenhofer, S.; Heukamp, L.C.; Gütgemann, I.; Walter, B.; Hofstädter, F.; Büttner, R.; Müller, S.C.; et al. Global levels of histone modifications predict prostate cancer recurrence. Prostate 2010, 70, 61–69. [Google Scholar] [CrossRef]
- Behbahani, T.E.; Kahl, P.; von der Gathen, J.; Heukamp, L.C.; Baumann, C.; Gütgemann, I.; Walter, B.; Hofstädter, F.; Bastian, P.J.; von Ruecker, A.; et al. Alterations of global histone H4K20 methylation during prostate carcinogenesis. BMC Urol. 2012, 12, 5. [Google Scholar] [CrossRef] [Green Version]
- Ellinger, J.; Kahl, P.; von der Gathen, J.; Heukamp, L.C.; Gütgemann, I.; Walter, B.; Hofstädter, F.; Bastian, P.J.; von Ruecker, A.; Müller, S.C.; et al. Global Histone H3K27 Methylation Levels are Different in Localized and Metastatic Prostate Cancer. Cancer Invest. 2012, 30, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Ngollo, M.; Lebert, A.; Daures, M.; Judes, G.; Rifai, K.; Dubois, L.; Kemeny, J.-L.; Penault-Llorca, F.; Bignon, Y.-J.; Guy, L.; et al. Global analysis of H3K27me3 as an epigenetic marker in prostate cancer progression. BMC Cancer 2017, 17, 261. [Google Scholar] [CrossRef] [PubMed]
- Ruggero, K.; Farran-Matas, S.; Martinez-Tebar, A.; Aytes, A. Epigenetic Regulation in Prostate Cancer Progression. Curr. Mol. Biol. Rep. 2018, 4, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Weichert, W.; Röske, A.; Gekeler, V.; Beckers, T.; Stephan, C.; Jung, K.; Fritzsche, F.R.; Niesporek, S.; Denkert, C.; Dietel, M.; et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br. J. Cancer 2008, 98, 604–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korkmaz, C.; Fronsdal, K.; Zhang, Y.; Lorenzo, P.; Saatcioglu, F. Potentiation of androgen receptor transcriptional activity by inhibition of histone deacetylation--rescue of transcriptionally compromised mutants. J. Endocrinol. 2004, 182, 377–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.H.; Roberts, C.W.M. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.A.B.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef]
- Wang, Q.; Carroll, J.S.; Brown, M. Spatial and Temporal Recruitment of Androgen Receptor and Its Coactivators Involves Chromosomal Looping and Polymerase Tracking. Mol. Cell 2005, 19, 631–642. [Google Scholar] [CrossRef]
- Wissmann, M.; Yin, N.; Müller, J.M.; Greschik, H.; Fodor, B.D.; Jenuwein, T.; Vogler, C.; Schneider, R.; Günther, T.; Buettner, R.; et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat. Cell Biol. 2007, 9, 347–353. [Google Scholar] [CrossRef]
- Keto, C.J.; Aronson, W.J.; Terris, M.K.; Presti, J.C.; Kane, C.J.; Amling, C.L.; Freedland, S.J. Obesity is associated with castration-resistant disease and metastasis in men treated with androgen deprivation therapy after radical prostatectomy: Results from the SEARCH database. BJU Int. 2012, 110, 492–498. [Google Scholar] [CrossRef] [Green Version]
- Discacciati, A.; Orsini, N.; Wolk, A. Body mass index and incidence of localized and advanced prostate cancer—a dose–response meta-analysis of prospective studies. Ann. Oncol. 2012, 23, 1665–1671. [Google Scholar] [CrossRef]
- Cao, Y.; Ma, J. Body Mass Index, Prostate Cancer–Specific Mortality, and Biochemical Recurrence: A Systematic Review and Meta-analysis. Cancer Prev. Res. 2011, 4, 486–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Hafe, P.; Pina, F.; Pérez, A.; Tavares, M.; Barros, H. Visceral Fat Accumulation as a Risk Factor for Prostate Cancer. Obes. Res. 2004, 12, 1930–1935. [Google Scholar] [CrossRef] [PubMed]
