
Intracellular Toxic Advanced Glycation End-Products in 1.4E7 Cell Line Induce Death with Reduction of Microtubule-Associated Protein 1 Light Chain 3 and p62



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. GA and Aminoguanidine (AG) Treatments
2.4. Viability of 1.4E7 Cells Treated with GA and AG
2.5. Measurement of Intracellular TAGE Levels in 1.4E7 Cells Treated with GA and AG Using a Slot Blot (SB) Analysis
2.6. Western Blot (WB) Analysis of LC3, p62, and GAPDH
2.7. Treatment of 1.4E7 Cells with NG-BSA and TAGE-BSA and Evaluation of Cell Viability
2.8. Statistical Analysis
3. Results
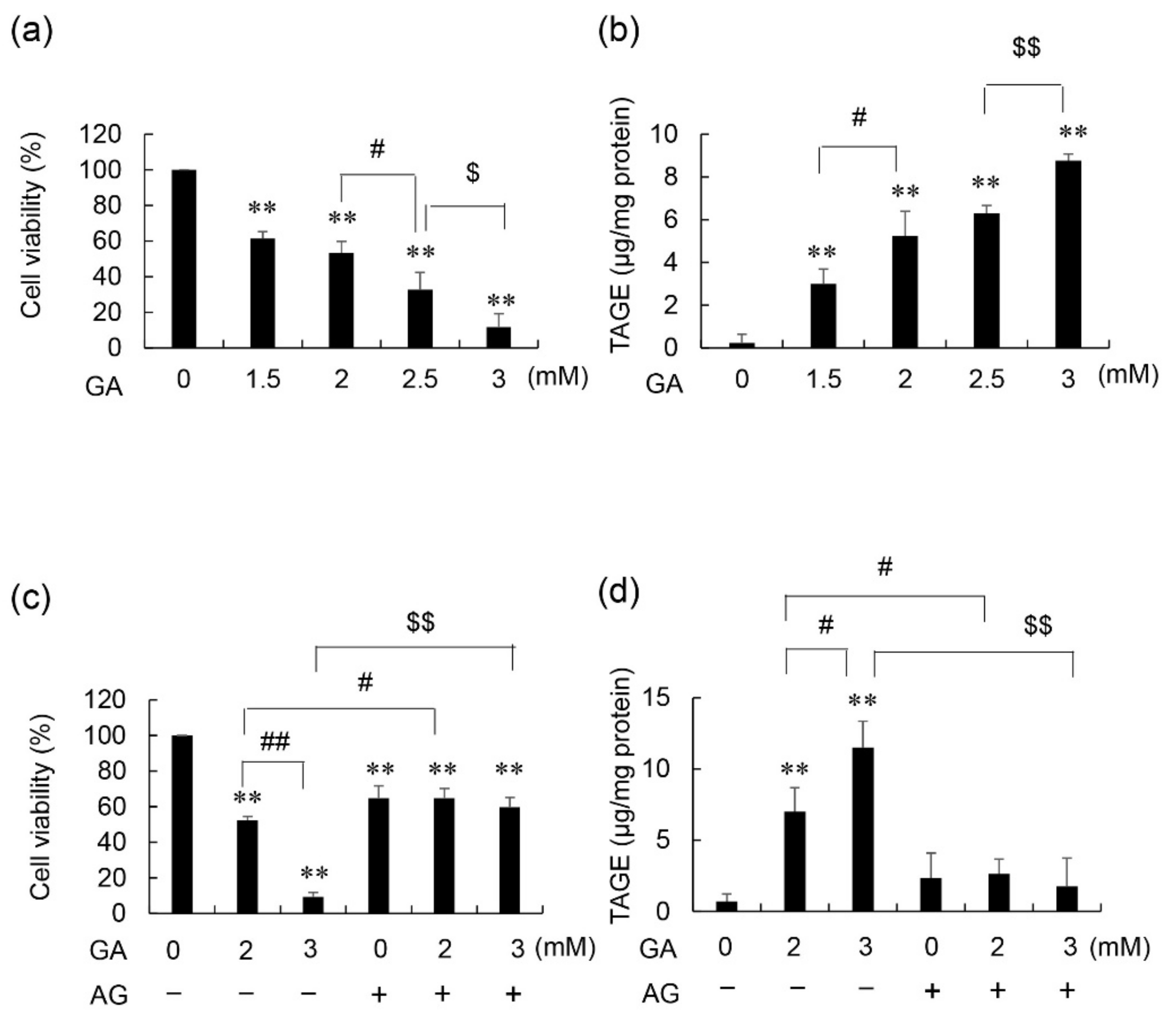
3.1. Cell Viability and Intracellular TAGE Levels in GA- and AG-Treated 1.4E7 Cells
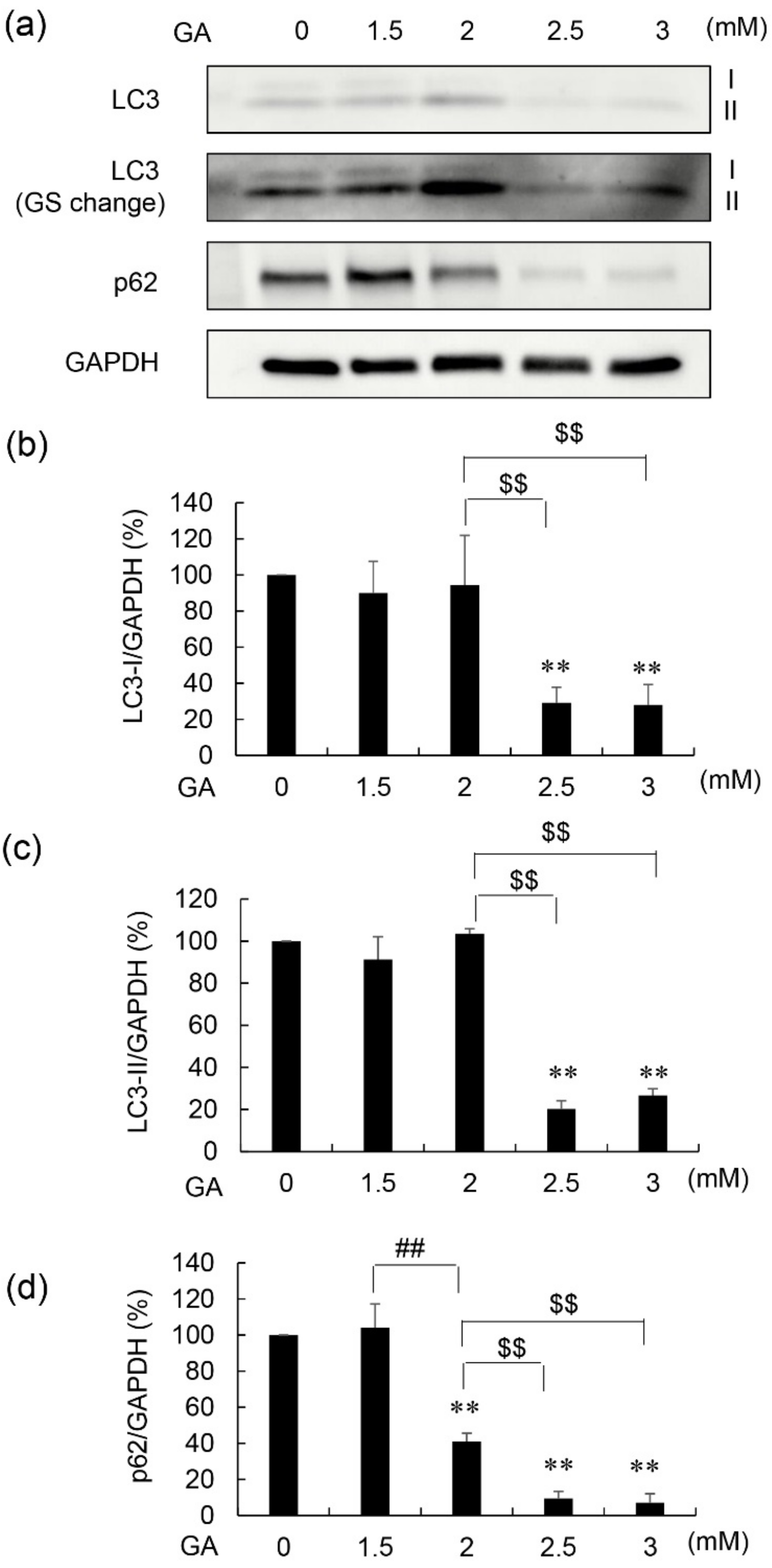
3.2. Protein Levels of LC3-I, LC3-II, and p62 in 1.4E7 Cells Treated with GA and AG
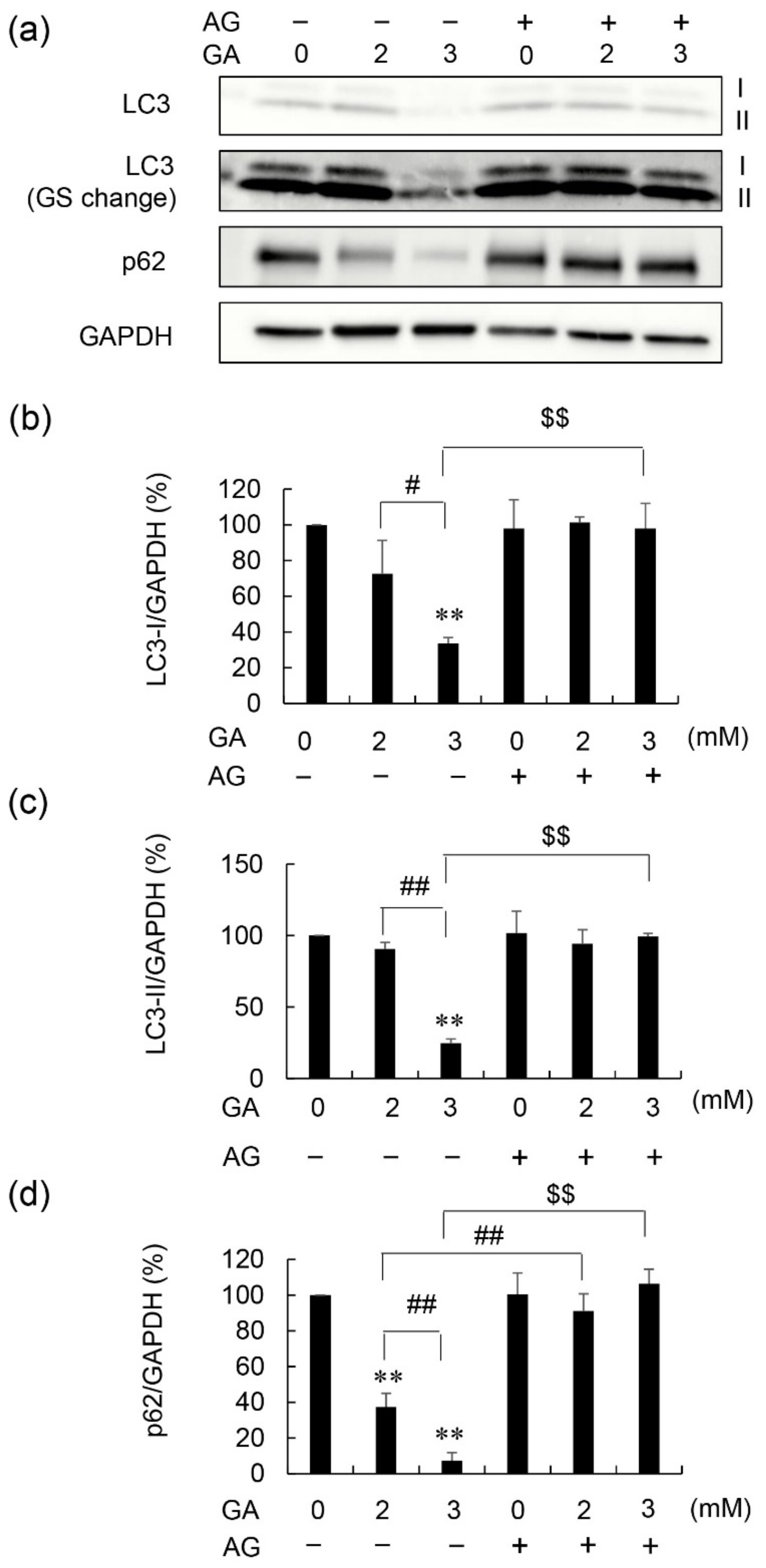
3.3. Effects of AG Pretreatment on LC3-I, LC3-II, and p62 Protein Levels in GA-Treated 1.4E7 Cells
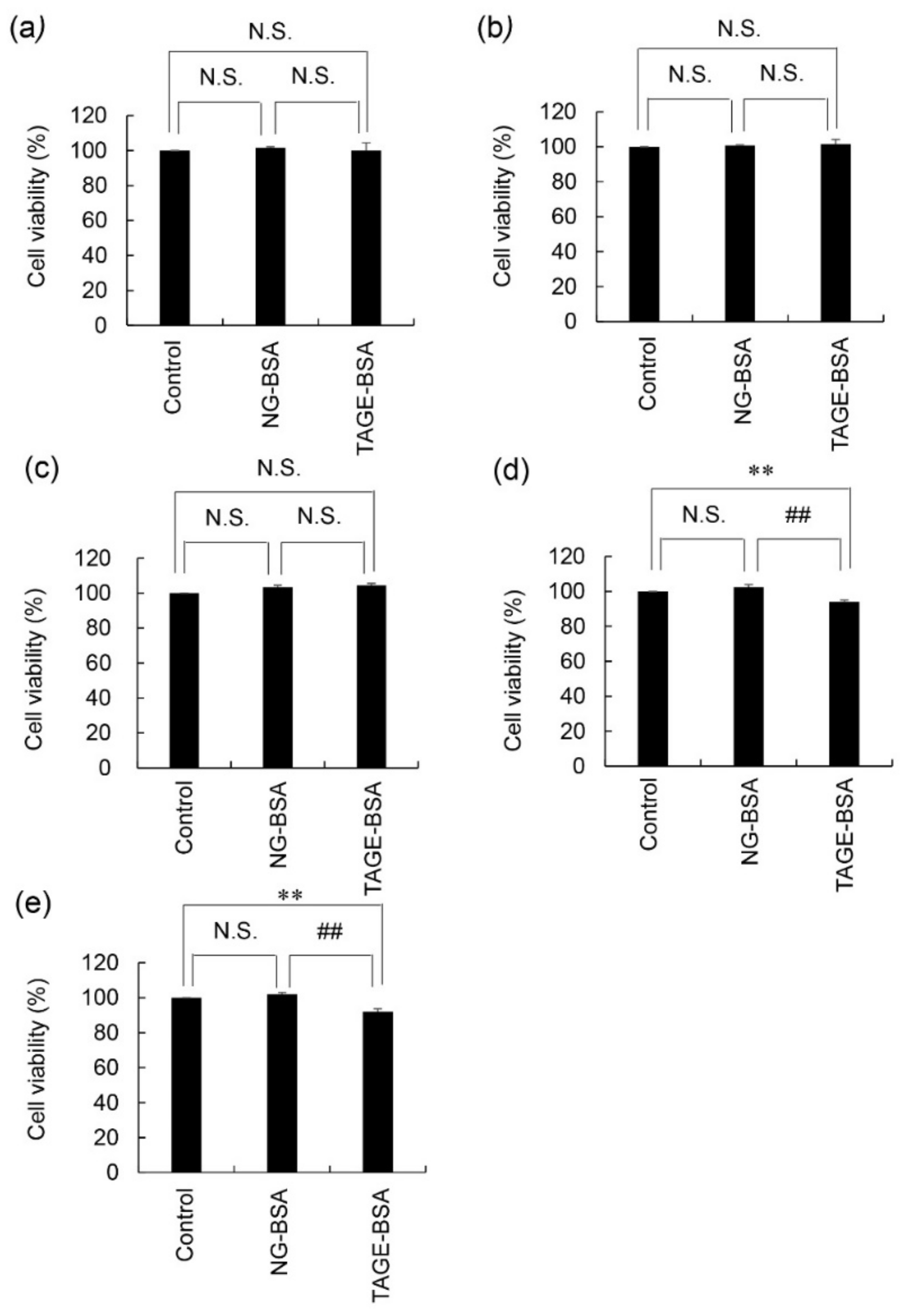
3.4. Viability of 1.4E7 Cells Treated with NG-BSA and TAGE-BSA
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AG | Aminoguanidine |
| AGEs | Advanced glycation end-products |
| GA | Glyceraldehyde |
| LC3 | Microtubule-associated protein 1 light chain 3 |
| NG-BSA | Non-glycated bovine serum albumin |
| RAGE | Receptor for advanced glycation end-products |
| TAGE | Toxic AGEs |
| TAGE-BSA | Toxic advanced glycation end-products-modified bovine serum albumin |
| T1DM | Type 1 diabetes mellitus |
| T2DM | Type 2 diabetes mellitus |
References
