
Role of Short Chain Fatty Acids in Epilepsy and Potential Benefits of Probiotics and Prebiotics: Targeting “Health” of Epileptic Patients


Abstract
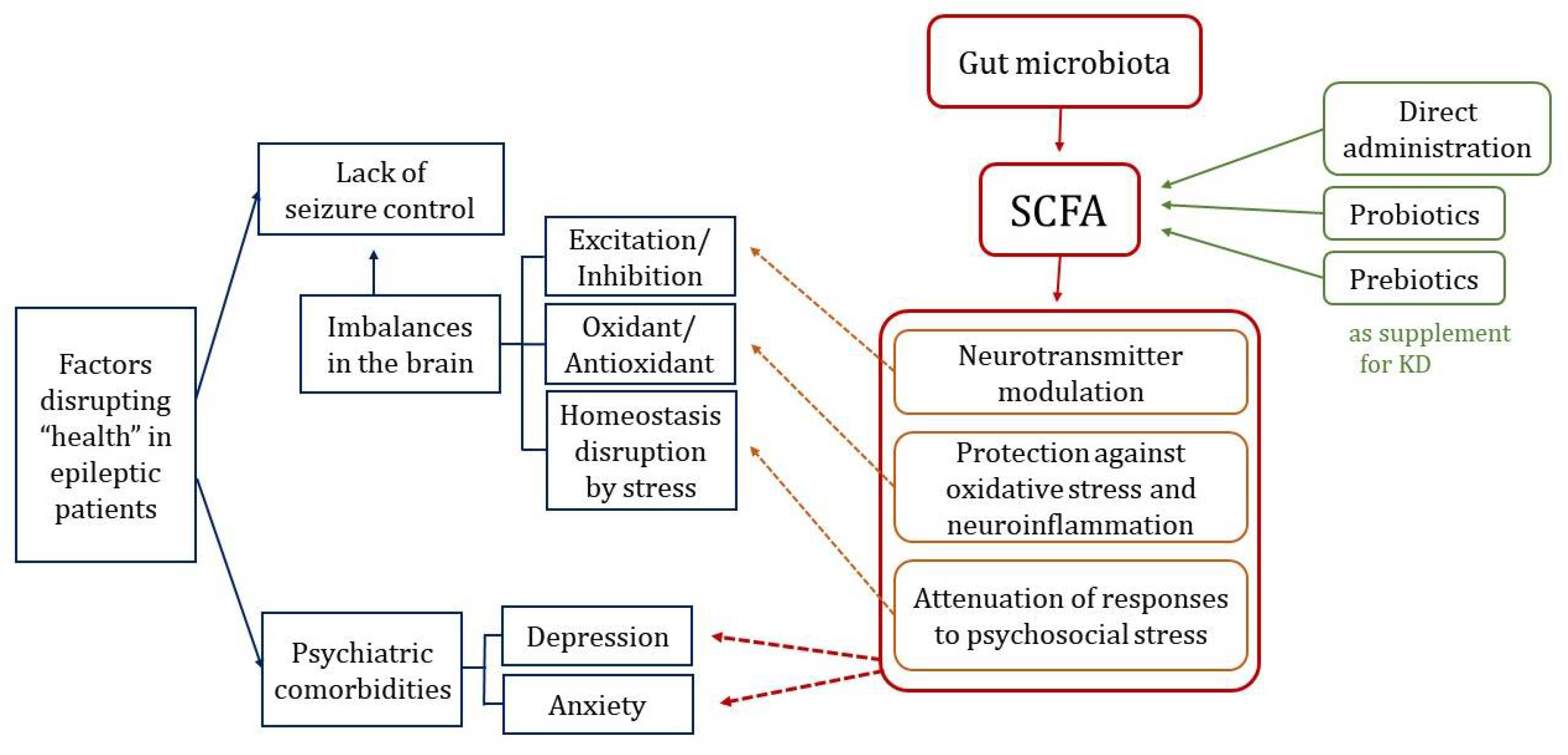
:1. Introduction
2. SCFA in the Gut–Brain Axis
3. Effects of Current Dietary Treatment on SCFA: Ketogenic Diet
4. SCFA in Seizure Control
4.1. Excitation/Inhibition Imbalance and Neurotransmitter Modulation
4.2. Oxidant/Antioxidant Imbalance and Neuroinflammation
4.2.1. Oxidative Stress
4.2.2. Neuroinflammation
4.2.3. Blood–Brain Barrier (BBB)
4.2.4. Studies Linking SCFA Administration, Epilepsy and Inflammation
4.3. Psychosocial Stress and the Hypothalamic-Pituitary-Adrenal Axis
5. SCFA in Psychiatric Comorbidities of Epilepsy
5.1. Depression in Epilepsy
5.1.1. Etiology
5.1.2. Possible Roles of GM and SCFA
5.2. Anxiety in Epilepsy
5.2.1. Etiology
5.2.2. Possible Roles of GM and SCFA
6. Potential Dietary Treatments: Pro/Prebiotics
6.1. Preclinical Studies
6.2. Clinical Studies
6.3. Clinical Potentiality of SCFA-Modulatory Nutritional Techniques
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hachinski, V.; Avan, A.; Gilliland, J.; Oveisgharan, S. A new definition of brain health. Lancet Neurol. 2021, 20, 335–336. [Google Scholar] [CrossRef]
- Carta, M.G.; Patten, S.; Nardi, A.E.; Bhugra, D. Mental health and chronic diseases: A challenge to be faced from a new perspective. Int. Rev. Psychiatry 2017, 29, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef] [PubMed]
- Malard, F.; Dore, J.; Gaugler, B.; Mohty, M. Introduction to host microbiome symbiosis in health and disease. Mucosal Immunol. 2020, 14, 547–554. [Google Scholar] [CrossRef]
- Hingray, C.; McGonigal, A.; Kotwas, I.; Micoulaud-Franchi, J.-A. The Relationship Between Epilepsy and Anxiety Disorders. Curr. Psychiatry Rep. 2019, 21, 40. [Google Scholar] [CrossRef]
- Dooling, S.W.; Costa-Mattioli, M. Gut Bacteria Seize Control of the Brain to Prevent Epilepsy. Cell Host Microbe 2018, 24, 3–5. [Google Scholar] [CrossRef] [Green Version]
- Jeżowska-Jurczyk, K.; Kotas, R.; Jurczyk, P.; Nowakowska-Kotas, M.; Budrewicz, S.; Pokryszko-Dragan, A. Mental disorders in patients with epilepsy. Psychiatr. Pol. 2020, 54, 51–68. [Google Scholar] [CrossRef]
- Akdemir, V.; Sut, N.; Guldiken, B. Factors affecting the quality of life in drug-resistant epilepsy patients. Acta Neurol. Belg. 2016, 116, 513–518. [Google Scholar] [CrossRef]
- Pitkänen, A.; Lukasiuk, K.; Dudek, F.E.; Staley, K.J. Epileptogenesis. Cold Spring Harb. Perspect. Med. 2015, 5, a022822. [Google Scholar] [CrossRef] [Green Version]
- Elger, C.E.; Johnston, S.A.; Hoppe, C. Diagnosing and treating depression in epilepsy. Seizure 2017, 44, 184–193. [Google Scholar] [CrossRef] [Green Version]
- Adelman, M.W.; Woodworth, M.H.; Langelier, C.; Busch, L.M.; Kempker, J.A.; Kraft, C.S.; Martin, G.S. The gut microbiome’s role in the development, maintenance, and outcomes of sepsis. Crit. Care 2020, 24, 278. [Google Scholar] [CrossRef] [PubMed]
- Ríos-Covián, D.; Ruas-Madiedo, P.; Margolles, A.; Gueimonde, M.; De Los Reyes-Gavilán, C.G.; Salazar, N. Intestinal Short Chain Fatty Acids and their Link with Diet and Human Health. Front. Microbiol. 2016, 7, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, P.; Scott, K.P.; Duncan, S.H.; Flint, H.J. Understanding the effects of diet on bacterial metabolism in the large intestine. J. Appl. Microbiol. 2007, 102, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.L.; Wolin, M.J. Pathways of acetate, propionate, and butyrate formation by the human fecal microbial flora. Appl. Environ. Microbiol. 1996, 62, 1589–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shilova, I.N.; Robidart, J.C.; Tripp, H.J.; Turk-Kubo, K.; Wawrik, B.; Post, A.F.; Thompson, A.W.; Ward, B.; Hollibaugh, J.T.; Millard, A.; et al. A microarray for assessing transcription from pelagic marine microbial taxa. ISME J. 2014, 8, 1476–1491. [Google Scholar] [CrossRef] [Green Version]
- Louis, P.; Young, P.; Holtrop, G.; Flint, H.J. Diversity of human colonic butyrate-producing bacteria revealed by analysis of the butyryl-CoA:acetate CoA-transferase gene. Environ. Microbiol. 2010, 12, 304–314. [Google Scholar] [CrossRef]
- Flint, H.J.; Duncan, S.H.; Scott, K.P.; Louis, P. Links between diet, gut microbiota composition and gut metabolism. Proc. Nutr. Soc. 2015, 74, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Cong, J.; Zhou, P.; Zhang, R. Intestinal Microbiota-Derived Short Chain Fatty Acids in Host Health and Disease. Nutrients 2022, 14, 1977. [Google Scholar] [CrossRef]
- Ohira, H.; Tsutsui, W.; Fujioka, Y. Are Short Chain Fatty Acids in Gut Microbiota Defensive Players for Inflammation and Atherosclerosis? J. Atheroscler. Thromb. 2017, 24, 660–672. [Google Scholar] [CrossRef] [Green Version]
- Blaak, E.E.; Canfora, E.E.; Theis, S.; Frost, G.; Groen, A.K.; Mithieux, G.; Nauta, A.; Scott, K.; Stahl, B.; Van Harsselaar, J.; et al. Short chain fatty acids in human gut and metabolic health. Benef. Microbes 2020, 11, 411–455. [Google Scholar] [CrossRef]
- Parada Venegas, D.; De La Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, E.S.; Preston, T.; Frost, G.; Morrison, D.J. Role of Gut Microbiota-Generated Short-Chain Fatty Acids in Metabolic and Cardiovascular Health. Curr. Nutr. Rep. 2018, 7, 198–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, Y.P.; Bernardi, A.; Frozza, R.L. The Role of Short-Chain Fatty Acids from Gut Microbiota in Gut-Brain Communication. Front. Endocrinol. 2020, 11, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Wang, Y.; Yang, G.; Zhang, Q.; Meng, L.; Xin, Y.; Jiang, X. The role of short-chain fatty acids in intestinal barrier function, inflammation, oxidative stress, and colonic carcinogenesis. Pharmacol. Res. 2021, 165, 105420. [Google Scholar] [CrossRef]
- Stilling, R.M.; van de Wouw, M.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. The neuropharmacology of butyrate: The bread and butter of the microbiota-gut-brain axis? Neurochem. Int. 2016, 99, 110–132. [Google Scholar] [CrossRef]
- Markowiak-Kopeć, P.; Śliżewska, K. The Effect of Probiotics on the Production of Short-Chain Fatty Acids by Human Intestinal Microbiome. Nutrients 2020, 12, 1107. [Google Scholar] [CrossRef]
- Verrotti, A.; Iapadre, G.; Di Francesco, L.; Zagaroli, L.; Farello, G. Diet in the Treatment of Epilepsy: What We Know So Far. Nutrients 2020, 12, 2645. [Google Scholar] [CrossRef]