- Di Sebastiano, K.; Mourtzakis, M. The Role of Dietary Fat throughout the Prostate Cancer Trajectory. Nutrients 2014, 6, 6095–6109. [Google Scholar] [CrossRef] [PubMed]
- Giovannucci, E.; Rimm, E.B.; Colditz, G.A.; Stampfer, M.J.; Ascherio, A.; Chute, C.C.; Willett, W.C. A Prospective Study of Dietary Fat and Risk of Prostate Cancer. JNCI J. Natl. Cancer Inst. 1993, 85, 1571–1579. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.W.-L.; Chapman, K. A systematic review of the effect of diet in prostate cancer prevention and treatment. J. Hum. Nutr. Diet. 2009, 22, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Gathirua-Mwangi, W.G.; Zhang, J. Dietary factors and risk for advanced prostate cancer. Eur. J. Cancer Prev. 2014, 23, 96–109. [Google Scholar] [CrossRef] [Green Version]
- Bairati, I.; Meyer, F.; Fradet, Y.; Moore, L. Dietary fat and advanced prostate cancer. J. Urol. 1998, 159, 1271–1275. [Google Scholar] [CrossRef]
- Richman, E.L.; Kenfield, S.A.; Chavarro, J.E.; Stampfer, M.J.; Giovannucci, E.L.; Willett, W.C.; Chan, J.M. Fat Intake After Diagnosis and Risk of Lethal Prostate Cancer and All-Cause Mortality. JAMA Intern. Med. 2013, 173, 1318. [Google Scholar] [CrossRef] [Green Version]
- Oczkowski, M.; Dziendzikowska, K.; Pasternak-Winiarska, A.; Włodarek, D.; Gromadzka-Ostrowska, J. Dietary Factors and Prostate Cancer Development, Progression, and Reduction. Nutrients 2021, 13, 496. [Google Scholar] [CrossRef]
- Narita, S.; Nara, T.; Sato, H.; Koizumi, A.; Huang, M.; Inoue, T.; Habuchi, T. Research Evidence on High-Fat Diet-Induced Prostate Cancer Development and Progression. J. Clin. Med. 2019, 8, 597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.; Koizumi, A.; Narita, S.; Inoue, T.; Tsuchiya, N.; Nakanishi, H.; Numakura, K.; Tsuruta, H.; Saito, M.; Satoh, S.; et al. Diet-induced alteration of fatty acid synthase in prostate cancer progression. Oncogenesis 2016, 5, e195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labbé, D.P.; Zadra, G.; Yang, M.; Reyes, J.M.; Lin, C.Y.; Cacciatore, S.; Ebot, E.M.; Creech, A.L.; Giunchi, F.; Fiorentino, M.; et al. High-fat diet fuels prostate cancer progression by rewiring the metabolome and amplifying the MYC program. Nat. Commun. 2019, 10, 4358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etchegaray, J.-P.; Mostoslavsky, R. Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol. Cell 2016, 62, 695–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izzo, L.T.; Affronti, H.C.; Wellen, K.E. The Bidirectional Relationship Between Cancer Epigenetics and Metabolism. Annu. Rev. Cancer Biol. 2021, 5, 235–257. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Tu, B.P. Sink into the Epigenome: Histones as Repositories That Influence Cellular Metabolism. Trends Endocrinol. Metab. 2018, 29, 626–637. [Google Scholar] [CrossRef]
