
mTORC1 and mTORC2 Complexes Regulate the Untargeted Metabolomics and Amino Acid Metabolites Profile through Mitochondrial Bioenergetic Functions in Pancreatic Beta Cells



Abstract
1. Introduction
2. Methods
2.1. Reagents
2.2. Antibodies
2.3. Cell Lysis for Western Blotting Analysis
2.4. Metabolites Extraction for the HRMS Untargeted Metabolomics Study
2.5. LC/MS/MS High-Resolution Mass Spectrometry (HRMS) Untargeted Metabolomics
2.6. Live Cells Bioenergetics
2.7. Live Cells Mitochondrial Functions
2.8. Data Processing and Statistical Analysis
3. Results
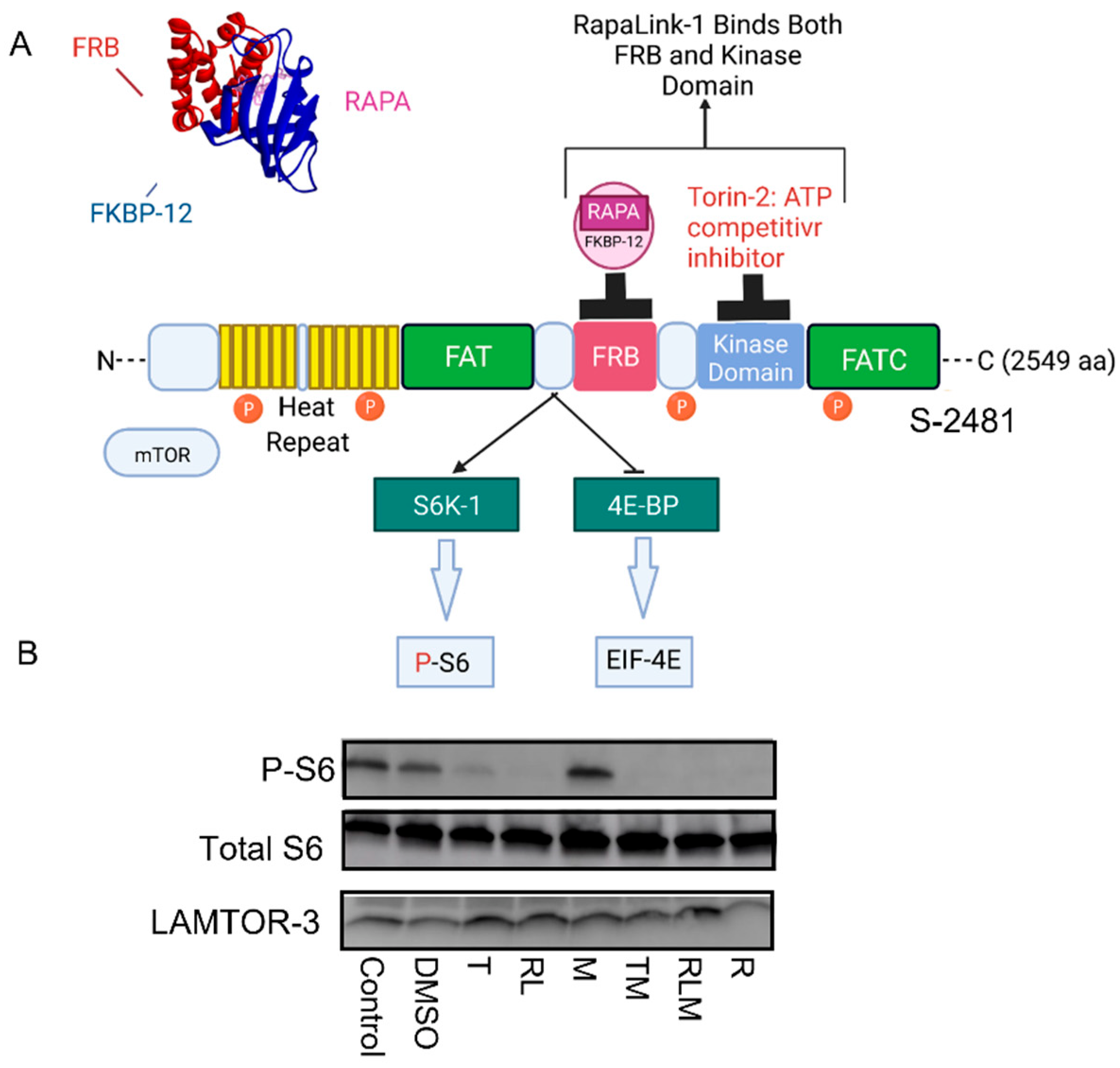
3.1. mTOR Complexes Module Cell Signaling
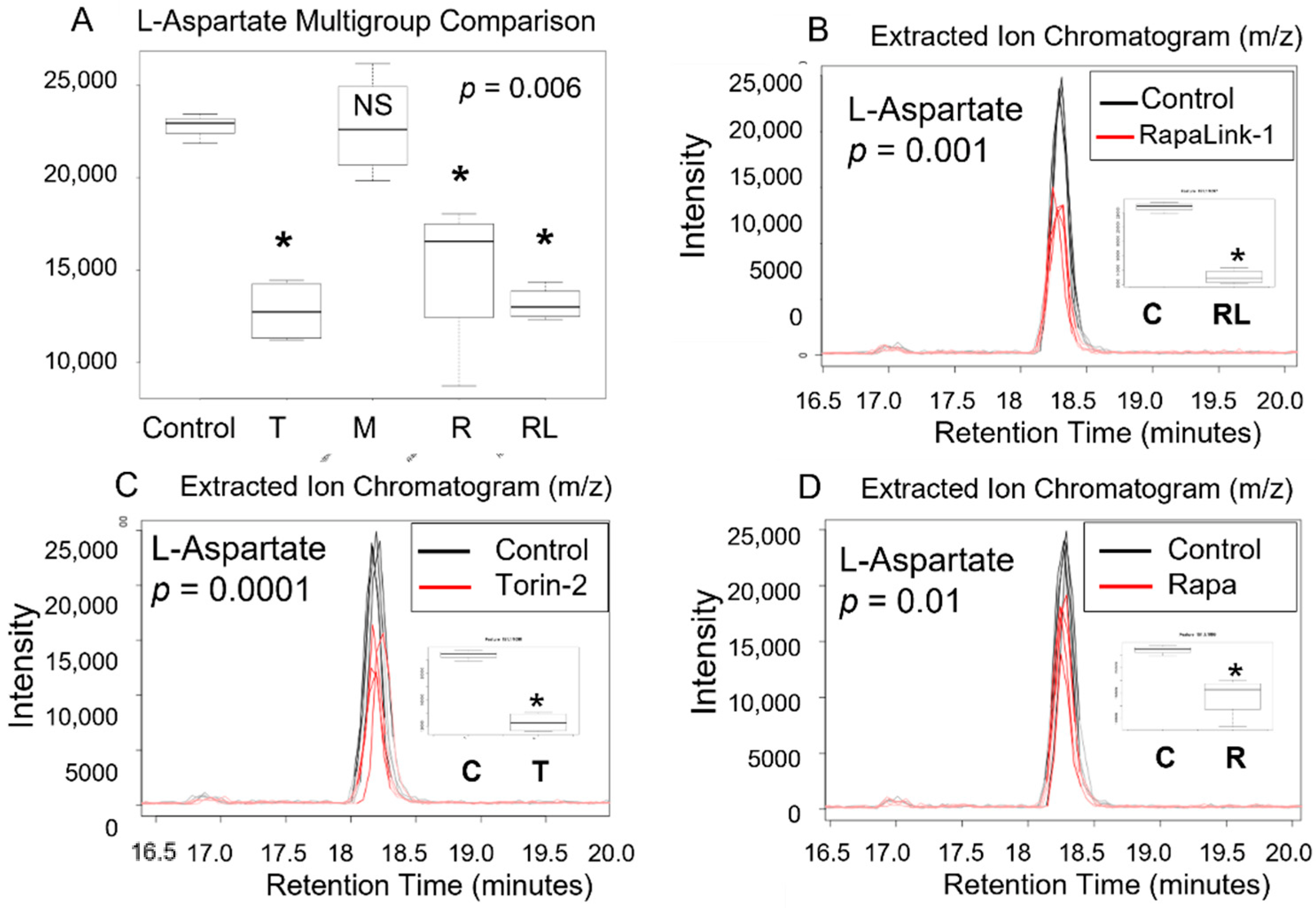
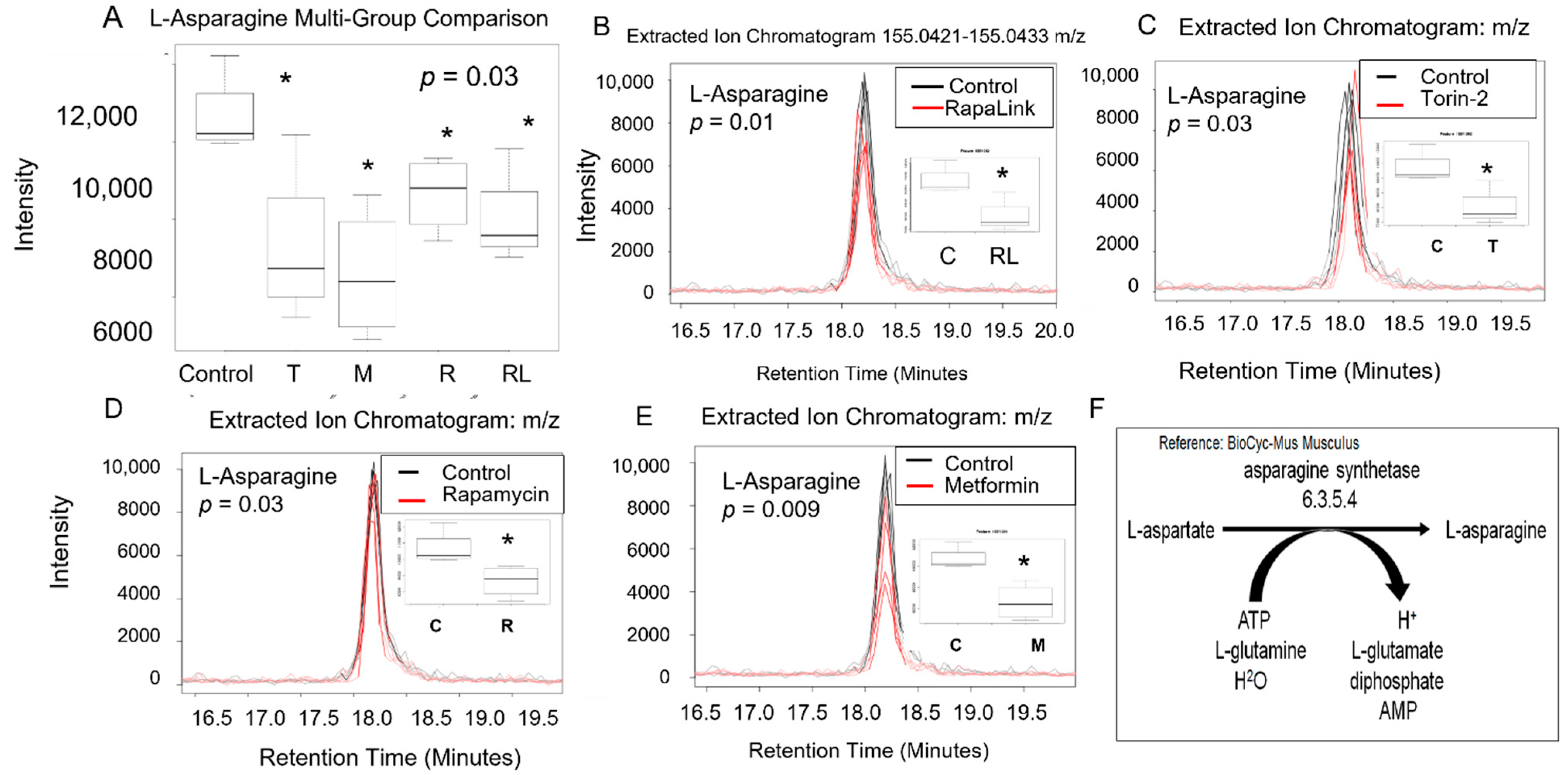
3.2. mTORC1 and mTORC2 and Amino Acid Levels
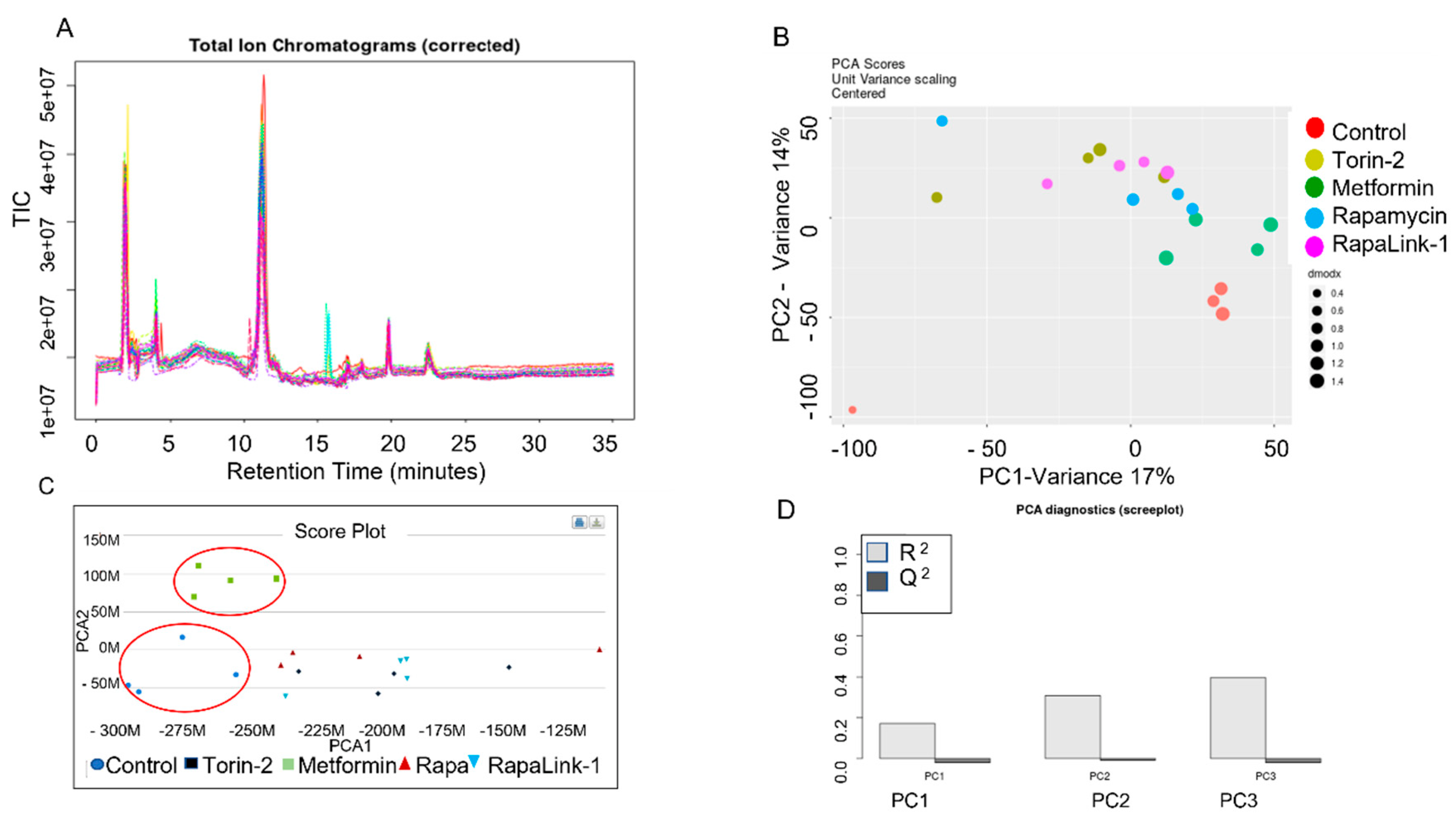
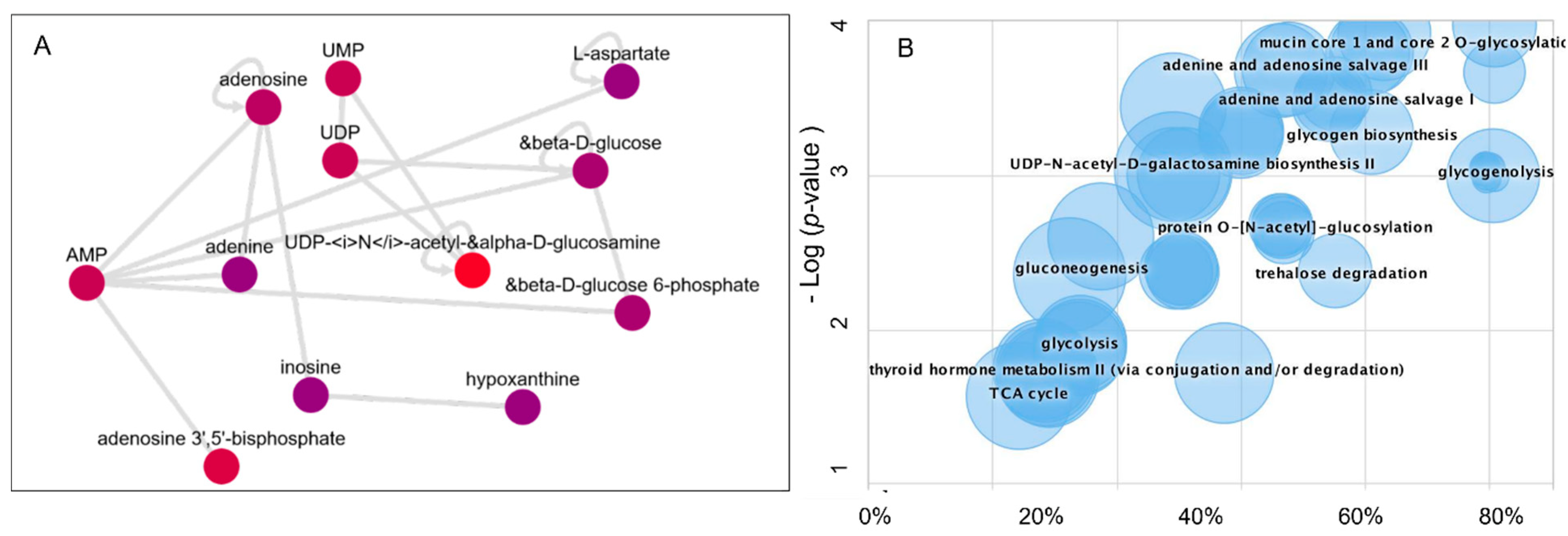
3.3. Dysregulated Metabolic Pathways and the Metabolic Activity Network Predictive Model
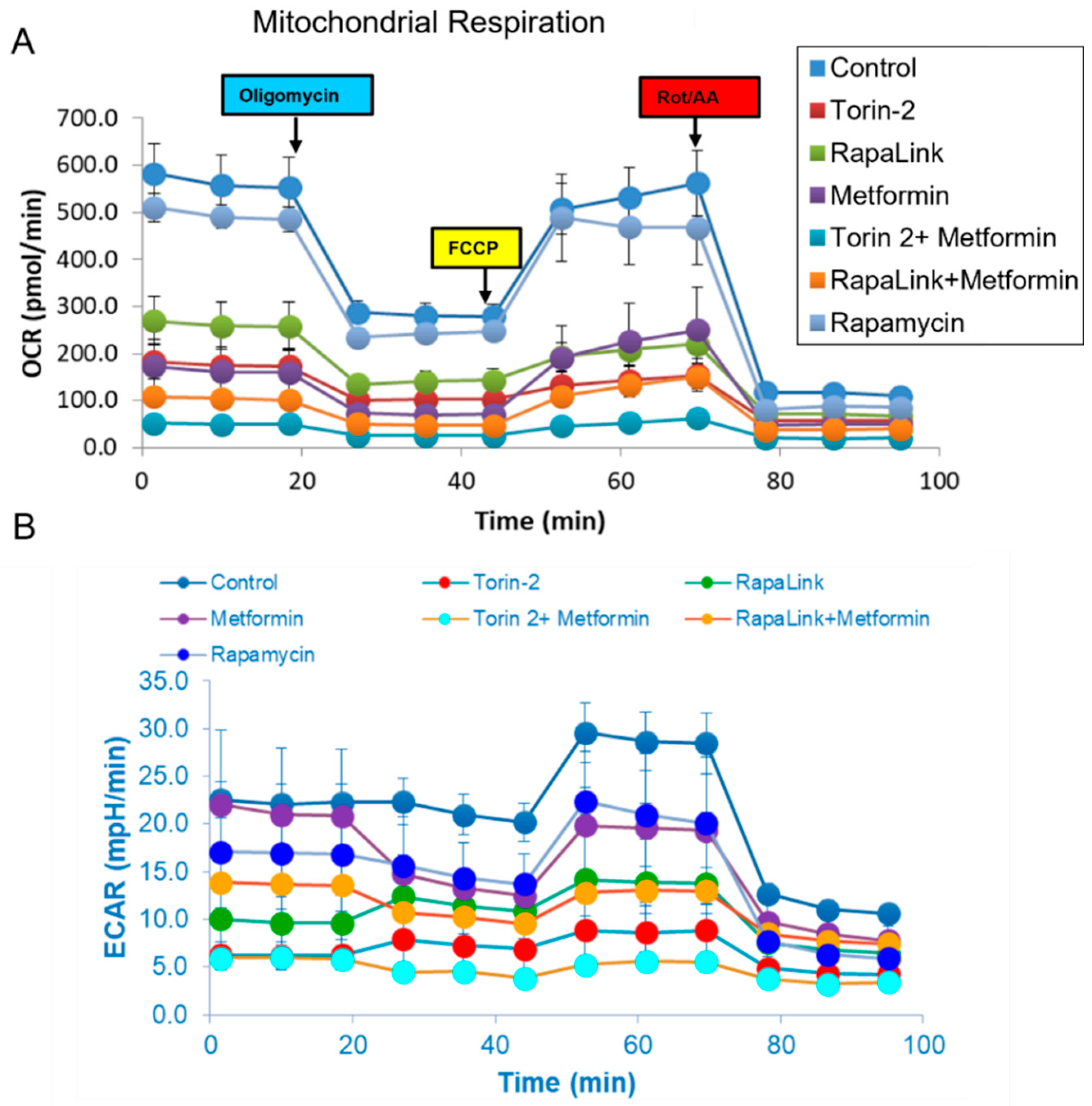
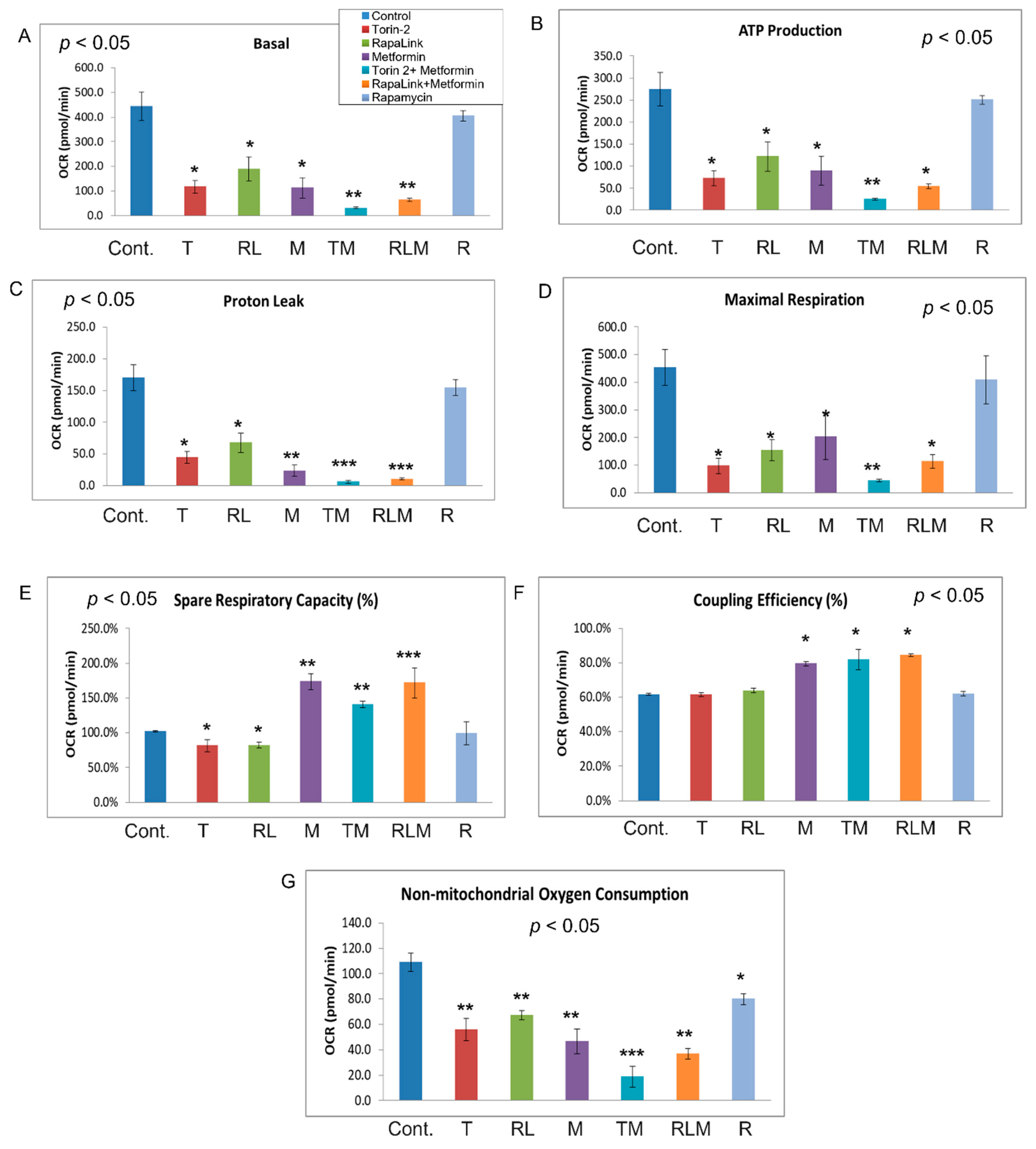
3.4. Live-Cell Bioenergetics
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Ben-Sahra, I.; Manning, B.D. mTORC1 signaling and the metabolic control of cell growth. Curr. Opin. Cell Biol. 2017, 45, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Soliman, G.A.; Acosta-Jaquez, H.A.; Dunlop, E.A.; Ekim, B.; Maj, N.E.; Tee, A.R.; Fingar, D.C. mTOR Ser-2481 autophosphorylation monitors mTORC-specific catalytic activity and clarifies rapamycin mechanism of action. J. Biol. Chem. 2010, 285, 7866–7879. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, R.M.; Seandel, M.; Falciatori, I.; Rafii, S.; Pandolfi, P.P. Plzf regulates germline progenitor self-renewal by opposing mTORC1. Cell 2010, 142, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Elghazi, L.; Blandino-Rosano, M.; Alejandro, E.; Cras-Meneur, C.; Bernal-Mizrachi, E. Role of nutrients and mTOR signaling in the regulation of pancreatic progenitors development. Mol. Metab. 