- Rho, J.M. How does the ketogenic diet induce anti-seizure effects? Neurosci. Lett. 2017, 637, 4–10. [Google Scholar] [CrossRef]
- Murakami, M.; Tognini, P. Molecular Mechanisms Underlying the Bioactive Properties of a Ketogenic Diet. Nutrients 2022, 14, 782. [Google Scholar] [CrossRef]
- Xie, G.; Zhou, Q.; Qiu, C.-Z.; Dai, W.-K.; Wang, H.-P.; Li, Y.-H.; Liao, J.-X.; Lu, X.-G.; Lin, S.-F.; Ye, J.-H.; et al. Ketogenic diet poses a significant effect on imbalanced gut microbiota in infants with refractory epilepsy. World J. Gastroenterol. 2017, 23, 6164–6171. [Google Scholar] [CrossRef]
- Gong, X.; Cai, Q.; Liu, X.; An, D.; Zhou, D.; Luo, R.; Peng, R.; Hong, Z. Gut flora and metabolism are altered in epilepsy and partially restored after ketogenic diets. Microb. Pathog. 2021, 155, 104899. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.A.; Vuong, H.E.; Yano, J.M.; Liang, Q.Y.; Nusbaum, D.J.; Hsiao, E.Y. The Gut Microbiota Mediates the Anti-Seizure Effects of the Ketogenic Diet. Cell 2018, 173, 1728–1741.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhou, S.; Zhou, Y.; Yu, L.; Zhang, L.; Wang, Y. Altered gut microbiome composition in children with refractory epilepsy after ketogenic diet. Epilepsy Res. 2018, 145, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Lindefeldt, M.; Eng, A.; Darban, H.; Bjerkner, A.; Zetterström, C.K.; Allander, T.; Andersson, B.; Borenstein, E.; Dahlin, M.; Prast-Nielsen, S. The ketogenic diet influences taxonomic and functional composition of the gut microbiota in children with severe epilepsy. NPJ Biofilms Microbiomes 2019, 5, 5. [Google Scholar] [CrossRef]
- Ferraris, C.; Meroni, E.; Casiraghi, M.C.; Tagliabue, A.; De Giorgis, V.; Erba, D. One Month of Classic Therapeutic Ketogenic Diet Decreases Short Chain Fatty Acids Production in Epileptic Patients. Front. Nutr. 2021, 8, 613100. [Google Scholar] [CrossRef]
- Eor, J.Y.; Son, Y.J.; Kim, J.-Y.; Kang, H.-C.; Youn, S.E.; Kim, J.H.; Kim, S.H. Neuroprotective effect of both synbiotics and ketogenic diet in a pentylenetetrazol-induced acute seizure murine model. Epilepsy Res. 2021, 174, 106668. [Google Scholar] [CrossRef]
- Cai, Q.-Y.; Zhou, Z.-J.; Luo, R.; Gan, J.; Li, S.-P.; Mu, D.Z.; Wan, C.-M. Safety and tolerability of the ketogenic diet used for the treatment of refractory childhood epilepsy: A systematic review of published prospective studies. World J. Pediatr. 2017, 13, 528–536. [Google Scholar] [CrossRef]
- Martin-McGill, K.J.; Bresnahan, R.; Levy, R.G.; Cooper, P.N. Ketogenic diets for drug-resistant epilepsy. Cochrane Database Syst. Rev. 2020, 2020, CD001903. [Google Scholar] [CrossRef]
- Akyuz, E.; Polat, A.K.; Eroglu, E.; Kullu, I.; Angelopoulou, E.; Paudel, Y.N. Revisiting the role of neurotransmitters in epilepsy: An updated review. Life Sci. 2021, 265, 118826. [Google Scholar] [CrossRef]
- Sarlo, G.L.; Holton, K.F. Brain Concentrations of Glutamate and GABA in Human Epilepsy: A Review. Seizure 2021, 91, 213–227. [Google Scholar] [CrossRef]
- Shao, L.-R.; Habela, C.W.; Stafstrom, C.E. Pediatric Epilepsy Mechanisms: Expanding the Paradigm of Excitation/Inhibition Imbalance. Children 2019, 6, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philip, V.; Newton, D.F.; Oh, H.; Collins, S.M.; Bercik, P.; Sibille, E. Transcriptional markers of excitation-inhibition balance in germ-free mice show region-specific dysregulation and rescue after bacterial colonization. J. Psychiatr. Res. 2021, 135, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Luo, Z.-Y.; Hu, Y.-Y.; Bi, Y.-W.; Yang, J.-M.; Zou, W.-J.; Song, Y.-L.; Li, S.; Shen, T.; Li, S.-J.; et al. The gut microbiota regulates autism-like behavior by mediating vitamin B6 homeostasis in EphB6-deficient mice. Microbiome 2020, 8, 120. [Google Scholar] [CrossRef] [PubMed]
- Bravo, J.A.; Forsythe, P.; Chew, M.V.; Escaravage, E.; Savignac, H.M.; Dinan, T.G.; Bienenstock, J.; Cryan, J.F. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 16050–16055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strandwitz, P. Neurotransmitter modulation by the gut microbiota. Brain Res. 2018, 1693, 128–133. [Google Scholar] [CrossRef]
- Skonieczna-Żydecka, K.; Jakubczyk, K.; Maciejewska-Markiewicz, D.; Janda, K.; Kaźmierczak-Siedlecka, K.; Kaczmarczyk, M.; Łoniewski, I.; Marlicz, W. Gut Biofactory—Neurocompetent Metabolites within the Gastrointestinal Tract. A Scoping Review. Nutrients 2020, 12, 3369. [Google Scholar] [CrossRef]
- Amlerova, J.; Šroubek, J.; Angelucci, F.; Hort, J. Evidences for a Role of Gut Microbiota in Pathogenesis and Management of Epilepsy. Int. J. Mol. Sci. 2021, 22, 5576. [Google Scholar] [CrossRef]
- Frost, G.; Sleeth, M.L.; Sahuri-Arisoylu, M.; Lizarbe, B.; Cerdan, S.; Brody, L.; Anastasovska, J.; Ghourab, S.; Hankir, M.; Zhang, S.; et al. The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat. Commun. 2014, 5, 3611. [Google Scholar] [CrossRef] [Green Version]
- Giuseppe, D.G. Role of Serotonin in Epilepsy. Front. Hum. Neurosci. 2013, 7. [Google Scholar] [CrossRef]
- Evans, J.M.; Morris, L.S.; Marchesi, J.R. The gut microbiome: The role of a virtual organ in the endocrinology of the host. J. Endocrinol. 2013, 218, R37–R47. [Google Scholar] [CrossRef] [Green Version]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous Bacteria from the Gut Microbiota Regulate Host Serotonin Biosynthesis. Cell 2015, 161, 264–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reigstad, C.S.; Salmonson, C.E.; Rainey, J.F., III; Szurszewski, J.H.; Linden, D.R.; Sonnenburg, J.L.; Farrugia, G.; Kashyap, P.C. Gut microbes promote colonic serotonin production through an effect of short-chain fatty acids on enterochromaffin cells. FASEB J. 2015, 29, 1395–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nankova, B.B.; Agarwal, R.; Macfabe, D.F.; La Gamma, E.F. Enteric Bacterial Metabolites Propionic and Butyric Acid Modulate Gene Expression, Including CREB-Dependent Catecholaminergic Neurotransmission, in PC12 Cells - Possible Relevance to Autism Spectrum Disorders. PLoS ONE 2014, 9, e103740. [Google Scholar] [CrossRef]
- Mazarati, A.; Sankar, R. Common Mechanisms Underlying Epileptogenesis and the Comorbidities of Epilepsy. Cold Spring Harb. Perspect. Med. 2016, 6, a022798. [Google Scholar] [CrossRef]