- Takeuchi, M.; Sakasai-Sakai, A.; Takata, T.; Takino, J.; Koriyama, Y.; Kikuchi, C.; Furukawa, A.; Nagamine, K.; Hori, T.; Matsunaga, T. Intracellular toxic (TAGE) triggers numerous type of cell damage. Biomolecules 2021, 11, 387. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M. Serum levels of toxic AGEs (TAGE) may be a promising novel biomarker for the onset/progression of lifestyle-related diseases. Diagnostics 2016, 6, 23. [Google Scholar] [CrossRef]
- Bohuslavova, R.; Smolik, O.; Malfatti, J.; Berkova, Z.; Novakova, Z.; Saudek, F.; Pavlinkova, G. NUEROD1 is required for the early α and β endocrine differentiation in the pancreas. Int. J. Mol. Sci. 2021, 22, 6713. [Google Scholar] [CrossRef]
- Ji, S.; Zhu, C.; Gao, S.; Shao, X.; Zhang, H.; Tang, D. Morus alba leaves ethanol extract protects pancreatic islet cells against dysfunction and death by inducing autophagy in type 2 diabetes. Phytomedicine 2021, 83, 153478. [Google Scholar] [CrossRef] [PubMed]
- Costes, S.; Bertrand, G.; Raier, M.A. Mechanisms of beta-cell apoptosis in type 2 diabetes-prone situations and potential protection by GLP-1-based therapies. Int. J. Mol. Sci. 2021, 22, 5303. [Google Scholar] [CrossRef]
- Suh, K.S.; Choi, E.M.; Jung, W.-W.; Kim, Y.J.; Hong, S.M.; Park, S.Y.; Rhee, S.Y.; Chon, S. Deoxyactein protects pancreatic β-cells againt methylglyoxal-induced oxidative cell damage by the upregulation of mitochondrial biogenesis. Int. J. Mol. Med. 2017, 40, 539–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borg, D.J.; Yap, F.Y.T.; Keshvari, S.; Simmons, D.G.; Gallo, L.A.; Fotheringham, A.K.; Zhuang, A.; Slattery, R.M.; Hasnain, S.Z.; Coughlan, M.T.; et al. Perinatal exposure to high dietary advanced glycation end products in transgenic NOD8.3 mice leads to pancreatic beta cell dysfunction. Islets 2018, 10, 10–24. [Google Scholar] [CrossRef]