2017, 6, 560–573. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Lupse, B.; Maedler, K.; Ardestani, A. mTORC2 Signaling: A Path for Pancreatic beta Cell’s Growth and Function. J. Mol. Biol. 2018, 430, 904–918. [Google Scholar] [CrossRef]
- Goodwin, P.J.; Stambolic, V.; Lemieux, J.; Chen, B.E.; Parulekar, W.R.; Gelmon, K.A.; Hershman, D.L.; Hobday, T.J.; Ligibel, J.A.; Mayer, I.A.; et al. Evaluation of metformin in early breast cancer: A modification of the traditional paradigm for clinical testing of anti-cancer agents. Breast Cancer Res. Treat. 2011, 126, 215–220. [Google Scholar] [CrossRef]
- Sabatini, D.M. Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc. Natl. Acad. Sci. USA 2017, 114, 11818–11825. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Chantranupong, L.; Scaria, S.M.; Saxton, R.A.; Gygi, M.P.; Shen, K.; Wyant, G.A.; Wang, T.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. The CASTOR Proteins Are Arginine Sensors for the mTORC1 Pathway. Cell 2016, 165, 153–164. [Google Scholar] [CrossRef]
- Chantranupong, L.; Wolfson, R.L.; Orozco, J.M.; Saxton, R.A.; Scaria, S.M.; Bar-Peled, L.; Spooner, E.; Isasa, M.; Gygi, S.P.; Sabatini, D.M. The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep. 2014, 9, 1–8. [Google Scholar] [CrossRef]
- Lei, H.T.; Ma, J.; Sanchez Martinez, S.; Gonen, T. Crystal structure of arginine-bound lysosomal transporter SLC38A9 in the cytosol-open state. Nat. Struct. Mol. Biol. 2018, 25, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.W.S.; Sim, A.Y.L.; Long, Y.C. Glutamine metabolism regulates autophagy-dependent mTORC1 reactivation during amino acid starvation. Nat. Commun. 