- Ren, E.; Curia, G. Synaptic Reshaping and Neuronal Outcomes in the Temporal Lobe Epilepsy. Int. J. Mol. Sci. 2021, 22, 3860. [Google Scholar] [CrossRef]
- Kao, A.C.-C.; Chan, K.W.; Anthony, D.C.; Lennox, B.R.; Burnet, P.W. Prebiotic reduction of brain histone deacetylase (HDAC) activity and olanzapine-mediated weight gain in rats, are acetate independent. Neuropharmacology 2019, 150, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Nash, M.S.; Selkirk, J.V.; Gaymer, C.E.; Challiss, R.A.J.; Nahorski, S.R. Enhanced inducible mGlu1α receptor expression in Chinese hamster ovary cells. J. Neurochem. 2001, 77, 1664–1667. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Huang, Q.; Xu, H.; Niu, L.; Zhou, J.-N. Antidepressant-like effects of sodium butyrate in combination with estrogen in rat forced swimming test: Involvement of 5-HT1A receptors. Behav. Brain Res. 2009, 196, 200–206. [Google Scholar] [CrossRef]
- Bonansco, C.; Fuenzalida, M. Plasticity of Hippocampal Excitatory-Inhibitory Balance: Missing the Synaptic Control in the Epileptic Brain. Neural Plast. 2016, 2016, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Jarero-Basulto, J.J.; Gasca-Martínez, Y.; Rivera-Cervantes, M.C.; Ureña-Guerrero, M.E.; Feria-Velasco, A.I.; Beas-Zarate, C. Interactions Between Epilepsy and Plasticity. Pharmaceuticals 2018, 11, 17. [Google Scholar] [CrossRef] [Green Version]
- Lupori, L.; Cornuti, S.; Mazziotti, R.; Borghi, E.; Ottaviano, E.; Cas, M.D.; Sagona, G.; Pizzorusso, T.; Tognini, P. The gut microbiota of environmentally enriched mice regulates visual cortical plasticity. Cell Rep. 2022, 38, 110212. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Q.; Wu, D.; Zhang, D.-W.; Li, S.-C.; Zhang, S.-W.; Chen, X.; Li, W. The effect of a novel anticonvulsant chemical Q808 on gut microbiota and hippocampus neurotransmitters in pentylenetetrazole-induced seizures in rats. BMC Neurosci. 2022, 23, 7. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.D.; Clossen, B.L.; Reddy, D.S. Epigenetic Histone Deacetylation Inhibition Prevents the Development and Persistence of Temporal Lobe Epilepsy. J. Pharmacol. Exp. Ther. 2017, 364, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Borowicz-Reutt, K.K.; Czuczwar, S.J. Role of oxidative stress in epileptogenesis and potential implications for therapy. Pharmacol. Rep. 2020, 72, 1218–1226. [Google Scholar] [CrossRef]
- Parsons, A.L.M.; Bucknor, E.M.V.; Castroflorio, E.; Soares, T.R.; Oliver, P.L.; Rial, D. The Interconnected Mechanisms of Oxidative Stress and Neuroinflammation in Epilepsy. Antioxidants 2022, 11, 157. [Google Scholar] [CrossRef]
- Cheng, Y.; Luo, F.; Zhang, Q.; Sang, Y.; Chen, X.; Zhang, L.; Liu, Y.; Li, X.; Li, J.; Ding, H.; et al. α-Lipoic acid alleviates pentetrazol-induced neurological deficits and behavioral dysfunction in rats with seizures via an Nrf2 pathway. RSC Adv. 2018, 8, 4084–4092. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.-Y.; Zhou, H.-H.; Jin, W.-L. Redox-Related Neuronal Death and Crosstalk as Drug Targets: Focus on Epilepsy. Front. Neurosci. 2019, 13, 512. [Google Scholar] [CrossRef] [Green Version]
- González-Bosch, C.; Boorman, E.; Zunszain, P.A.; Mann, G.E. Short-chain fatty acids as modulators of redox signaling in health and disease. Redox Biol. 2021, 47, 102165. [Google Scholar] [CrossRef]
- Matt, S.M.; Allen, J.M.; Lawson, M.A.; Mailing, L.J.; Woods, J.A.; Johnson, R.W. Butyrate and Dietary Soluble Fiber Improve Neuroinflammation Associated with Aging in Mice. Front. Immunol. 2018, 9, 1832. [Google Scholar] [CrossRef]
- Guo, W.; Liu, J.; Sun, J.; Gong, Q.; Ma, H.; Kan, X.; Cao, Y.; Wang, J.; Fu, S. Butyrate alleviates oxidative stress by regulating NRF2 nuclear accumulation and H3K9/14 acetylation via GPR109A in bovine mammary epithelial cells and mammary glands. Free Radic. Biol. Med. 2020, 152, 728–742. [Google Scholar] [CrossRef]
- Hamer, H.M.; Jonkers, D.M.A.E.; Bast, A.; Vanhoutvin, S.A.L.W.; Fischer, M.A.J.G.; Kodde, A.; Troost, F.J.; Venema, K.; Brummer, R.-J.M. Butyrate modulates oxidative stress in the colonic mucosa of healthy humans. Clin. Nutr. 2009, 28, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Milder, J.; Patel, M. Modulation of oxidative stress and mitochondrial function by the ketogenic diet. Epilepsy Res. 2012, 100, 295–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, L.-C.; Wang, Y.; Wang, Z.-B.; Liu, W.-Y.; Sun, S.; Li, L.; Su, D.-F.; Zhang, L.-C. Propionate Ameliorates Dextran Sodium Sulfate-Induced Colitis by Improving Intestinal Barrier Function and Reducing Inflammation and Oxidative Stress. Front. Pharmacol. 2016, 7, 253. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Cui, C. The role of short-chain fatty acids in central nervous system diseases. Mol. Cell. Biochem. 2022, 477, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Valvassori, S.S.; Dal-Pont, G.C.; Steckert, A.V.; Varela, R.B.; Lopes-Borges, J.; Mariot, E.; Resende, W.R.; Arent, C.O.; Carvalho, A.F.; Quevedo, J. Sodium butyrate has an antimanic effect and protects the brain against oxidative stress in an animal model of mania induced by ouabain. Psychiatry Res. 2015, 235, 154–159. [Google Scholar] [CrossRef]
- Arena, A.; Zimmer, T.S.; van Scheppingen, J.; Korotkov, A.; Anink, J.J.; Mühlebner, A.; Jansen, F.E.; van Hecke, W.; Spliet, W.G.; van Rijen, P.C.; et al. Oxidative stress and inflammation in a spectrum of epileptogenic cortical malformations: Molecular insights into their interdependence. Brain Pathol. 2019, 29, 351–365. [Google Scholar] [CrossRef]
- Pracucci, E.; Pillai, V.; Lamers, D.; Parra, R.; Landi, S. Neuroinflammation: A Signature or a Cause of Epilepsy? Int. J. Mol. Sci. 2021, 22, 6981. [Google Scholar] [CrossRef]
- Vezzani, A. Epilepsy and Inflammation in the Brain: Overview and Pathophysiology. Epilepsy Curr. 2014, 14, 3–7. [Google Scholar] [CrossRef]
- Haghikia, A.; Jörg, S.; Duscha, A.; Berg, J.; Manzel, A.; Waschbisch, A.; Hammer, A.; Lee, D.-H.; May, C.; Wilck, N.; et al. Dietary Fatty Acids Directly Impact Central Nervous System Autoimmunity via the Small Intestine. Immunity 2015, 43, 817–829. [Google Scholar] [CrossRef] [Green Version]
- Sajdel-Sulkowska, E.M. Neuropsychiatric Ramifications of COVID-19: Short-Chain Fatty Acid Deficiency and Disturbance of Microbiota-Gut-Brain Axis Signaling. BioMed Res. Int. 2021, 2021, 1–15. [Google Scholar] [CrossRef]