- Takeuchi, M.; Takino, J.; Sakasai-Sakai, A.; Takata, T.; Tsutsumi, M. Toxic AGE (TAGE) theory for the pathophysiology of the onset/progression of NAFLD and ALD. Nutrients 2017, 9, 634. [Google Scholar] [CrossRef] [Green Version]
- Jeffery, J.; Jörnvall, H. Enzyme relationships in a sorbitol pathway that bypasses glycolysis and pentose phosphate in glucose metabolism. Proc. Natl. Acad. Sci. USA 1983, 80, 901–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayward, B.E.; Fantes, J.A.; Warner, J.P.; Intody, S.; Leek, J.P.; Mrkham, A.F.; Bonthron, D.T. Co-localization of the ketohexokinase and glucokinase regulator genes to a 500-kb region of Chromosome 2p23. Mamm. Genome 1996, 7, 454–458. [Google Scholar] [CrossRef]
- Gerst, F.; Jaghutriz, B.A.; Staiger, H.; Schulte, A.M.; Lorza-Gil, E.; Kaiser, G.; Panse, M.; Haug, S.; Heni, M.; Schütz, M. The expression of aldolase B in islets is negatively associated with insulin secretion in humans. J. Clin. Endocrinol. Metab. 2018, 103, 4373–4383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takata, T.; Sakasai-Sakai, A.; Takino, J.; Takeuchi, M. Evidence for toxic advanced glycation end-products generated in the normal rat Liver. Nutrients 2019, 11, 1612. [Google Scholar] [CrossRef] [Green Version]