2017, 8, 338. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Shi, T.; Cui, X.; Yan, L.; Wang, Q.; Xu, X.; Zhao, Q.; Xu, X.; Tang, Q.Q.; Tang, H.; et al. Asparagine reinforces mTORC1 signaling to boost thermogenesis and glycolysis in adipose tissues. EMBO J. 2021, 40, e108069. [Google Scholar] [CrossRef] [PubMed]
- Kikani, C.K.; Wu, X.; Fogarty, S.; Kang, S.A.W.; Dephoure, N.; Gygi, S.P.; Sabatini, D.M.; Rutter, J. Activation of PASK by mTORC1 is required for the onset of the terminal differentiation program. Proc. Natl. Acad. Sci. USA 2019, 116, 10382–10391. [Google Scholar] [CrossRef] [PubMed]
- Kuroshima, K.; Yoshino, H.; Okamura, S.; Tsuruda, M.; Osako, Y.; Sakaguchi, T.; Sugita, S.; Tatarano, S.; Nakagawa, M.; Enokida, H. Potential new therapy of Rapalink-1, a new generation mammalian target of rapamycin inhibitor, against sunitinib-resistant renal cell carcinoma. Cancer Sci. 2020, 111, 1607–1618. [Google Scholar] [CrossRef]
- Lawlor, N.; Youn, A.; Kursawe, R.; Ucar, D.; Stitzel, M.L. Alpha TC1 and Beta-TC-6 genomic profiling uncovers both shared and distinct transcriptional regulatory features with their primary islet counterparts. Sci. Rep. 2017, 7, 11959. [Google Scholar] [CrossRef]
- Gautam, N.; Roy, U.; Balkundi, S.; Puligujja, P.; Guo, D.; Smith, N.; Liu, X.M.; Lamberty, B.; Morsey, B.; Fox, H.S.; et al. Preclinical pharmacokinetics and tissue distribution of long-acting nanoformulated antiretroviral therapy. Antimicrob. Agents Chemother. 2013, 57, 3110–3120. [Google Scholar] [CrossRef]
- Soliman, G.A.; Steenson, S.M.; Etekpo, A.H. Effects of Metformin and a Mammalian Target of Rapamycin (mTOR) ATP-Competitive Inhibitor on Targeted Metabolomics in Pancreatic Cancer Cell Line. Metabolomics 2016, 6, 183. [Google Scholar]