- Profaci, C.P.; Munji, R.N.; Pulido, R.S.; Daneman, R. The blood–brain barrier in health and disease: Important unanswered questions. J. Exp. Med. 2020, 217, e20190062. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Friedman, A. Structural, Molecular, and Functional Alterations of the Blood-Brain Barrier during Epileptogenesis and Epilepsy: A Cause, Consequence, or Both? Int. J. Mol. Sci. 2020, 21, 591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rempe, R.G.; Hartz, A.M.; Soldner, E.L.; Sokola, B.S.; Alluri, S.R.; Abner, E.L.; Kryscio, R.J.; Pekcec, A.; Schlichtiger, J.; Bauer, B. Matrix Metalloproteinase-Mediated Blood-Brain Barrier Dysfunction in Epilepsy. J. Neurosci. 2018, 38, 4301–4315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Vliet, E.; Aronica, E.; Gorter, J. Blood–brain barrier dysfunction, seizures and epilepsy. Semin. Cell Dev. Biol. 2014, 38, 26–34. [Google Scholar] [CrossRef]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Tóth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 6, 263ra158. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Sun, J.; Wang, F.; Ding, G.; Chen, W.; Fang, R.; Yao, Y.; Pang, M.; Lu, Z.; Liu, J. Sodium butyrate exerts neuroprotective effects by restoring the blood-brain barrier in traumatic brain injury mice. Brain Res. 2016, 1642, 70–78. [Google Scholar] [CrossRef]
- Liu, J.; Wang, F.; Liu, S.; Du, J.; Hu, X.; Xiong, J.; Fang, R.; Chen, W.; Sun, J. Sodium butyrate exerts protective effect against Parkinson’s disease in mice via stimulation of glucagon like peptide-1. J. Neurol. Sci. 2017, 381, 176–181. [Google Scholar] [CrossRef]
- Park, M.J.; Sohrabji, F. The histone deacetylase inhibitor, sodium butyrate, exhibits neuroprotective effects for ischemic stroke in middle-aged female rats. J. Neuroinflammation 2016, 13, 300. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.; Ding, Y.; Wang, L.; Xiao, Y. Gut microbiome improves postoperative cognitive function by decreasing permeability of the blood-brain barrier in aged mice. Brain Res. Bull. 2020, 164, 249–256. [Google Scholar] [CrossRef]
- Hoyles, L.; Snelling, T.; Umlai, U.-K.; Nicholson, J.K.; Carding, S.R.; Glen, R.C.; McArthur, S. Microbiome–host systems interactions: Protective effects of propionate upon the blood–brain barrier. Microbiome 2018, 6, 55. [Google Scholar] [CrossRef] [Green Version]
- Erny, D.; Hrabě de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef]
- Wang, C.; Xia, G.; Li, G.; Wan, F.; Lv, Y.; Xiao, Z. Attenuation of kainic acid-induced epilepsy by butyrate is associated with inhibition of glial activation. Trop. J. Pharm. Res. 2018, 17, 1739. [Google Scholar] [CrossRef]
- De Caro, C.; Leo, A.; Nesci, V.; Ghelardini, C.; di Cesare Mannelli, L.; Striano, P.; Avagliano, C.; Calignano, A.; Mainardi, P.; Constanti, A.; et al. Intestinal inflammation increases convulsant activity and reduces antiepileptic drug efficacy in a mouse model of epilepsy. Sci. Rep. 2019, 9, 13983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Bai, X.; Jiang, Y.; Cheng, Y. Butyrate alleviates PTZ-induced mitochondrial dysfunction, oxidative stress and neuron apoptosis in mice via Keap1/Nrf2/HO-1 pathway. Brain Res. Bull. 2020, 168, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Mai, Q.; Zeng, X.; Wang, H.; Xiao, Y.; Tang, L.; Li, J.; Zhang, Y.; Ding, H. Propionate Relieves Pentylenetetrazol-Induced Seizures, Consequent Mitochondrial Disruption, Neuron Necrosis and Neurological Deficits in Mice. Biochem. Pharmacol. 2019, 169, 113607. [Google Scholar] [CrossRef]
- Koolhaas, J.; Bartolomucci, A.; Buwalda, B.; de Boer, S.; Flügge, G.; Korte, S.; Meerlo, P.; Murison, R.; Olivier, B.; Palanza, P.; et al. Stress revisited: A critical evaluation of the stress concept. Neurosci. Biobehav. Rev. 2011, 35, 1291–1301. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Garcia, C.; Zeleke, H.; Rojas, A. Impact of Stress on Epilepsy: Focus on Neuroinflammation—A Mini Review. Int. J. Mol. Sci. 2021, 22, 4061. [Google Scholar] [CrossRef]
- Maguire, J.; Salpekar, J.A. Stress, seizures, and hypothalamic–pituitary–adrenal axis targets for the treatment of epilepsy. Epilepsy Behav. 2013, 26, 352–362. [Google Scholar] [CrossRef] [Green Version]
- O’Toole, K.K.; Hooper, A.; Wakefield, S.; Maguire, J. Seizure-induced disinhibition of the HPA axis increases seizure susceptibility. Epilepsy Res. 2014, 108, 29–43. [Google Scholar] [CrossRef] [Green Version]
- Van De Wouw, M.; Boehme, M.; Lyte, J.M.; Wiley, N.; Strain, C.; O’Sullivan, O.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Short-chain fatty acids: Microbial metabolites that alleviate stress-induced brain-gut axis alterations. J. Physiol. 2018, 596, 4923–4944. [Google Scholar] [CrossRef]
- Tian, P.; Zhu, H.; Qian, X.; Chen, Y.; Wang, Z.; Zhao, J.; Zhang, H.; Wang, G.; Chen, W. Consumption of Butylated Starch Alleviates the Chronic Restraint Stress-Induced Neurobehavioral and Gut Barrier Deficits Through Reshaping the Gut Microbiota. Front. Immunol. 2021, 12, 755481. [Google Scholar] [CrossRef] [PubMed]
- Dalile, B.; Vervliet, B.; Bergonzelli, G.; Verbeke, K.; Van Oudenhove, L. Colon-delivered short-chain fatty acids attenuate the cortisol response to psychosocial stress in healthy men: A randomized, placebo-controlled trial. Neuropsychopharmacology 2020, 45, 2257–2266. [Google Scholar] [CrossRef] [PubMed]
- Maltz, R.M.; Keirsey, J.; Kim, S.C.; Mackos, A.; Gharaibeh, R.Z.; Moore, C.C.; Xu, J.; Somogyi, A.; Bailey, M. Social Stress Affects Colonic Inflammation, the Gut Microbiome, and Short-chain Fatty Acid Levels and Receptors. J. Pediatr. Gastroenterol. Nutr. 2019, 68, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Medel-Matus, J.-S.; Shin, N.; Dorfman, E.; Sankar, R.; Mazarati, A. Facilitation of kindling epileptogenesis by chronic stress may be mediated by intestinal microbiome. Epilepsia Open 2018, 3, 290–294. [Google Scholar] [CrossRef]
- Deutsch, S.I.; Rosse, R.B.; Long, K.D.; Gaskins, B.L.; Burket, J.A.; Mastropaolo, J. Sodium butyrate, an epigenetic interventional strategy, attenuates a stress-induced alteration of MK-801′s pharmacologic action. Eur. Neuropsychopharmacol. 2008, 18, 565–568. [Google Scholar] [CrossRef]