- Inoue, S.; Takata, T.; Nakazawa, Y.; Nakamura, Y.; Guo, X.; Yamada, S.; Ishigaki, Y.; Takeuchi, M.; Miyazawa, K. Potential of an interorgan network mediated by toxic advanced glycation end-products in a rat model. Nutrients 2021, 13, 80. [Google Scholar] [CrossRef]
- Takata, T.; Sakasai-Sakai, A.; Ueda, T.; Takeuchi, M. Intracellular toxic advanced glycation end-products in cardiomyocytes may cause cardiovascular disease. Sci. Rep. 2019, 9, 2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takata, T.; Ueda, T.; Sakasai-Sakai, A.; Takeuchi, M. Generation of glyceraldehyde-derived advanced glycation end-products in pancreatic cancer cells and the potential of tumor promotion. World J. Gastroenterol. 2017, 23, 4910–4919. [Google Scholar] [CrossRef]
- Takata, T.; Sakasai-Sakai, A.; Takeuchi, M. Impact of intracellular toxic advanced glycation end-products (TAGE) on murine myoblast cell death. Diabetol. Metab. Syndr. 2020, 12, 54. [Google Scholar] [CrossRef]
- Nasu, R.; Furukawa, A.; Suzuki, K.; Takeuchi, M.; Koriyama, Y. The Effect of glyceraldehyde-derived advanced glycation end products on β-tubulin-inhibited neurite outgrowth in SH-SY5Y human neuroblastoma cells. Nutrients 2020, 12, 2958. [Google Scholar] [CrossRef]
- Ilie, I.; Ilie, R.; Moccan, T.; Tabaran, F.; Iancu, C.; Mocan, L. Nicotinamide-functionalized multiwalled carbon nanotubes increase insulin production in pancreatic beta cell via MIF pathyway. Nanomedicine 2013, 8, 3345–3358. [Google Scholar]
- Habtemariam, S. Investigation into antioxidant and antidiabetic potential of Moringa stenopetala: Identification of the active priciples. Nat. Prod. Commun. 2015, 10, 475–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puddu, A.; Sanguineti, R.; Durante, A.; Viviani, G.L. Pioglitazone attenuates the detrimental effects of advancced glycation end-products in the pancreatic beta cell line HIT-T15. Regul. Rep. 2012, 177, 79–84. [Google Scholar]