- Soliman, G.A.; Acosta-Jaquez, H.A.; Fingar, D.C. mTORC1 inhibition via rapamycin promotes triacylglycerol lipolysis and release of free fatty acids in 3T3-L1 adipocytes. Lipids 2010, 45, 1089–1100. [Google Scholar] [CrossRef]
- Lorenz, M.A.; Burant, C.F.; Kennedy, R.T. Reducing time and increasing sensitivity in sample preparation for adherent mammalian cell metabolomics. Anal. Chem. 2011, 83, 3406–3414. [Google Scholar] [CrossRef]
- Tautenhahn, R.; Bottcher, C.; Neumann, S. Highly sensitive feature detection for high resolution LC/MS. BMC Bioinform. 2008, 9, 504. [Google Scholar] [CrossRef] [PubMed]
- Tautenhahn, R.; Patti, G.J.; Rinehart, D.; Siuzdak, G. XCMS Online: A web-based platform to process untargeted metabolomic data. Anal. Chem. 2012, 84, 5035–5039. [Google Scholar] [CrossRef] [PubMed]
- Gowda, H.; Ivanisevic, J.; Johnson, C.H.; Kurczy, M.E.; Benton, H.P.; Rinehart, D.; Nguyen, T.; Ray, J.; Kuehl, J.; Arevalo, B.; et al. Interactive XCMS Online: Simplifying advanced metabolomic data processing and subsequent statistical analyses. Anal. Chem. 2014, 86, 6931–6939. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Almenara, X.; Montenegro-Burke, J.R.; Ivanisevic, J.; Thomas, A.; Sidibe, J.; Teav, T.; Guijas, C.; Aisporna, A.E.; Rinehart, D.; Hoang, L.; et al. XCMS-MRM and METLIN-MRM: A cloud library and public resource for targeted analysis of small molecules. Nat. Methods 2018, 15, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Park, Y.; Duraisingham, S.; Strobel, F.H.; Khan, N.; Soltow, Q.A.; Jones, D.P.; Pulendran, B. Predicting network activity from high throughput metabolomics. PLoS Comput. Biol. 2013, 9, e1003123. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Aksoy, O.; Wong, R.A.; Ilkhanizadeh, S.; Novotny, C.J.; Gustafson, W.C.; Truong, A.Y.; Cayanan, G.; Simonds, E.F.; Haas-Kogan, D.; et al. A Kinase Inhibitor Targeted to mTORC1 Drives Regression in Glioblastoma. Cancer Cell 2017, 31, 424–435. [Google Scholar] [CrossRef]
- Ntranos, A.; Ntranos, V.; Bonnefil, V.; Liu, J.; Kim-Schulze, S.; He, Y.; Zhu, Y.; Brandstadter, R.; Watson, C.T.; Sharp, A.J.; et al. Fumarates target the metabolic-epigenetic interplay of brain-homing T cells in multiple sclerosis. Brain 2019, 142, 647–661. [Google Scholar] [CrossRef]
- He, Y.; Kim, J.Y.; Dupree, J.; Tewari, A.; Melendez-Vasquez, C.; Svaren, J.; Casaccia, P. Yy1 as a molecular link between neuregulin and transcriptional modulation of peripheral myelination. Nat. Neurosci. 2010, 13, 1472–1480. [Google Scholar] [CrossRef]
- Domingo-Almenara, X.; Siuzdak, G. Metabolomics Data Processing Using XCMS. Methods Mol. Biol. 2020, 2104, 11–24. [Google Scholar]
- Alboniga, O.E.; Gonzalez, O.; Alonso, R.M.; Xu, Y.; Goodacre, R. Optimization of XCMS parameters for LC-MS metabolomics: An assessment of automated versus manual tuning and its effect on the final results. Metabolomics 2020, 16, 14. [Google Scholar] [CrossRef]
- Forsberg, E.M.; Huan, T.; Rinehart, D.; Benton, H.P.; Warth, B.; Hilmers, B.; Siuzdak, G. Data processing, multi-omic pathway mapping, and metabolite activity analysis using XCMS Online. Nat. Protoc. 2018, 13, 633–651. [Google Scholar] [CrossRef] [PubMed]
- Huan, T.; Forsberg, E.M.; Rinehart, D.; Johnson, C.H.; Ivanisevic, J.; Benton, H.P.; Fang, M.; Aisporna, A.; Hilmers, B.; Poole, F.L.; et al. Systems biology guided by XCMS Online metabolomics. Nat. Methods 2017, 14, 461–462. [Google Scholar] [CrossRef] [PubMed]