- De Caro, C.; di Cesare Mannelli, L.; Branca, J.J.V.; Micheli, L.; Citraro, R.; Russo, E.; De Sarro, G.; Ghelardini, C.; Calignano, A.; Russo, R. Pain Modulation in WAG/Rij Epileptic Rats (A Genetic Model of Absence Epilepsy): Effects of Biological and Pharmacological Histone Deacetylase Inhibitors. Front. Pharmacol. 2020, 11, 549191. [Google Scholar] [CrossRef]
- Citraro, R.; Leo, A.; De Caro, C.; Nesci, V.; Cantafio, M.E.G.; Amodio, N.; Raso, G.M.; Lama, A.; Russo, R.; Calignano, A.; et al. Effects of Histone Deacetylase Inhibitors on the Development of Epilepsy and Psychiatric Comorbidity in WAG/Rij Rats. Mol. Neurobiol. 2020, 57, 408–421. [Google Scholar] [CrossRef]
- Kwon, O.-Y.; Park, S.-P. Depression and Anxiety in People with Epilepsy. J. Clin. Neurol. 2014, 10, 175–188. [Google Scholar] [CrossRef] [Green Version]
- Fiest, K.M.; Dykeman, J.; Patten, S.B.; Wiebe, S.; Kaplan, G.G.; Maxwell, C.J.; Bulloch, A.G.; Jette, N. Depression in epilepsy: A systematic review and meta-analysis. Neurology 2013, 80, 590–599. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.-W.; Kim, D.-H.; Kim, Y.-S.; Kim, J.; Kwon, O.-Y. Anxiety disorders in outpatient clinics of epilepsy in tertiary care hospitals: A meta-analysis. Seizure 2020, 75, 34–42. [Google Scholar] [CrossRef]
- Simpson, C.A.; Diaz-Arteche, C.; Eliby, D.; Schwartz, O.S.; Simmons, J.G.; Cowan, C.S. The gut microbiota in anxiety and depression—A systematic review. Clin. Psychol. Rev. 2021, 83, 101943. [Google Scholar] [CrossRef] [PubMed]
- Pineda, E.; Shin, N.; Sankar, R.; Mazarati, A.M. Comorbidity between epilepsy and depression: Experimental evidence for the involvement of serotonergic, glucocorticoid, and neuroinflammatory mechanisms. Epilepsia 2010, 51, 110–114. [Google Scholar] [CrossRef]
- Valente, K.D.R.; Filho, G.B. Depression and temporal lobe epilepsy represent an epiphenomenon sharing similar neural networks: Clinical and brain structural evidences. Arq. Neuro-Psiquiatria 2013, 71, 183–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arulsamy, A.; Shaikh, M.F. Epilepsy-associated comorbidities among adults: A plausible therapeutic role of gut microbiota. Neurobiol. Dis. 2022, 165, 105648. [Google Scholar] [CrossRef] [PubMed]
- A Pineda, E.; Hensler, J.G.; Sankar, R.; Shin, D.; Burke, T.F.; Mazarati, A.M. Plasticity of Presynaptic and Postsynaptic Serotonin 1A Receptors in an Animal Model of Epilepsy-Associated Depression. Neuropsychopharmacology 2011, 36, 1305–1316. [Google Scholar] [CrossRef] [Green Version]
- Lothe, A.; Didelot, A.; Hammers, A.; Costes, N.; Saoud, M.; Gilliam, F.; Ryvlin, P. Comorbidity between temporal lobe epilepsy and depression: A [18 F]MPPF PET study. Brain 2008, 131, 2765–2782. [Google Scholar] [CrossRef] [Green Version]
- Kanner, A.M. Depression and epilepsy: A bidirectional relation? Epilepsia 2011, 52, 21–27. [Google Scholar] [CrossRef]
- Lang, U.E.; Borgwardt, S. Molecular Mechanisms of Depression: Perspectives on New Treatment Strategies. Cell. Physiol. Biochem. 2013, 31, 761–777. [Google Scholar] [CrossRef] [PubMed]
- Rosenblat, J.D.; Cha, D.S.; Mansur, R.B.; McIntyre, R.S. Inflamed moods: A review of the interactions between inflammation and mood disorders. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2014, 53, 23–34. [Google Scholar] [CrossRef]
- Parletta, N. The Gut-Brain-Microbe Interaction: Relevance in Inflammation and Depression. In Inflammation and Immunity in Depression; Elsevier: Amsterdam, The Netherlands, 2018; pp. 241–252. [Google Scholar]
- Pineda, E.A.; Hensler, J.G.; Sankar, R.; Shin, D.; Burke, T.F.; Mazarati, A.M. Interleukin-1beta Causes Fluoxetine Resistance in an Animal Model of Epilepsy-Associated Depression. Neurotherapeutics 2012, 9, 477–485. [Google Scholar] [CrossRef] [Green Version]
- Singh, T.; Goel, R.K. Epilepsy Associated Depression: An Update on Current Scenario, Suggested Mechanisms, and Opportunities. Neurochem. Res. 2021, 46, 1305–1321. [Google Scholar] [CrossRef] [PubMed]
- Mazarati, A.M.; Lewis, M.L.; Pittman, Q.J. Neurobehavioral comorbidities of epilepsy: Role of inflammation. Epilepsia 2017, 58, 48–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaikh, M.F.; Lee, C.Y.; Chen, W.N.; Shaikh, F.A. The Gut-Brain-Axis on the Manifestation of Depressive Symptoms in Epilepsy: An Evidence-Driven Hypothesis. Front. Pharmacol. 2020, 11, 465. [Google Scholar] [CrossRef]
- Strandwitz, P.; Kim, K.H.; Terekhova, D.; Liu, J.K.; Sharma, A.; Levering, J.; McDonald, D.; Dietrich, D.; Ramadhar, T.R.; Lekbua, A.; et al. GABA-modulating bacteria of the human gut microbiota. Nat. Microbiol. 2019, 4, 396–403. [Google Scholar] [CrossRef]
- Kazemi, A.; Noorbala, A.A.; Azam, K.; Eskandari, M.H.; Djafarian, K. Effect of probiotic and prebiotic vs placebo on psychological outcomes in patients with major depressive disorder: A randomized clinical trial. Clin. Nutr. 2019, 38, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Skonieczna-Żydecka, K.; Grochans, E.; Maciejewska, D.; Szkup, M.; Schneider-Matyka, D.; Jurczak, A.; Łoniewski, I.; Kaczmarczyk, M.; Marlicz, W.; Czerwińska-Rogowska, M.; et al. Faecal Short Chain Fatty Acids Profile is Changed in Polish Depressive Women. Nutrients 2018, 10, 1939. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Tian, T.; Mao, Q.; Zou, T.; Zhou, C.-J.; Xie, J.; Chen, J.-J. Associations between disordered gut microbiota and changes of neurotransmitters and short-chain fatty acids in depressed mice. Transl. Psychiatry 2020, 10, 350. [Google Scholar] [CrossRef]
- Leo, A.; De Caro, C.; Mainardi, P.; Tallarico, M.; Nesci, V.; Marascio, N.; Striano, P.; Russo, E.; Constanti, A.; De Sarro, G.; et al. Increased efficacy of combining prebiotic and postbiotic in mouse models relevant to autism and depression. Neuropharmacology 2021, 198, 108782. [Google Scholar] [CrossRef]
- Kratsman, N.; Getselter, D.; Elliott, E. Sodium butyrate attenuates social behavior deficits and modifies the transcription of inhibitory/excitatory genes in the frontal cortex of an autism model. Neuropharmacology 2016, 102, 136–145. [Google Scholar] [CrossRef]
- Rode, J.; Yang, L.; König, J.; Hutchinson, A.N.; Wall, R.; Venizelos, N.; Brummer, R.-J.; Rangel, I.; Vumma, R. Butyrate Rescues Oxidative Stress-Induced Transport Deficits of Tryptophan: Potential Implication in Affective or Gut-Brain Axis Disorders. Neuropsychobiology 2021, 80, 253–263. [Google Scholar] [CrossRef]