- Pejnovic, N.N.; Pantic, J.M.; Jovanovic, I.P.; Randosavaljevic, G.D.; Milovanovic, M.Z.; Nikolic, I.G.; Zdravkovic, N.S.; Djukic, A.L.; Arsenijevic, N.N.; Lukic, M.L. Galectin-3 deficientry accelerates high-fat diet-induced obesity and amplifies inflammation in adipose tissue and pancreatic islets. Diabetes 2013, 62, 1932–1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, M.; Makita, Z.; Bucala, R.; Suzuki, T.; Koike, T.; Kameda, Y. Immunological evidence that non-carboxymethyllysine advanced glycation end-products are produced from short chain sugars and dicarbonyl compounds in vitro. Mol. Med. 2000, 6, 114–125. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, S.; Okinaka, M.; Tanigawa, K.; Miwa, I. Difference in mechanism between glyceraldehyde- and glucose-induced insulin secretion from isolated rat pancreatic islets. J. Biochem. 2000, 127, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Tran, P.O.T.; LeRoy, E.; Harmon, J.S.; Tanaka, Y.; Robertson, R.P. D-Glyceraldehyde causes production of intracellular peroxide in pancreatic islets, oxidative stress, and defective beta cell function via non-mitochondrial pathways. J. Biol. Chem. 2004, 279, 37316–37323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazachkov, M.; Chen, K.; Babiy, S.; Peter, H.Y. Evidence for in vivo scavenging by aminoguanidine of formaldehyde producted via semicarbazide-senstive amine oxidase-mediated deamination. J. Pharmacol. Exp. Ther. 2007, 322, 1201–1207. [Google Scholar] [CrossRef] [PubMed]