- Benton, H.P.; Ivanisevic, J.; Mahieu, N.G.; Kurczy, M.E.; Johnson, C.H.; Franco, L.; Rinehart, D.; Valentine, E.; Gowda, H.; Ubhi, B.K.; et al. Autonomous metabolomics for rapid metabolite identification in global profiling. Anal. Chem. 2015, 87, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Kurczy, M.E.; Ivanisevic, J.; Johnson, C.H.; Uritboonthai, W.; Hoang, L.; Fang, M.; Hicks, M.; Aldebot, A.; Rinehart, D.; Mellander, L.J.; et al. Determining conserved metabolic biomarkers from a million database queries. Bioinformatics 2015, 31, 3721–3724. [Google Scholar] [CrossRef][Green Version]
- Najdekr, L.; Friedecky, D.; Tautenhahn, R.; Pluskal, T.; Wang, J.; Huang, Y.; Adam, T. Influence of Mass Resolving Power in Orbital Ion-Trap Mass Spectrometry-Based Metabolomics. Anal. Chem. 2016, 88, 11429–11435. [Google Scholar] [CrossRef]
- Conley, C.J.; Smith, R.; Torgrip, R.J.; Taylor, R.M.; Tautenhahn, R.; Prince, J.T. Massifquant: Open-source Kalman filter-based XC-MS isotope trace feature detection. Bioinformatics 2014, 30, 2636–2643. [Google Scholar] [CrossRef]
- Tautenhahn, R.; Patti, G.J.; Kalisiak, E.; Miyamoto, T.; Schmidt, M.; Lo, F.Y.; McBee, J.; Baliga, N.S.; Siuzdak, G. metaXCMS: Second-order analysis of untargeted metabolomics data. Anal. Chem. 2011, 83, 696–700. [Google Scholar] [CrossRef]
- Patti, G.J.; Tautenhahn, R.; Rinehart, D.; Cho, K.; Shriver, L.P.; Manchester, M.; Nikolskiy, I.; Johnson, C.H.; Mahieu, N.G.; Siuzdak, G. A view from above: Cloud plots to visualize global metabolomic data. Anal. Chem. 2013, 85, 798–804. [Google Scholar] [CrossRef]
- Ramanathan, A.; Schreiber, S.L. Direct control of mitochondrial function by mTOR. Proc. Natl. Acad. Sci. USA 2009, 106, 22229–22232. [Google Scholar] [CrossRef]
