
α-Lipoic Acid Inhibits Apoptosis by Suppressing the Loss of Ku Proteins in Helicobacter pylori-Infected Human Gastric Epithelial Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell line and Culture Conditions
2.2. Bacterial Strain and Growth Conditions
2.3. Reagents
2.4. Infection of AGS Cells with H. pylori
2.5. Experimental Protocols
2.6. Preparation of Whole-Cell Extracts and Nuclear Extracts
2.7. Measurement of Cell Viability and DNA Fragmentation
2.8. Measurement of Intracellular ROS Levels
2.9. Electrophoretic Mobility Shift Assay (EMSA)
2.10. Western Blotting
2.11. Immunofluorescence Staining
2.12. Co-Immunoprecipitation Assay
2.13. Statistical Analysis
3. Results
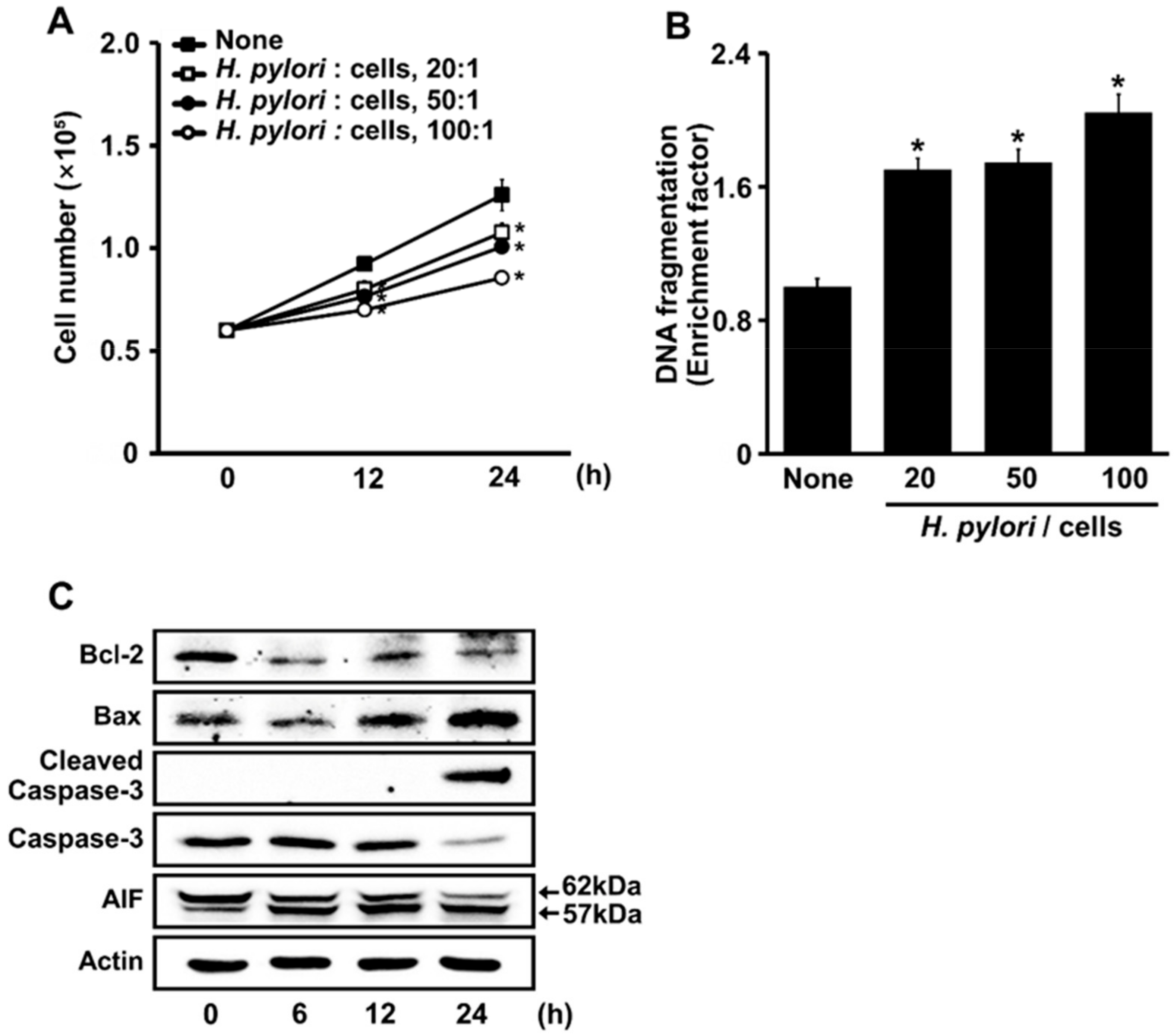
3.1. H. pylori Induces DNA Fragmentation and Apoptosis in AGS Cells
3.2. α-LA Inhibits H. pylori-Induced DNA Fragmentation and Apoptosis in AGS Cells
3.3. α-LA Inhibits H. pylori-Induced Increases in ROS and γ-H2AX in AGS Cells
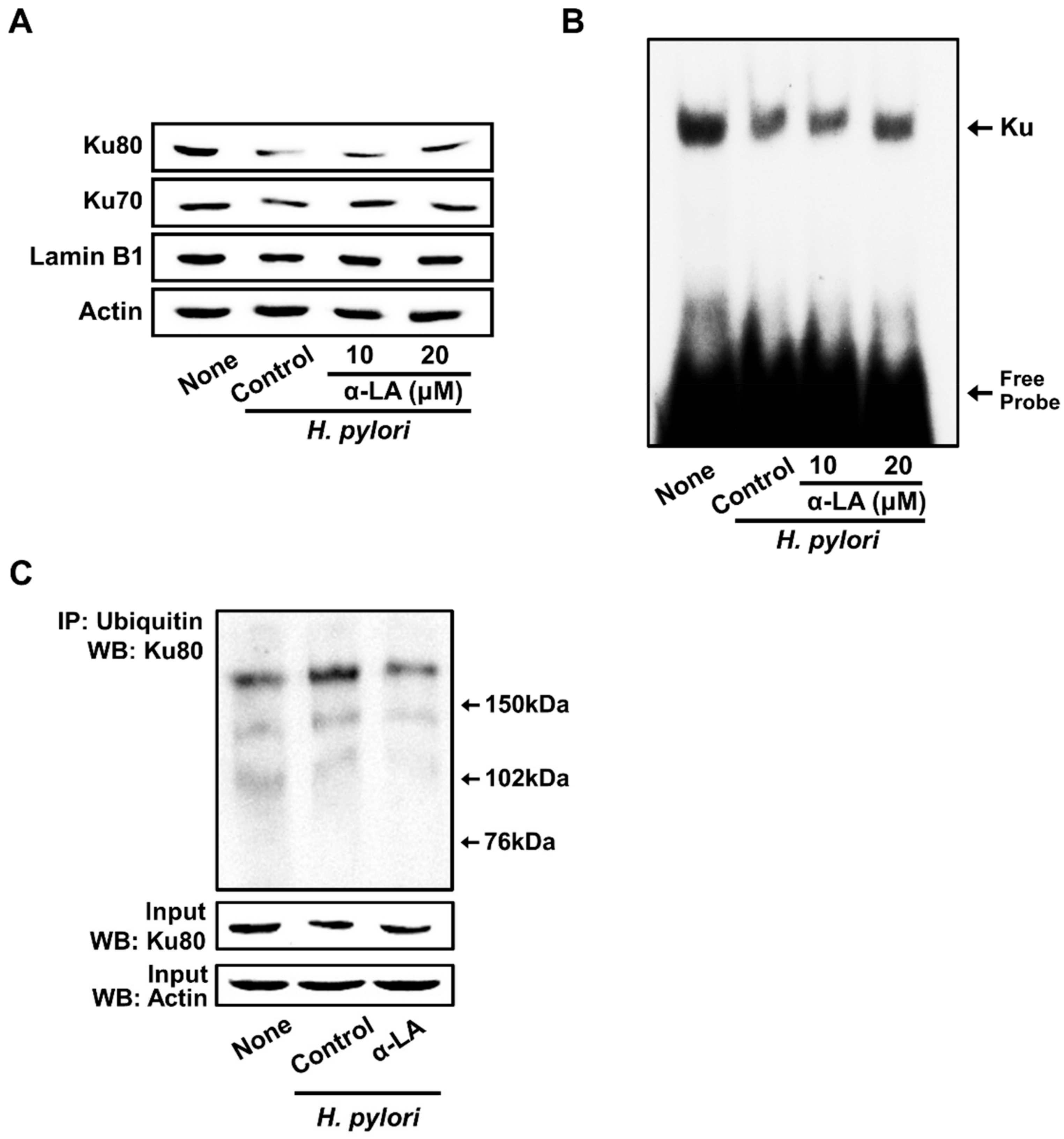
3.4. α-LA Inhibits H. pylori-Induced Decreases in Nuclear Ku70/80 in AGS Cells
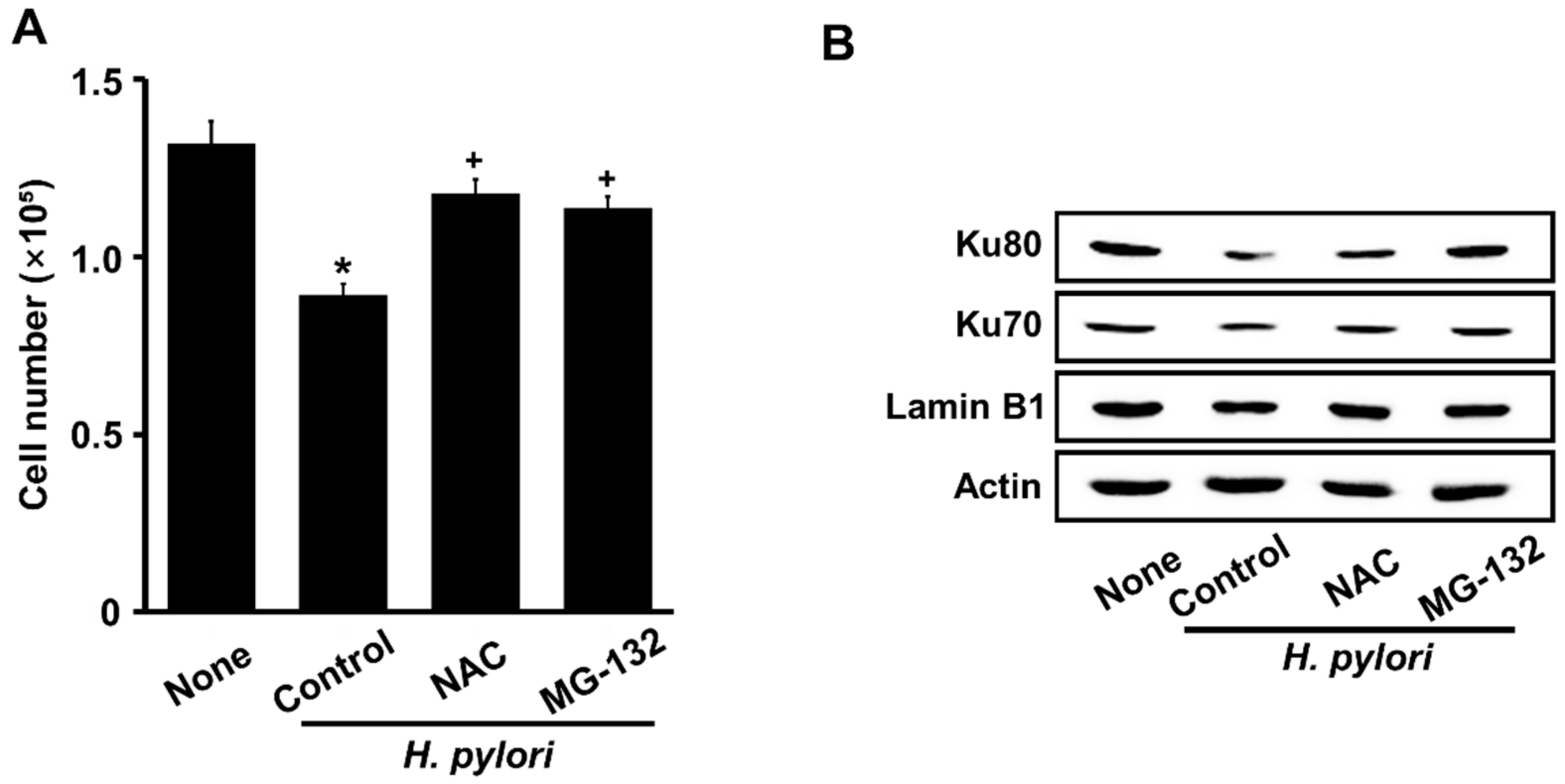
3.5. Both NAC and MG-132 Inhibited Cell Death and Loss of Nuclear Ku Induced by H. pylori in AGS Cells
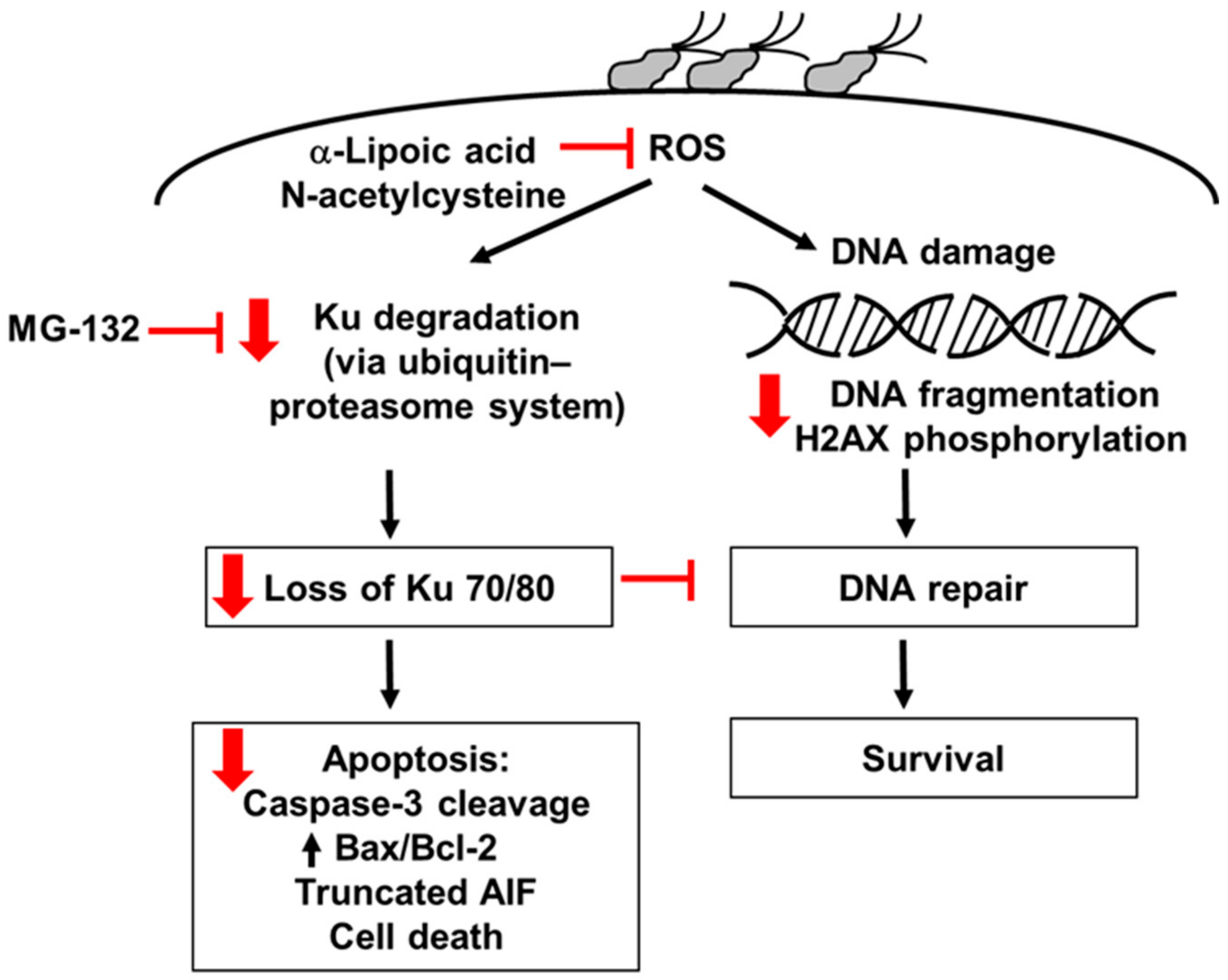
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AGS | gastric epithelial adenocarcinoma cell line |
| α-LA | α lipoic acid |
| AIF | apoptosis-inducing factor |
| H. pylori | Helicobacter pylori |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DCF | dichlorofluorescein |
| DCFH-DA | dichlorofluorescein diacetate |
| DDRs | DNA damage responses |
| DMSO | dimethyl sulfoxide |
| DNA-PKcs | DNA-dependent protein kinase catalytic subunit |
| DSBs | double-strand breaks |
| ECL | enhanced chemiluminescence |
| EDTA | ethylene diaminetetraacetic acid |
| EGTA | ethylene glycol tetraacetic acid |