- Donadel, G.; Pastore, D.; Della-Morte, D.; Capuani, B.; Lombardo, M.F.; Pacifici, F.; Bugaliani, M.; Grieco, F.A.; Marchetti, P.; Lauro, D. FGF-2b and h-PL transform duct and non-endocrine human pancreatic cells into endocrine insulin secreting cells by modulating differentiating genes. Int. J. Mol. Sci. 2017, 18, 2234. [Google Scholar] [CrossRef] [Green Version]
- Ebato, C.; Uchida, T.; Arakawa, M.; Komatsu, M.; Ueno, T.; Komiya, K.; Azuma, K.; Hirose, T.; Tanaka, K.; Kominami, E.; et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat. Cell. Metab. 2008, 8, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Drieβen, S.; Berleth, N.; Fiesen, O.; Löffler, A.; Böhler, P.; Hieke, N.; Stuhldreier, F.; Peter, C.; Schink, K.O.; Schultz, S.W.; et al. Deubiquitinase inhibiton by WP1130 leads to ULK1 aggregation and blockade of autophagy. Autophagy 2015, 11, 1458–1470. [Google Scholar]
- Su, L.-Y.; Li, H.; Feng, Y.-M.; Li, G.-D.; Luo, R.; Zho, H.-J.; Lei, X.-G.; Ma, L.; Li, J.-L.; Xu, L. Melatonin attenuates MPTP-induced neurotoxicity via preventing CDK5-mediated autophagy and SNCA/α-synuclein aggregation. Autophagy 2015, 11, 1745–1759. [Google Scholar] [CrossRef] [Green Version]
- Darwish, M.A.; Abdel-Bakky, M.S.; Mssiha, B.A.S.; Abo-Saif, A.A.; Abo-Youssef, A.M. Resveratrol mitigates pancreatic TF activation and autophagy-mediated beta cell death via inhibition of CXCL16/ox-LDL pathway: A novel protective mechanism against type 1 diabetes mellitus. Eur. J. Pharmacol. 2021, 901, 174059. [Google Scholar] [CrossRef]