- Bin Wei, M.Y.; Melas, P.A.; Wegener, G.; Mathé, A.A.; Lavebratt, C. Antidepressant-Like Effect of Sodium Butyrate is Associated with an Increase in TET1 and in 5-Hydroxymethylation Levels in the Bdnf Gene. Int. J. Neuropsychopharmacol. 2015, 18, pyu032. [Google Scholar] [CrossRef] [Green Version]
- Valvassori, S.S.; Resende, W.R.; Budni, J.; Dal-Pont, G.C.; Bavaresco, D.V.; Réus, G.Z.; Carvalho, A.F.; Gonçalves, C.L.; Furlanetto, C.B.; Streck, E.L.; et al. Sodium Butyrate, a Histone Deacetylase Inhibitor, Reverses Behavioral and Mitochondrial Alterations in Animal Models of Depression Induced by Early- or Late-life Stress. Curr. Neurovascular Res. 2015, 12, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wang, F.; Hong, G.; Pang, M.; Xu, H.; Li, H.; Tian, F.; Fang, R.; Yao, Y.; Liu, J. Antidepressant-like effects of sodium butyrate and its possible mechanisms of action in mice exposed to chronic unpredictable mild stress. Neurosci. Lett. 2016, 618, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Burokas, A.; Arboleya, S.; Moloney, R.D.; Peterson, V.L.; Murphy, K.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Targeting the Microbiota-Gut-Brain Axis: Prebiotics Have Anxiolytic and Antidepressant-like Effects and Reverse the Impact of Chronic Stress in Mice. Biol. Psychiatry 2017, 82, 472–487. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Shaikh, M.F.; Shah, S.; Kumari, Y.; Othman, I. Role of inflammation in epilepsy and neurobehavioral comorbidities: Implication for therapy. Eur. J. Pharmacol. 2018, 837, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Bartolomeil, F.; Guye, M.; Wendling, F.; Gavaret, M.; Régis, J.; Chauvel, P. Fear, anger and compulsive behavior during seizure: Involvement of large scale fronto-temporal neural networks. Epileptic Disord. 2002, 4, 235–241. [Google Scholar] [PubMed]
- Shaikh, M.F.; Arulsamy, A. The impact of epilepsy on the manifestation of anxiety disorder. Int. J. Nutr. Pharmacol. Neurol. Dis. 2016, 6, 3. [Google Scholar] [CrossRef]
- Hovatta, I.; Juhila, J.; Donner, J. Oxidative stress in anxiety and comorbid disorders. Neurosci. Res. 2010, 68, 261–275. [Google Scholar] [CrossRef]
- Jin, X.; Zhang, Y.; Celniker, S.E.; Xia, Y.; Mao, J.-H.; Snijders, A.M.; Chang, H. Gut microbiome partially mediates and coordinates the effects of genetics on anxiety-like behavior in Collaborative Cross mice. Sci. Rep. 2021, 11, 270. [Google Scholar] [CrossRef]
- Neufeld, K.M.; Kang, N.; Bienenstock, J.; Foster, J.A. Reduced anxiety-like behavior and central neurochemical change in germ-free mice. Neurogastroenterol. Motil. 2010, 23, 255–264. [Google Scholar] [CrossRef]
- Liu, Y.; Ju, Y.; Cui, L.; Liu, T.; Hou, Y.; Wu, Q.; Ojo, O.; Du, X.; Wang, X. Association between Dietary Fiber Intake and Incidence of Depression and Anxiety in Patients with Essential Hypertension. Nutrients 2021, 13, 4159. [Google Scholar] [CrossRef] [PubMed]
- Müller, B.; Rasmusson, A.J.; Just, D.; Jayarathna, S.; Moazzami, A.; Novicic, Z.K.; Cunningham, J.L. Fecal Short-Chain Fatty Acid Ratios as Related to Gastrointestinal and Depressive Symptoms in Young Adults. Psychosom. Med. 2021, 83, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.S.; Gibson, G.R.; Hutkins, R.; Reimer, R.A.; Reid, G.; Verbeke, K.; Scott, K.P.; Holscher, H.D.; Azad, M.B.; Delzenne, N.M.; et al. The International Scientific Association for Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of synbiotics. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 687–701. [Google Scholar] [CrossRef] [PubMed]
- Azad, M.A.K.; Sarker, M.; Li, T.; Yin, J. Probiotic Species in the Modulation of Gut Microbiota: An Overview. BioMed Res. Int. 2018, 2018, 9478630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davani-Davari, D.; Negahdaripour, M.; Karimzadeh, I.; Seifan, M.; Mohkam, M.; Masoumi, S.; Berenjian, A.; Ghasemi, Y. Prebiotics: Definition, Types, Sources, Mechanisms, and Clinical Applications. Foods 2019, 8, 92. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Shi, B. Gut microbiota as a potential target of metabolic syndrome: The role of probiotics and prebiotics. Cell Biosci. 2017, 7, 48. [Google Scholar] [CrossRef] [Green Version]
- Slavin, J. Fiber and Prebiotics: Mechanisms and Health Benefits. Nutrients 2013, 5, 1417–1435. [Google Scholar] [CrossRef] [Green Version]
- Akkol, S. Effects of Probiotic Consumption On Absence Seizures. J. Turk. Epilepsi Soc. 2017. [Google Scholar] [CrossRef]
- Bagheri, S.; Heydari, A.; Alinaghipour, A.; Salami, M. Effect of probiotic supplementation on seizure activity and cognitive performance in PTZ-induced chemical kindling. Epilepsy Behav. 2019, 95, 43–50. [Google Scholar] [CrossRef]
- Tahmasebi, S.; Oryan, S.; Mohajerani, H.R.; Akbari, N.; Palizvan, M.R. Probiotics and Nigella sativa extract supplementation improved behavioral and electrophysiological effects of PTZ-induced chemical kindling in rats. Epilepsy Behav. 2020, 104, 106897. [Google Scholar] [CrossRef]
- Eor, J.Y.; Tan, P.L.; Son, Y.J.; Kwak, M.J.; Kim, S.H. Gut microbiota modulation by both Lactobacillus fermentum MSK 408 and ketogenic diet in a murine model of pentylenetetrazole-induced acute seizure. Epilepsy Res. 2021, 169, 106506. [Google Scholar] [CrossRef] [PubMed]
- Sabouri, S.; Kangi, S.; Najimi, S.; Rahimi, H.-R. Effects of probiotics on pentylenetetrazol-induced convulsions in mice. Epilepsy Res. 2021, 176, 106723. [Google Scholar] [CrossRef] [PubMed]
- Kilinc, E.; Ankarali, S.; Ayhan, D.; Ankarali, H.; Torun, I.E.; Cetinkaya, A. Protective effects of long-term probiotic mixture supplementation against pentylenetetrazole-induced seizures, inflammation and oxidative stress in rats. J. Nutr. Biochem. 2021, 98, 108830. [Google Scholar] [CrossRef] [PubMed]
- Mu, C.; Nikpoor, N.; Tompkins, T.A.; Rho, J.M.; Scantlebury, M.H.; Shearer, J. Probiotics counteract hepatic steatosis caused by ketogenic diet and upregulate AMPK signaling in a model of infantile epilepsy. eBioMedicine 2022, 76, 103838. [Google Scholar] [CrossRef]
- Aygun, H.; Akin, A.T.; Kızılaslan, N.; Sumbul, O.; Karabulut, D. Probiotic supplementation alleviates absence seizures and anxiety- and depression-like behavior in WAG/Rij rat by increasing neurotrophic factors and decreasing proinflammatory cytokines. Epilepsy Behav. 2022, 128, 108588. [Google Scholar] [CrossRef]