- Rosario, F.J.; Gupta, M.B.; Myatt, L.; Powell, T.L.; Glenn, J.P.; Cox, L.; Jansson, T. Mechanistic Target of Rapamycin Complex 1 Promotes the Expression of Genes Encoding Electron Transport Chain Proteins and Stimulates Oxidative Phosphorylation in Primary Human Trophoblast Cells by Regulating Mitochondrial Biogenesis. Sci. Rep. 2019, 9, 246. [Google Scholar] [CrossRef]
- Shigeyama, Y.; Kobayashi, T.; Kido, Y.; Hashimoto, N.; Asahara, S.; Matsuda, T.; Takeda, A.; Inoue, T.; Shibutani, Y.; Koyanagi, M.; et al. Biphasic response of pancreatic beta-cell mass to ablation of tuberous sclerosis complex 2 in mice. Mol. Cell Biol. 2008, 28, 2971–2979. [Google Scholar] [CrossRef] [PubMed]
- Ghomlaghi, M.; Yang, G.; Shin, S.Y.; James, D.E.; Nguyen, L.K. Dynamic modelling of the PI3K/MTOR signalling network uncovers biphasic dependence of mTORC1 activity on the mTORC2 subunit SIN1. PLoS Comput. Biol. 2021, 17, e1008513. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Varamini, B.; Lamming, D.W.; Sabatini, D.M.; Baur, J.A. Rapamycin has a biphasic effect on insulin sensitivity in C2C12 myotubes due to sequential disruption of mTORC1 and mTORC2. Front. Genet. 2012, 3, 177. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amino Acid | Mass/Ion | Retention Time (RT) | Intensity | p-Value | ||||
|---|---|---|---|---|---|---|---|---|
| (m/z) | Control | Torin-2 | Metformin | RAPA | RapaLink-1 | |||
| Glycine | 98.02123 | 18.29 | 0.034 | |||||
| L-Alanine | 90.05473 | 9.08 | 0.077 | |||||
| L-Serine | 106.0497 | 18.34 | 0.279 | |||||
| L-Proline | 116.0705 | 16.76 | 0.056 | |||||
| L-Valine | 118.086 | 19.97 | 0.307 | |||||
| L-Threonine | 120.0654 | 18.53 | 0.296 | |||||
| L-Cysteine | 122.0268 | 16.83 | 0.154 | |||||
| L-Leucine | 132.1018 | 13.51 | 0.676 | |||||