- Zhao, Y.; Song, W.; Wang, Z.; Wang, Z.; Jin, X.; Xu, J.; Bai, L.; Li, Y.; Cui, J.; Cai, L. Resveratrol attenuates testicular apoptosis in type 1 diabetic mice: Role of Akt-mediated Nrf2 activation and p62-dependent Keap1 degradation. Redox Biol. 2018, 14, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Tsunosue, M.; Mashiko, N.; Ohta, Y.; Matsuo, Y.; Ueda, K.; Ninomiya, M.; Tanaka, S.; Hoshiko, M.; Yoshiyama, Y.; Takeuchi, M.; et al. An α-glucosidase inhibitor, acarbose treatment decreases serum levels of glyceraldehyde-derived advanced glycation end products (AGEs) in patients with type 2 diabetes. Clin. Exp. Med. 2010, 10, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, I.; Oyama, J.; Komoda, H.; Shiraki, A.; Sakamoto, Y.; Taguchi, I.; Hiwatashi, A.; Komatsu, A.; Takeuchi, M.; Yamagishi, S.; et al. Possible effects of glimepiride beyond glycemic control in patients with type 2 diabetes: A preliminary report. Cardiovasc. Diabetol. 2014, 13, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinnouchi, Y.; Yamagishi, S.; Takeuchi, M.; Ishiga, S.; Jinnouchi, Y.; Jinnouchi, J.; Imaizumi, T. Atrovastatin decrease serum levels of advanced glycation end products (AGEs) in patients with type 2 diabetes. Clin. Exp. Med. 2006, 6, 191–193. [Google Scholar] [CrossRef]
- Yanagisawa, K.; Ashihara, J.; Obara, S.; Wada, N.; Takeuchi, M.; Nishino, Y.; Maeda, S.; Ishibashi, Y.; Yamagishi, S. Switching to multiple daily injection therapy with glulisine improves glycemic control, vascular damage and treatment statisfaction in basal insulin glargine-injected diabetic patients. Diabetes Metab. Res. Rev. 2013, 30, 693–700. [Google Scholar] [CrossRef]