- Aygun, H.; Akin, A.T.; Kızılaslan, N.; Sumbul, O.; Karabulut, D. Electrophysiological, histopathological, and biochemical evaluation of the protective effect of probiotic supplementation against pentylenetetrazole-induced seizures in rats. Eur. J. Neurol. 2022. [Google Scholar] [CrossRef]
- Kızılaslan, N.; Sumbul, O.; Aygun, H. The Beneficial Effect of Probiotics Supplementation on Penicillin-Induced Focal Seizure in Rats. Neurochem. Res. 2022, 47, 1395–1404. [Google Scholar] [CrossRef]
- Mu, C.; Nikpoor, N.; Tompkins, T.A.; Choudhary, A.; Wang, M.; Marks, W.N.; Rho, J.M.; Scantlebury, M.H.; Shearer, J. Targeted gut microbiota manipulation attenuates seizures in a model of infantile spasms syndrome. JCI Insight 2022, 7. [Google Scholar] [CrossRef]
- Gómez-Eguílaz, M.; Ramón-Trapero, J.; Pérez-Martínez, L.; Blanco, J.-R. The beneficial effect of probiotics as a supplementary treatment in drug-resistant epilepsy: A pilot study. Benef. Microbes 2018, 9, 875–881. [Google Scholar] [CrossRef]
- Yeom, J.S.; Park, J.S.; Kim, Y.-S.; Kim, R.B.; Choi, D.-S.; Chung, J.-Y.; Han, T.H.; Seo, J.-H.; Park, E.S.; Lim, J.-Y.; et al. Neonatal seizures and white matter injury: Role of rotavirus infection and probiotics. Brain Dev. 2019, 41, 19–28. [Google Scholar] [CrossRef]
- Lemos, V.R.; Aires, R.; Côco, L.Z.; Domingues, R.B.; Meyrelles, S.S.; Vasquez, E.C.; Pereira, T.M.C.; Campagnaro, B.P. Benefits of multi-day supplementation with probiotic kefir in Rasmussen encephalitis: The first case report. Nutr. Neurosci. 2021, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Li, S.; Sun, B. Curative Effect of Prebiotics/Probiotics-Assisted Ketogenic Diet on Children with Refractory Epilepsy. Emerg. Med. Int. 2022, 2022, 1–6. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Cui, B.-T.; Zhang, T.; Li, P.; Long, C.; Ji, G.-Z.; Zhang, F. Fecal microbiota transplantation cured epilepsy in a case with Crohn’s disease: The first report. World J. Gastroenterol. 2017, 23, 3565–3568. [Google Scholar] [CrossRef]
- Gudan, A. Effects of dietary components on intestinal short-chain fatty acids (SCFAs) synthesis in healthy adult persons following a ketogenic diet. Roczniki Państwowego Zakładu Higieny 2022, 73, 51–69. [Google Scholar] [CrossRef]
- Gerhardt, S.; Mohajeri, M.H. Changes of Colonic Bacterial Composition in Parkinson’s Disease and Other Neurodegenerative Diseases. Nutrients 2018, 10, 708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greathouse, K.L.; Sinha, R.; Vogtmann, E. DNA extraction for human microbiome studies: The issue of standardization. Genome Biol. 2019, 20, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shabbir, U.; Arshad, M.; Sameen, A.; Oh, D.-H. Crosstalk between Gut and Brain in Alzheimer’s Disease: The Role of Gut Microbiota Modulation Strategies. Nutrients 2021, 13, 690. [Google Scholar] [CrossRef]
- Weinstein, N.; Garten, B.; Vainer, J.; Minaya, D.; Czaja, K. Managing the Microbiome: How the Gut Influences Development and Disease. Nutrients 2020, 13, 74. [Google Scholar] [CrossRef]
- Cai, T.; Shi, X.; Yuan, L.-Z.; Tang, D.; Wang, F. Fecal microbiota transplantation in an elderly patient with mental depression. Int. Psychogeriatr. 2019, 31, 1525–1526. [Google Scholar] [CrossRef] [Green Version]
- Doll, J.P.K.; Vázquez-Castellanos, J.F.; Schaub, A.-C.; Schweinfurth, N.; Kettelhack, C.; Schneider, E.; Yamanbaeva, G.; Mählmann, L.; Brand, S.; Beglinger, C.; et al. Fecal Microbiota Transplantation (FMT) as an Adjunctive Therapy for Depression—Case Report. Front. Psychiatry 2022, 13, 815422. [Google Scholar] [CrossRef]
- Lee, B.H.; Smith, T.; Paciorkowski, A.R. Autism spectrum disorder and epilepsy: Disorders with a shared biology. Epilepsy Behav. 2015, 47, 191–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vendrik, K.E.W.; Ooijevaar, R.E.; De Jong, P.R.C.; Laman, J.D.; van Oosten, B.W.; Van Hilten, J.J.; Ducarmon, Q.R.; Keller, J.J.; Kuijper, E.J.; Contarino, M.F. Fecal Microbiota Transplantation in Neurological Disorders. Front. Cell. Infect. Microbiol. 2020, 10, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| SCFAs | Estimated Proportion in the Gut [13,18] | Pathways [12] | Main Producers [12,19] | Receptors [18] |
|---|---|---|---|---|
| Acetate (C2) | 60–75% | Carbohydrate fermentation | Most GM bacteria, including mainly: Akkermansia municiphilia. Bacteroidetes; Bacteroides, Prevotella. Firmicutes; Ruminococcus. Actinobacteria; Bifidobacterium. | FFAR2, FFAR3 |
| Wood–Ljungdahl pathway; Reductive acetogenesis | Acetogenic bacteria from Firmicutes; Blautia hydrogenotrophica, Clostridium, Streptococcus | |||
| Propionate (C3) | 15–20% | Succinate pathway | Bacteriodetes; Bacteroides. Firmicutes; Phascolarctobacterium succinatutens, Veilonella. | FFAR2, FFAR3 |
| Acrylate pathway | Firmicutes; Megasphaera, Coprococcus catus | |||
| Propanediol pathway | Firmicutes; Roseburia, Ruminocossus | |||
| Butyrate (C4) | 15–20% | Acetyl-coA-transferase pathway | Firmicutes; Faecalibacterium, Eubacterium, Roseburia, Anaerostipes, Coprococcus | GPR109A, FFAR2, FFAR3 |
| Butyrate kinase pathway | Firmicutes; Coprococcus |
| Sections | Species | SCFAs | Treatments | Major Outcomes | References |
|---|---|---|---|---|---|
| 4.1 | C57BL/6 mice | Butyrate | NaB, 600 mg/kg, (1) twice daily for 7 weeks, (2) twice daily for 14 days, and (3) twice daily for 14 days without kindling stimulations | (1) ↓HDAC activity in hippocampus and cortex, rate of kindling (1), (2) Suppressed seizure stages (3) ↑Mean afterdischarge duration | [63] |
| 4.2 | Sprague-Dawley rats | Butyrate | NaB 1.5/3.0 g/kg, once as pretreatment | ↓Seizure-related p-ERK and glial fibrillary acidic protein, IL-1β | [92] |
| 4.2 | BALB/c AnNHsd mice | Butyrate | NaB 100 mg/kg a day, 9 days | ↑CD50, occludin ↓IκBα, NF-κB, COX-2, iNOS in DSS-induced colitis | [93] |
| 4.2 | Kunming mice | Propionate | Propionate 37.5/50/75 mg/kg a day, 40 days | ↑ATP, mitochondrial CAT, SOD, GSH-Px ↓8-OHdG, hippocampal neuronal loss | [95] |
| 4.2 | Kunming mice | Butyrate | NaB 5/10/20 mg/kg a day, 40 days | ↑NAD+, ATP, CAT, SOD, GSH-Px, Keap1/Nrf2/HO-1 signals (↑Nrf2&HO-1,↓Keap1) ↓ROS accumulation | [94] |
| 4.3 | NIH Swiss mice | Butyrate | NaB, 1.5 g/kg, once as pretreatment | ↑H3, H4 acetylation in hippocampus and cerebral cortex | [105] |