- Shi, L.; Tu, B.P. Acetyl-CoA and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Verdone, L. Histone acetylation in gene regulation. Brief. Funct. Genom. Proteom. 2006, 5, 209–221. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.; Sutter, B.M.; Li, B.; Tu, B.P. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol. Cell 2011, 42, 426–437. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; McCaffery, J.M.; Irizarry, R.A.; Boeke, J.D. Nucleocytosolic Acetyl-Coenzyme A Synthetase Is Required for Histone Acetylation and Global Transcription. Mol. Cell 2006, 23, 207–217. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-Citrate Lyase Links Cellular Metabolism to Histone Acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.V.; Carrer, A.; Shah, S.; Snyder, N.W.; Wei, S.; Venneti, S.; Worth, A.J.; Yuan, Z.-F.; Lim, H.-W.; Liu, S.; et al. Akt-Dependent Metabolic Reprogramming Regulates Tumor Cell Histone Acetylation. Cell Metab. 2014, 20, 306–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, R.A.; Kuo, Y.-M.; Bhattacharjee, V.; Yen, T.J.; Andrews, A.J. Changing the Selectivity of p300 by Acetyl-CoA Modulation of Histone Acetylation. ACS Chem. Biol. 2015, 10, 146–156. [Google Scholar] [CrossRef] [Green Version]
- Sivanand, S.; Viney, I.; Wellen, K.E. Spatiotemporal Control of Acetyl-CoA Metabolism in Chromatin Regulation. Trends Biochem. Sci. 2018, 43, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Guccini, I.; Di Mitri, D.; Brina, D.; Revandkar, A.; Sarti, M.; Pasquini, E.; Alajati, A.; Pinton, S.; Losa, M.; et al. Compartmentalized activities of the pyruvate dehydrogenase complex sustain lipogenesis in prostate cancer. Nat. Genet. 2018, 50, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-L.; Hsu, S.-C.; Chung, T.-Y.; Chu, C.-Y.; Wang, H.-J.; Hsiao, P.-W.; Yeh, S.-D.; Ann, D.K.; Yen, Y.; Kung, H.-J. Arginine is an epigenetic regulator targeting TEAD4 to modulate OXPHOS in prostate cancer cells. Nat. Commun. 2021, 12, 2398. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Snyder, N.W. Should we consider subcellular compartmentalization of metabolites, and if so, how do we measure them? Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Berndsen, C.E.; Denu, J.M. Assays for mechanistic investigations of protein/histone acetyltransferases. Methods 2005, 36, 321–331. [Google Scholar] [CrossRef]
- Dillon, B.J.; Prieto, V.G.; Curley, S.A.; Ensor, C.M.; Holtsberg, F.W.; Bomalaski, J.S.; Clark, M.A. Incidence and distribution of argininosuccinate synthetase deficiency in human cancers. Cancer 2004, 100, 826–833. [Google Scholar] [CrossRef]
- Xu, Q.; Liu, X.; Zhu, S.; Hu, X.; Niu, H.; Zhang, X.; Zhu, D.; Nesa, E.U.; Tian, K.; Yuan, H. Hyper-acetylation contributes to the sensitivity of chemo-resistant prostate cancer cells to histone deacetylase inhibitor Trichostatin A. J. Cell. Mol. Med. 2018, 22, 1909–1922. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 Histone Marks and Histone Lysine Crotonylation as a New Type of Histone Modification. Cell 2011, 146, 1016–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabari, B.R.; Zhang, D.; Allis, C.D.; Zhao, Y. Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol. 2017, 18, 90–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, C.; Park, S.; Oh, S.; Choi, J.; Kim, E.-K.; Youn, H.-D.; Cho, E.-J. Histone acylation marks respond to metabolic perturbations and enable cellular adaptation. Exp. Mol. Med. 2020, 52, 2005–2019. [Google Scholar] [CrossRef] [PubMed]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Zhang, D.; Chung, D.; Tang, Z.; Huang, H.; Dai, L.; Qi, S.; Li, J.; Colak, G.; Chen, Y.; et al. Metabolic Regulation of Gene Expression by Histone Lysine β-Hydroxybutyrylation. Mol. Cell 2016, 62, 194–206. [Google Scholar] [CrossRef] [Green Version]