| L-Isoleucine | 132.1019 | 3.31 | 0.056 | |||||
| L-Asparagine | 133.0609 | 18.19 | 0.042 | |||||
| L-Aspartate | 134.0448 | 17.01 | 0.009 | |||||
| L-Glutamine | 147.0763 | 17.94 | 0.307 | |||||
| L-Lysine | 147.1133 | 2.188 | 0.182 | |||||
| L-Glutamate | 148.0604 | 17 | 0.012 | |||||
| L-Methionine | 172.0402 | 14.32 | 0.22 | |||||
| L-Histidine | 156.0766 | 19.62 | 0.731 | |||||
| L-Phenylalanine | 166.0862 | 12.9 | 0.253 | |||||
| L-Arginine | 175.1188 | 22.19 | 0.057 | |||||
| L-Tyrosine | 182.0811 | 15.21 | 0.089 | |||||
| L-Tryptophan | 205.0972 | 13.55 | 0.329 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soliman, G.A.; Abzalimov, R.R.; He, Y. mTORC1 and mTORC2 Complexes Regulate the Untargeted Metabolomics and Amino Acid Metabolites Profile through Mitochondrial Bioenergetic Functions in Pancreatic Beta Cells. Nutrients 2022, 14, 3022. https://doi.org/10.3390/nu14153022
Soliman GA, Abzalimov RR, He Y. mTORC1 and mTORC2 Complexes Regulate the Untargeted Metabolomics and Amino Acid Metabolites Profile through Mitochondrial Bioenergetic Functions in Pancreatic Beta Cells. Nutrients. 2022; 14(15):3022. https://doi.org/10.3390/nu14153022
Chicago/Turabian StyleSoliman, Ghada A., Rinat R. Abzalimov, and Ye He. 2022. "mTORC1 and mTORC2 Complexes Regulate the Untargeted Metabolomics and Amino Acid Metabolites Profile through Mitochondrial Bioenergetic Functions in Pancreatic Beta Cells" Nutrients 14, no. 15: 3022. https://doi.org/10.3390/nu14153022
APA StyleSoliman, G. A., Abzalimov, R. R., & He, Y. (2022). mTORC1 and mTORC2 Complexes Regulate the Untargeted Metabolomics and Amino Acid Metabolites Profile through Mitochondrial Bioenergetic Functions in Pancreatic Beta Cells. Nutrients, 14(15), 3022. https://doi.org/10.3390/nu14153022