| EMSA | electrophoretic mobility shift assay |
| FBS | fetal bovine serum |
| HR | homologous recombination |
| H2AX | H2A histone family member X |
| MOI | multiplicity-of-infection |
| NAC | N-acetycysteine |
| NHEJ | non-homologous end joining |
| NP-40 | nonidet P-40 |
| 8-OH-dG | 8-hydroxy-2′-deoxyguanosine |
| PMSF | phenylmethylsulfonylfluoride |
| ROS | reactive oxygen species |
| RPMI | Roswell Park Memorial Institute |
| SDS | sodium dodecylsulfate |
| TBST | Tris-buffered saline and 0.2% Tween-20 |
References
- Marshall, B.J. Helicobacter pylori: The etiologic agent for peptic ulcer. JAMA 1995, 274, 1064–1066. [Google Scholar] [CrossRef] [PubMed]
- Zamani, M.; Ebrahimtabar, F.; Zamani, V.; Miller, W.H.; Alizadeh-Navaei, R.; Shokri-Shirvani, J.; Derakhshan, M.H. Systematic review with meta-analysis: The worldwide prevalence of Helicobacter pylori infection. Aliment. Pharmacol. Ther. 2018, 47, 868–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, B.E.; Cohen, H.; Blaser, M.J. Helicobacter pylori. Clin. Microbiol. Rev. 1997, 10, 720–741. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Peek, R.M.; Blaser, M.J. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer 2002, 2, 28–37. [Google Scholar] [CrossRef]
- Blaser, M.J.; Atherton, J.C. Helicobacter pylori rersistence: Biology and disease. J. Clin. Investig. 2004, 113, 321–333. [Google Scholar] [CrossRef] [Green Version]
- Khansari, N.; Shakiba, Y.; Mahmoudi, M. Chronic inflammation and oxidative stress as a major cause of age-related diseases and cancer. Recent Pat. Inflamm. Allergy Drug Discov. 2009, 3, 73–80. [Google Scholar] [CrossRef]
- Handa, O.; Naito, Y.; Yoshikawa, T. Helicobacter pylori: A ROS-inducing bacterial species in the stomach. Inflamm. Res. 2010, 59, 997–1003. [Google Scholar] [CrossRef]