- Sabari, B.R.; Tang, Z.; Huang, H.; Yong-Gonzalez, V.; Molina, H.; Kong, H.E.; Dai, L.; Shimada, M.; Cross, J.R.; Zhao, Y.; et al. Intracellular Crotonyl-CoA Stimulates Transcription through p300-Catalyzed Histone Crotonylation. Mol. Cell 2015, 58, 203–215. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Zhu, X.; Liu, F.; Lu, W.; Wang, Y.; Yu, J. The effects of histone crotonylation and bromodomain protein 4 on prostate cancer cell lines. Transl. Androl. Urol. 2021, 10, 900–914. [Google Scholar] [CrossRef]
- Suenaga, M.; Soda, H.; Oka, M.; Yamaguchi, A.; Nakatomi, K.; Shiozawa, K.; Kawabata, S.; Kasai, T.; Yamada, Y.; Kamihira, S.; et al. Histone deacetylase inhibitors suppress telomerase reverse transcriptase mRNA expression in prostate cancer cells. Int. J. cancer 2002, 97, 621–625. [Google Scholar] [CrossRef]
- Kim, J.; Park, H.; Im, J.Y.; Choi, W.S.; Kim, H.S. Sodium butyrate regulates androgen receptor expression and cell cycle arrest in human prostate cancer cells. Anticancer Res. 2007, 27, 3285–3292. [Google Scholar]
- Maier, S.; Reich, E.; Martin, R.; Bachem, M.; Altug, V.; Hautmann, R.E.; Gschwend, J.E. Tributyrin induces differentiation, growth arrest and apoptosis in androgen-sensitive and androgen-resistant human prostate cancer cell lines. Int. J. Cancer 2000, 88, 245–251. [Google Scholar] [CrossRef]
- Paskova, L.; Smesny Trtkova, K.; Fialova, B.; Benedikova, A.; Langova, K.; Kolar, Z. Different effect of sodium butyrate on cancer and normal prostate cells. Toxicol. Vitr. 2013, 27, 1489–1495. [Google Scholar] [CrossRef]
- Jing, H.; Lin, H. Sirtuins in Epigenetic Regulation. Chem. Rev. 2015, 115, 2350–2375. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Sauve, A.A. Regulatory Effects of NAD + Metabolic Pathways on Sirtuin Activity. Prog. Mol. Biol. Transl. Sci. 2018, 154, 71–104. [Google Scholar]
- Martinez-Redondo, P.; Vaquero, A. The Diversity of Histone Versus Nonhistone Sirtuin Substrates. Genes Cancer 2013, 4, 148–163. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Khan, S.; Wang, Y.; Charron, G.; He, B.; Sebastian, C.; Du, J.; Kim, R.; Ge, E.; Mostoslavsky, R.; et al. SIRT6 regulates TNF-α secretion through hydrolysis of long-chain fatty acyl lysine. Nature 2013, 496, 110–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H.; et al. Sirt5 Is a NAD-Dependent Protein Lysine Demalonylase and Desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawant Dessai, A.; Dominguez, M.P.; Chen, U.-I.; Hasper, J.; Prechtl, C.; Yu, C.; Katsuta, E.; Dai, T.; Zhu, B.; Jung, S.Y.; et al. Transcriptional Repression of SIRT3 Potentiates Mitochondrial Aconitase Activation to Drive Aggressive Prostate Cancer to the Bone. Cancer Res. 2021, 81, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Kosciuk, T.; Wang, M.; Hong, J.Y.; Lin, H. Updates on the epigenetic roles of sirtuins. Curr. Opin. Chem. Biol. 2019, 51, 18–29. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Ryall, J.G.; Dell’Orso, S.; Derfoul, A.; Juan, A.; Zare, H.; Feng, X.; Clermont, D.; Koulnis, M.; Gutierrez-Cruz, G.; Fulco, M.; et al. The NAD+-Dependent SIRT1 Deacetylase Translates a Metabolic Switch into Regulatory Epigenetics in Skeletal Muscle Stem Cells. Cell Stem Cell 2015, 16, 171–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD+ metabolism: Pathophysiologic mechanisms and therapeutic potential. Signal Transduct. Target. Ther. 2020, 5, 227. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Deik, A.; Gonzalez, C.; Gonzalez, M.E.; Fu, F.; Ferrari, M.; Churchhouse, C.L.; Florez, J.C.; Jacobs, S.B.R.; Clish, C.B.; et al. Polyunsaturated Fatty Acid Desaturation Is a Mechanism for Glycolytic NAD+ Recycling. Cell Metab. 2019, 29, 856–870.e7. [Google Scholar] [CrossRef] [Green Version]
- Vriens, K.; Christen, S.; Parik, S.; Broekaert, D.; Yoshinaga, K.; Talebi, A.; Dehairs, J.; Escalona-Noguero, C.; Schmieder, R.; Cornfield, T.; et al. Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature 2019, 566, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Bruno, J.; Easlon, E.; Lin, S.-J.; Cheng, H.-L.; Alt, F.W.; Guarente, L. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 2008, 22, 1753–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korotkov, A.; Seluanov, A.; Gorbunova, V. Sirtuin 6: Linking longevity with genome and epigenome stability. Trends Cell Biol. 2021, 31, 994–1006. [Google Scholar] [CrossRef]
- Feldman, J.L.; Baeza, J.; Denu, J.M. Activation of the Protein Deacetylase SIRT6 by Long-chain Fatty Acids and Widespread Deacylation by Mammalian Sirtuins. J. Biol. Chem. 2013, 288, 31350–31356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carreño, M.; Bresque, M.; Machado, M.R.; Santos, L.; Durán, R.; Vitturi, D.A.; Escande, C.; Denicola, A. Nitro-fatty acids as activators of hSIRT6 deacetylase activity. J. Biol. Chem. 2020, 295, 18355–18366. [Google Scholar] [CrossRef]
- Caldas, A.P.S.; Rocha, D.M.U.P.; Bressan, J.; Hermsdorff, H.H.M. Dietary fatty acids as nutritional modulators of sirtuins: A systematic review. Nutr. Rev. 2021, 79, 235–246. [Google Scholar] [CrossRef]
- Markham, G.D. S -Adenosylmethionine. In Encyclopedia of Life Sciences; John Wiley & Sons, Ltd.: Chichester, UK, 2010. [Google Scholar]
- Ye, C.; Sutter, B.M.; Wang, Y.; Kuang, Z.; Tu, B.P. A Metabolic Function for Phospholipid and Histone Methylation. Mol. Cell 2017, 66, 180–193.e8. [Google Scholar] [CrossRef] [Green Version]
- Madigan, A.A.; Rycyna, K.J.; Parwani, A.V.; Datiri, Y.J.; Basudan, A.M.; Sobek, K.M.; Cummings, J.L.; Basse, P.H.; Bacich, D.J.; O’Keefe, D.S. Novel Nuclear Localization of Fatty Acid Synthase Correlates with Prostate Cancer Aggressiveness. Am. J. Pathol. 2014, 184, 2156–2162. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Yin, X.; Jin, Y.; Liu, F.; Gao, J. Identification of aberrantly methylated differentially expressed genes in prostate carcinoma using integrated bioinformatics. Cancer Cell Int. 2019, 19, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oatman, N.; Dasgupta, N.; Arora, P.; Choi, K.; Gawali, M.V.; Gupta, N.; Parameswaran, S.; Salomone, J.; Reisz, J.A.; Lawler, S.; et al. Mechanisms of stearoyl CoA desaturase inhibitor sensitivity and acquired resistance in cancer. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Slieker, R.C.; Relton, C.L.; Gaunt, T.R.; Slagboom, P.E.; Heijmans, B.T. Age-related DNA methylation changes are tissue-specific with ELOVL2 promoter methylation as exception. Epigenetics Chromatin 2018, 11, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wang, J.; Wang, L.; Feng, G.; Li, G.; Yu, M.; Li, Y.; Liu, C.; Yuan, X.; Zang, G.; et al. Impaired lipid metabolism by age-dependent DNA methylation alterations accelerates aging. Proc. Natl. Acad. Sci. USA 2020, 117, 4328–4336. [Google Scholar] [CrossRef]