- Sandu, O.; Song, K.; Cai, W.; Zheng, F.; Uribarri, J.; Vlassara, H. Insulin resistance and type 2 diabetes in high-fat-fed mice are linked to high glycotoxin intake. Diabetes 2005, 54, 2314–2319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuzan, A. Toxicity advanced glycation end products (Review). Biomed. Rep. 2021, 14, 46. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takata, T.; Sakasai-Sakai, A.; Takeuchi, M. Intracellular Toxic Advanced Glycation End-Products in 1.4E7 Cell Line Induce Death with Reduction of Microtubule-Associated Protein 1 Light Chain 3 and p62. Nutrients 2022, 14, 332. https://doi.org/10.3390/nu14020332
Takata T, Sakasai-Sakai A, Takeuchi M. Intracellular Toxic Advanced Glycation End-Products in 1.4E7 Cell Line Induce Death with Reduction of Microtubule-Associated Protein 1 Light Chain 3 and p62. Nutrients. 2022; 14(2):332. https://doi.org/10.3390/nu14020332
Chicago/Turabian StyleTakata, Takanobu, Akiko Sakasai-Sakai, and Masayoshi Takeuchi. 2022. "Intracellular Toxic Advanced Glycation End-Products in 1.4E7 Cell Line Induce Death with Reduction of Microtubule-Associated Protein 1 Light Chain 3 and p62" Nutrients 14, no. 2: 332. https://doi.org/10.3390/nu14020332
APA StyleTakata, T., Sakasai-Sakai, A., & Takeuchi, M. (2022). Intracellular Toxic Advanced Glycation End-Products in 1.4E7 Cell Line Induce Death with Reduction of Microtubule-Associated Protein 1 Light Chain 3 and p62. Nutrients, 14(2), 332. https://doi.org/10.3390/nu14020332