- De Bont, R.; van Larebeke, N. Endogenous DNA damage in humans: A review of quantitative data. Mutagenesis 2004, 19, 169–185. [Google Scholar] [CrossRef] [Green Version]
- Marnett, L.J. Oxyradicals and DNA damage. Carcinogenesis 2000, 21, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Krokan, H.E.; Bjørås, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Norbury, C.J.; Hickson, I.D. Cellular responses to DNA damage. Annu. Rev. Pharmacol. Toxicol. 2003, 41, 367–401. [Google Scholar] [CrossRef] [Green Version]
- Friedberg, E.C. DNA damage and repair. Nature 2003, 421, 436–440. [Google Scholar] [CrossRef]
- Khanna, K.K.; Jackson, S.P. DNA double-strand breaks: Signaling, repair and the cancer connection. Nat. Genet. 2001, 27, 247–254. [Google Scholar] [CrossRef]
- Jackson, S.P. Sensing and repairing DNA double-strand breaks. Carcinogenesis 2002, 23, 687–696. [Google Scholar] [CrossRef]
- Van Gent, D.C.; Hoeijmakers, J.H.J.; Kanaar, R. Chromosomal stability and the DNA double-stranded break connection. Nat. Rev. Genet. 2001, 2, 196–206. [Google Scholar] [CrossRef]
- Chiruvella, K.K.; Liang, Z.; Wilson, T.E. Repair of double-strand breaks by end joining. Cold Spring Harb. Perspect. Biol. 2013, 5, a012757. [Google Scholar] [CrossRef]
- Fell, V.L.; Schild-Poulter, C. The Ku heterodimer: Function in DNA repair and beyond. Mutat. Res. Rev. Mutat. Res. 2015, 763, 15–29. [Google Scholar] [CrossRef]
- Blier, P.R.; Griffith, A.J.; Craft, J.; Hardin, J.A. Binding of Ku protein to DNA. measurement of affinity for ends and demonstration of binding to nicks. J. Biol. Chem. 1993, 268, 7594–7601. [Google Scholar] [CrossRef]
- Downs, J.A.; Jackson, S.P. A Means to a DNA end: The many roles of Ku. Nat. Rev. Mol. Cell Biol. 2004, 5, 367–378. [Google Scholar] [CrossRef]
- Gullo, C.; Au, M.; Feng, G.; Teoh, G. The biology of Ku and its potential oncogenic role in cancer. Biochim. Biophys. Acta Rev. Cancer 2006, 1765, 223–234. [Google Scholar] [CrossRef]