- Niculite, C.-M.; Enciu, A.-M.; Hinescu, M.E. CD 36: Focus on Epigenetic and Post-Transcriptional Regulation. Front. Genet. 2019, 10, 680. [Google Scholar] [CrossRef]
- Sun, Q.; Zhang, W.; Wang, L.; Guo, F.; Song, D.; Zhang, Q.; Zhang, D.; Fan, Y.; Wang, J. Hypermethylated CD36 gene affected the progression of lung cancer. Gene 2018, 678, 395–406. [Google Scholar] [CrossRef]
- Alfaqih, M.A.; Nelson, E.R.; Liu, W.; Safi, R.; Jasper, J.S.; Macias, E.; Geradts, J.; Thompson, J.W.; Dubois, L.G.; Freeman, M.R.; et al. CYP27A1 Loss Dysregulates Cholesterol Homeostasis in Prostate Cancer. Cancer Res. 2017, 77, 1662–1673. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.H.; Taylor, M.G.; Robinet, P.; Smith, J.D.; Schweitzer, J.; Sehayek, E.; Falzarano, S.M.; Magi-Galluzzi, C.; Klein, E.A.; Ting, A.H. Dysregulation of Cholesterol Homeostasis in Human Prostate Cancer through Loss of ABCA1. Cancer Res. 2013, 73, 1211–1218. [Google Scholar] [CrossRef] [Green Version]
- Ettinger, S.L.; Sobel, R.; Whitmore, T.G.; Akbari, M.; Bradley, D.R.; Gleave, M.E.; Nelson, C.C. Dysregulation of Sterol Response Element-Binding Proteins and Downstream Effectors in Prostate Cancer during Progression to Androgen Independence. Cancer Res. 2004, 64, 2212–2221. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Kong, M.; Li, M.; Hong, W.; Fan, X.; Xu, Y. Brahma Related Gene 1 (Brg1) Regulates Cellular Cholesterol Synthesis by Acting as a Co-factor for SREBP2. Front. Cell Dev. Biol. 2020, 8, 259. [Google Scholar] [CrossRef]
- Li, N.; Li, M.; Hong, W.; Shao, J.; Xu, H.; Shimano, H.; Lu, J.; Xu, Y. Brg1 regulates pro-lipogenic transcription by modulating SREBP activity in hepatocytes. Biochim. Biophys. Acta-Mol. Basis Dis. 2018, 1864, 2881–2889. [Google Scholar] [CrossRef] [PubMed]
- Gang, X.; Yang, Y.; Zhong, J.; Jiang, K.; Pan, Y.; Karnes, R.J.; Zhang, J.; Xu, W.; Wang, G.; Huang, H. P300 acetyltransferase regulates fatty acid synthase expression, lipid metabolism and prostate cancer growth. Oncotarget 2016, 7, 15135–15149. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, L.; Li, M.; Cheng, X.; Fang, M.; Zeng, Q.; Xu, Y. The chromatin remodeling protein BRG1 links ELOVL3 trans-activation to prostate cancer metastasis. Biochim. Biophys. Acta-Gene Regul. Mech. 2019, 1862, 834–845. [Google Scholar] [CrossRef]
- Muthuswami, R.; Bailey, L.; Rakesh, R.; Imbalzano, A.N.; Nickerson, J.A.; Hockensmith, J.W. BRG1 is a prognostic indicator and a potential therapeutic target for prostate cancer. J. Cell. Physiol. 2019, 234, 15194–15205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Lin, S.-H.; Ren, F.; Li, J.-T.; Chen, J.-J.; Yao, C.-B.; Yang, H.-B.; Jiang, S.-X.; Yan, G.-Q.; Wang, D.; et al. Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat. Commun. 2016, 7, 11960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitro, N.; Godio, C.; De Fabiani, E.; Scotti, E.; Galmozzi, A.; Gilardi, F.; Caruso, D.; Chacon, A.B.V.; Crestani, M. Insights in the regulation of cholesterol 7α-hydroxylase gene reveal a target for modulating bile acid synthesis. Hepatology 2007, 46, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Xiong, X.; DePinho, R.A.; Deng, C.-X.; Dong, X.C. Hepatic SREBP-2 and cholesterol biosynthesis are regulated by FoxO3 and Sirt6. J. Lipid Res. 2013, 54, 2745–2753. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Pathway | Focus | Drug | Disease | Phase | Patients | Objective | Status | Results | Idenitifier |
|---|---|---|---|---|---|---|---|---|---|
| Cholesterol | HMGCR | Rosuvastatin | Metastatic PCa | Phase 4 | 70 | Agressive parameters | Completed | Not published | NCT04776889 |
| Atorvastatin | Localized PCa | Phase 2 | 160 | Agressive parameters | Completed | [119] | NCT01821404 | ||
| Localized PCa | Phase 2 | 354 | Recurrence rate | Completed | [120] | NCT01759836 | |||
| Metastatic PCa | Phase 3 | 400 | Recurrence rate | Recruiting | Not published | NCT04026230 | |||
| Atorvastatin + celecoxib | Localized PCa | Phase 2 | 27 | PSA response | Completed | Not published | NCT01220973 | ||
| Atorvastatin + AAS Acetylsalicylic Acid | Castration Resistant | Phase 3 | 1210 | Overall Survival | Recruiting | Not published | NCT03819101 | ||
| Simvastatin | Localizaed PCa | WOP | 42 | Changes in Mevalonate Pathway | Completed | Not published | NCT00572468 | ||
| Simvastatin + Ezetimibe | Localized PCa | WOP | 63 | Agressive parameters | Completed | Not published | NCT02534376 | ||
| Fluvastatin + Pimonidazole | Localized PCa | WOP | 33 | Agressive parameters | Completed | [121] | NCT01992042 | ||
| Fatty acid | FASN | Omeprazole | Metastatic PCa | Phase 2 | 20 | Response rate | Recruiting | Not published | NCT04337580 |
| TVB-2640 | Metastatic Solid tumour * | Phase 1 | 180 | MTD | Completed | [122] | NCT02223247 | ||
| ACSS2 | MTB-9655 | Metastatic Solid tumour * | Phase 1 | 30 | MTD | Recruiting | Not published | NCT04990739 | |
| LXR | RGX-104 | Metastatic Solid tumour * | Phase 1 | 135 | MTD | Recruiting | Not published | NCT02922764 | |
| OXPHOS | IACS-010759 | Metastatic Solid tumour * | Phase 1 | 29 | MTD | Completed | JCO2019_37:15_sup | NCT03291938 | |
| CD36 | VT1021 | Metastatic Solid tumour * | Phase 1 | 116 | MTD | Active | Not published | NCT03364400 | |
| CVX-045 | Metastatic Solid tumour * | Phase 1 | 40 | MTD | Completed | Not published | NCT00879554 | ||
| ABT-510 | Metastatic Solid tumour * | Phase 1 | 45 | MTD | Completed | Not published | NCT00586092 | ||
| LDLR | ANG1005 | Metastatic Solid tumour * | Phase 1 | 56 | MTD | Completed | JCO2014 32:15_sup | NCT00539383 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pardo, J.C.; Ruiz de Porras, V.; Gil, J.; Font, A.; Puig-Domingo, M.; Jordà, M. Lipid Metabolism and Epigenetics Crosstalk in Prostate Cancer. Nutrients 2022, 14, 851. https://doi.org/10.3390/nu14040851
Pardo JC, Ruiz de Porras V, Gil J, Font A, Puig-Domingo M, Jordà M. Lipid Metabolism and Epigenetics Crosstalk in Prostate Cancer. Nutrients. 2022; 14(4):851. https://doi.org/10.3390/nu14040851
Chicago/Turabian StylePardo, Juan C., Vicenç Ruiz de Porras, Joan Gil, Albert Font, Manel Puig-Domingo, and Mireia Jordà. 2022. "Lipid Metabolism and Epigenetics Crosstalk in Prostate Cancer" Nutrients 14, no. 4: 851. https://doi.org/10.3390/nu14040851
APA StylePardo, J. C., Ruiz de Porras, V., Gil, J., Font, A., Puig-Domingo, M., & Jordà, M. (2022). Lipid Metabolism and Epigenetics Crosstalk in Prostate Cancer. Nutrients, 14(4), 851. https://doi.org/10.3390/nu14040851