- Amsel, A.D.; Rathaus, M.; Kronman, N.; Cohen, H.Y. Regulation of the proapoptotic factor Bax by Ku70-dependent deubiquitylation. Proc. Natl. Acad. Sci. USA 2008, 105, 5117–5122. [Google Scholar] [CrossRef] [Green Version]
- Sawada, M.; Sun, W.; Hayes, P.; Leskov, K.; Boothman, D.A.; Matsuyama, S. Ku70 suppresses the apoptotic translocation of Bax to mitochondria. Nat. Cell Biol. 2003, 5, 320–329. [Google Scholar] [CrossRef]
- Cohen, H.Y.; Lavu, S.; Bitterman, K.J.; Hekking, B.; Imahiyerobo, T.A.; Miller, C.; Frye, R.; Ploegh, H.; Kessler, B.M.; Sinclair, D.A. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol. Cell 2004, 13, 627–638. [Google Scholar] [CrossRef]
- De Zio, D.; Cianfanelli, V.; Cecconi, F. New insights into the link between DNA damage and apoptosis. Antioxid. Redox Signal. 2013, 19, 559–571. [Google Scholar] [CrossRef] [Green Version]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006, 12, 440–450. [Google Scholar] [CrossRef]
- Qi, D.; Hu, Y.; Li, J.; Peng, T.; Su, J.; He, Y.; Ji, W. Hyperthermia induces apoptosis of 786-O cells through suppressing Ku80 expression. PLoS ONE 2015, 10, e0122977. [Google Scholar] [CrossRef] [Green Version]
- Li, J.J.; Gu, Q.H.; Li, M.; Yang, H.P.; Cao, L.M.; Hu, C.P. Role of Ku70 and Bax in epigallocatechin-3-gallate-induced apoptosis of A549 Cells in vivo. Oncol. Lett. 2013, 5, 101–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.Y.; Lim, J.W.; Kim, H.; Morio, T.; Kim, K.H. Oxidative stress induces nuclear loss of DNA repair proteins Ku70 and Ku80 and apoptosis in pancreatic acinar AR42J cells. J. Biol. Chem. 2003, 278, 36676–36687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.; Choi, J.; Lim, J.W.; Kim, H. β-Carotene-induced apoptosis is mediated with loss of Ku proteins in gastric cancer AGS cells. Genes Nutr. 2015, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase Chromatin domains involved in DNA double-strand breaks in vivo. J. Cell. Biol. 1999, 146, 905–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shay, K.P.; Moreau, R.F.; Smith, E.J.; Hagen, T.M. Is alpha-lipoic acid a scavenger of reactive oxygen species in vivo? Evidence for its initiation of stress signaling pathways that promote endogenous antioxidant capacity. IUBMB Life 2008, 60, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Lodge, J.K.; Packer, L. Chapter 9—Natural sources of lipoic acid in plant and animal tissues. In Antioxidant Food Supplements in Human Health; Academic Press: Cambridge, MA, USA, 1999; pp. 121–134. [Google Scholar]
- Reed, L.J. Multienzyme complexes. Acc. Chem. Res. 1974, 7, 40–46. [Google Scholar] [CrossRef]
- Packer, L.; Witt, E.H.; Tritschler, H.J. Alpha-lipoic acid as a biological antioxidant. Free Radic. Biol. Med. 1995, 19, 227–250. [Google Scholar] [CrossRef]
- Shay, K.P.; Moreau, R.F.; Smith, E.J.; Smith, A.R.; Hagen, T.M. Alpha-lipoic acid as a dietary supplement: Molecular mechanisms and therapeutic potential. Biochim. Biophys. Acta. 2009, 1790, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Salehi, B.; Berkay Yılmaz, Y.; Antika, G.; Boyunegmez Tumer, T.; Fawzi Mahomoodally, M.; Lobine, D.; Akram, M.; Riaz, M.; Capanoglu, E.; Sharopov, F.; et al. Insights on the use of α-lipoic acid for therapeutic purposes. Biomolecules 2019, 9, 356. [Google Scholar] [CrossRef] [Green Version]
- Mottola, F.; Santonastaso, M.; Iovine, C.; Rossetti, C.; Ronga, V.; Rocco, L. DNA damage in human amniotic cells: Antigenotoxic potential of curcumin and α-lipoic acid. Antioxidants 2021, 10, 1137. [Google Scholar] [CrossRef]
- Shirpoor, A.; Minassian, S.; Salami, S.; Khadem-Ansari, M.H.; Yeghiazaryan, M. Alpha--lipoic acid decreases DNA damage and oxidative stress induced by alcohol in the developing hippocampus and cerebellum of rat. Cell Physiol. Biochem. 2008, 22, 769–776. [Google Scholar] [CrossRef]
- Uemura, N.; Okamoto, S.; Yamamoto, S.; Matsumura, N.; Yamaguchi, S.; Yamakido, M.; Taniyama, K.; Sasaki, N.; Schlemper, R.J. Helicobacter pylori infection and the development of gastric cancer. N. Engl. Med. 2001, 345, 784–789. [Google Scholar] [CrossRef]
- Kim, S.H.; Lim, J.W.; Kim, H. Astaxanthin inhibits mitochondrial dysfunction and interleukin-8 expression in Helicobacter pylori-infected gastric epithelial cells. Nutrients 2018, 10, 1320. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Lim, J.W.; Kim, H. Korean red ginseng extract inhibits IL-8 expression via Nrf2 activation in Helicobacter pylori-infected gastric epithelial cells. Nutrients 2022, 14, 1044. [Google Scholar] [CrossRef]
- Kyung, S.; Lim, J.W.; Kim, H. α-Lipoic acid inhibits IL-8 expression by activating Nrf2 signaling in Helicobacter pylori-infected gastric epithelial cells. Nutrients 2019, 11, 2524. [Google Scholar] [CrossRef] [Green Version]
- Davies, G.R.; Simmonds, N.J.; Stevens, T.R.J.; Sheaff, M.T.; Banatvala, N.; Laurenson, I.F.; Blake, D.R.; Rampton, D.S. Helicobacter pylori stimulates antral mucosal reactive oxygen metabolite production in vivo. Gut 1994, 35, 179–185. [Google Scholar] [CrossRef]
- Ernst, P. The Role of Inflammation in the pathogenesis of gastric cancer. Aliment. Pharmacol. Ther. 1999, 13, 13–18. [Google Scholar] [CrossRef]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 31, 118. [Google Scholar] [CrossRef]
- Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Multiple pathways of cytochrome c release from mitochondria in apoptosis. Biochim. Biophys. Acta. Bioenerg. 2006, 1757, 639–647. [Google Scholar] [CrossRef] [Green Version]
- Cohen, G.M. Caspases: The executioners of apoptosis. Biochem. J. 1997, 326, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Porter, A.G.; Jänicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef]
- Jänicke, R.U.; Sprengart, M.L.; Wati, M.R.; Porter, A.G. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J. Biol. Chem. 1998, 273, 9357–9360. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Yan, C.; Schor, N.F. Apoptosis in the absence of caspase 3. Oncogene 2001, 20, 6570–6578. [Google Scholar] [CrossRef] [Green Version]
- Cregan, S.P.; Dawson, V.L.; Slack, R.S. Role of AIF in caspase-dependent and caspase-independent cell death. Oncogene 2004, 23, 2785–2796. [Google Scholar] [CrossRef] [Green Version]
- Maeda, S.; Yoshida, H.; Mitsuno, Y.; Hirata, Y.; Ogura, K.; Shiratori, Y.; Omata, M. Analysis of apoptotic and antiapoptotic signalling pathways induced by Helicobacter pylori. Gut 2002, 50, 771–778. [Google Scholar] [CrossRef]
- Ashktorab, H.; Dashwood, R.H.; Dashwood, M.M.; Zaidi, S.I.; Hewitt, S.M.; Green, W.R.; Lee, E.L.; Daremipouran, M.; Nouraie, M.; Malekzadeh, R.; et al. pylori-induced apoptosis in human gastric cancer cells mediated via the release of apoptosis-inducing factor from mitochondria. Helicobacter 2008, 13, 506–517. [Google Scholar] [CrossRef]
- Kontizas, E.; Tastsoglou, S.; Karamitros, T.; Karayiannis, Y.; Kollia, P.; Hatzigeorgiou, A.G.; Sgouras, D.N. Impact of Helicobacter pylori infection and its major virulence factor CagA on DNA damage repair. Microorganisms 2020, 8, 2007. [Google Scholar] [CrossRef]
- Tonotsuka, N.; Hosoi, Y.; Miyazaki, S.; Miyata, G.; Sugawara, K.; Mori, T.; Ouchi, N.; Satomi, S.; Matsumoto, Y.; Nakagawa, K.; et al. Heterogeneous expression of DNA-dependent protein kinase in esophageal cancer and normal epithelium. Int. J. Mol. Med. 2006, 18, 441–447. [Google Scholar] [CrossRef] [Green Version]
- Pucci, S.; Paola, M.; Fabiola, S.; David, B.A.; Luigi, S.G. Bax-Ku70-clusterin interactions in human colon cancer progression. Cell Cycle 2009, 8, 473–481. [Google Scholar] [CrossRef] [Green Version]
- López-Burillo, S.; Tan, D.X.; Mayo, J.C.; Sainz, R.M.; Manchester, L.C.; Reiter, R.J. Melatonin, xanthurenic acid, resveratrol, EGCG, vitamin C and alpha-lipoic acid differentially reduce oxidative DNA damage induced by fenton reagents: A study of their individual and synergistic actions. J. Pineal. Res. 2003, 34, 269–277. [Google Scholar] [CrossRef]
- Trivedi, P.P.; Jena, G.B. Role of α-lipoic acid in dextran sulfate sodium-induced ulcerative colitis in mice: Studies on inflammation, oxidative stress, DNA damage and fibrosis. Food Chem. Toxicol. 2013, 59, 339–355. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, D.; Lim, J.W.; Kim, H. α-Lipoic Acid Inhibits Apoptosis by Suppressing the Loss of Ku Proteins in Helicobacter pylori-Infected Human Gastric Epithelial Cells. Nutrients 2022, 14, 3206. https://doi.org/10.3390/nu14153206
Park D, Lim JW, Kim H. α-Lipoic Acid Inhibits Apoptosis by Suppressing the Loss of Ku Proteins in Helicobacter pylori-Infected Human Gastric Epithelial Cells. Nutrients. 2022; 14(15):3206. https://doi.org/10.3390/nu14153206
Chicago/Turabian StylePark, Dayong, Joo Weon Lim, and Hyeyoung Kim. 2022. "α-Lipoic Acid Inhibits Apoptosis by Suppressing the Loss of Ku Proteins in Helicobacter pylori-Infected Human Gastric Epithelial Cells" Nutrients 14, no. 15: 3206. https://doi.org/10.3390/nu14153206
APA StylePark, D., Lim, J. W., & Kim, H. (2022). α-Lipoic Acid Inhibits Apoptosis by Suppressing the Loss of Ku Proteins in Helicobacter pylori-Infected Human Gastric Epithelial Cells. Nutrients, 14(15), 3206. https://doi.org/10.3390/nu14153206