| 4.3 | WAG/Rij rats | Butyrate | NaB, 30 mg/kg/day, 6 months | ↓Hypersensitivity to stimuli, NFκB, protein oxidation ↑Glutathione reductase | [106] |
| 5.1 | WAG/Rij rats | Butyrate | NaB, 30 mg/kg/day, (1) 17 weeks or (2) 27 weeks | (1) ↑H3, H4 acetylation ↓HDAC1, HDAC3 expression (2) ↓Number and duration of SWDs ↓Depressive-like activities when combined with valproic acid (forced swimming test) No anxiolytic effects found | [107] |
| Species | Pro/Prebiotics | Treatments | Major Outcomes | References |
|---|---|---|---|---|
| GAERS rats, absence epilepsy | Enterococcus faecium, Lactobacillus acidophillus, L.rhamnosus, Bifidobacterium longum, B. bifidum, fructooligosaccharide, polydextrose | 1 month, bottles replaced twice a week | No significant effect on duration and number of SWDs | [149] |
| Swiss Webster mice, DRE | Akkermansia muciniphila, Parabacteroides merdae | (1) 14 days, Abx treatment + 109 CFU with KD or CD (2) 28 days, 109 CFU twice a day with CD | (1) ↑6-Hz seizure threshold, GABA/glutamate ratio, glutamine ↓Ketogenic gamma-glutamylated amino acids (2) ↑6-Hz seizure threshold | [32] |
| Kcna1-/- C3HeB/FeJ mice, TLE, and SUDEP | A. muciniphila, Parabacteroides | 3 weeks, Abx treatment + 109 CFU with KD | ↓Seizure incidence and duration compared to CD-fed controls | |
| Wistar rats, PTZ-induced kindling | Lactobacillus rhamnosus, L. reuteri, B. infantis | 3 weeks, 1 mL solution a day | ↓Seizure severity (Racine scale), NO, MDA ↑TAC, GABA, spatial learning and memory (water maze) | [150] |
| Wistar rats, PTZ-induced kindling | Lactobacillus casei, L. acidophilus, Bifidobacterium bifidum | 6 weeks, 1 mL solution a day | ↓Rate of kindling development ↑spatial learning and memory (water maze) Altered synaptic plasticity;↑PS amplitude (when with Nigella Sativa), ↓LTP | [151] |
| ICR mice, PTZ-induced kindling | Lactobacillus fermentum MSK 408 | 4 weeks, 4 × 109 CFU/mL (1) without or (2) with KD | ↓Seizure frequency and duration (1) ↑GLUT-1, TJ proteins ZO-1, claudin, occludin in brain ↓Glucose, cholesterol, TNF-α in serum (2) ↑GABAa1, b1b, acetate, isobutyrate ↓Propionate, butyrate | [152] |
| ICR mice, PTZ-induced kindling | Lactobacillus fermentum MSK 408, Galactooligosaccharide | 8 weeks, 5 × 108 CFU/g/day with ① CD ② CD + GOS ③ KD ④ KD + GOS | ↓Seizure numbers (③,④) ↑TJ proteins ZO-1, occludin, mucin-2 in gut barrier (②,④) ↓GABA receptor subunits GABAγ2, GABAa1a, GABAδ (③, ④), ↑NMDA receptor subunits NR2A, NR2B (②) ↓Acetate, propionate, butyrate | [36] |
| NMRI mice, PTZ-induced kindling | Lactobacillus casei, L. acidophilus, Bifidobacterium bifidum | 14 or 28 days, 109 CFU, 10 mL/kg/day | ↓Intensity of tonic-clonic movements Significant seizure prevention only when used with DZP Preserves protective effects of DZP in the presence of flumazenil | [153] |
| Wistar weaner rats, PTZ-induced kindling | Bifidobacterium lactis, B. breve, B. longum, B. bifidum, Lactobacillus acidophilus, L. casei, L. plantarum, L. salivarius, L. rhamnosus, L. bulgaricus, L. paracasei, Streptococcus thermophilus, Ascophyllum nodosum | 60 days, 109 CFU/mL/day | ↓Seizure duration and onset ↓IL-1β, IL-6, IL-17A and total oxidant status in plasma and brain tissue, disulfide in plasma ↑ Total thiol | [154] |
| Sprague-Dawley rats, infantile spasms | Streptococcus thermophilus HA-110, Lactococcus lactis subsp. lactis HA-136 | 5 postnatal days, 100µL total of 1010 CFU/mL with KD or CD | ↑PPARα, HDAC activity, carnitines, caspase 1 and IL-18 (activate AMPK to drive lipid oxidation), IL-1β, IL-6 ↓Triglyceride, MDA, polyunsaturated fatty acids | [155] |
| WAG/Rij rats, absence epilepsy | VSL#3; Lactobacillus plantarum, L. acidophilus, L. delbrueckii subsp. bulgaricus, L. casei, Bifidobacterium longum, B. breve, B infantis, Streptococcus salivarius subsp. Thermophilus | 30 days, 12.86 billion live bacteria/kg/day | ↓Duration and number of SWDs, anxiety- and depression-like behaviors (open-field test, forced swimming test), TNF-α, IL-6, NO ↑NGF immunoreactivity | [156] |
| Wistar albino rats, PTZ-induced kindling | VSL#3 | 6 weeks, 12.86 billion live bacteria/kg/day | ↓Seizure severity (Racine scale), TNF-α, IL-6, NO, total oxidant status in brain ↑ NGF, BDNF | [157] |
| Wistar Albino rats, penicillin G induced focal seizures | VSL#3 | 30 days, 12.86 billion live bacteria/kg/day | Increased latency and decreased spike frequency of focal seizures ↓IL-6, TNF-α, NO | [158] |
| Sprague-Dawley rats, infantile spasms | Streptococcus thermophilus HA-110, Lactococcus lactis subsp. lactis HA-136, Ligilactobacillus salivarius HA-118 | 100 μL, 1010 CFU/mL a day with KD | ↓Seizure frequency, IL-18, IL-6, TNF-α ↑Serum and hippocampal metabolites in antioxidant pathways Enhanced neurobehavior (surface righting time) and locomotor activity (open field test) | [159] |
| Patient Group | Pro/Prebiotics | Treatments | Major Outcomes | References |
|---|---|---|---|---|
| ≥18 years, DRE (n = 45) | Lactobacillus acidophilus, L. plantarum, L. casei, L. helveticus, L. brevis, Bifidobacterium lactis, Streptococcus salivarius subsp. Thermophilus | 4 months, twice a day | >50% reduction in seizures in 28.9% (n = 13) patients ↑QoL (QOLIE-10), Serum GABA ↓IL-6, CD-14 | [160] |
| ≥34 weeks, neonatal seizures (n = 228) | Saccharomyces boulardii, Lactobacillus casei | Within 24 h of birth | ↓Risk for seizures; Rotavirus infection remained a risk factor only in those who did not take probiotics | [161] |
| 18 years male, DRE with Rasmussen encephalitis | Kefir (fermented milk); Lactobacillus kefiranofaciens, L. kefiri, L. helveticus, L. lactis | 90 days, 2 mL/Kg/day | ↓Seizure numbers, superoxide anion/hydrogen peroxide/peroxynitrite, TNF/IL-1β/IL-6/IL-8 ↑Lactobacillus, Bifidobacterium, NO bioavailability, anti-inflammatory IL-10 | [162] |
| <9 years children, DRE (n = 44) | Bacillus subtilis | 1 year, taken as granules with KD | When compared to group only taken KD (n = 36): ↑Clinical efficacy, QoL (QOLCE-55), cognitive function (verbal intelligence quotient, performance intelligence quotient), 5-HT ↓Incidence of adverse reactions | [163] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.; Park, S.; Choi, T.G.; Kim, S.S. Role of Short Chain Fatty Acids in Epilepsy and Potential Benefits of Probiotics and Prebiotics: Targeting “Health” of Epileptic Patients. Nutrients 2022, 14, 2982. https://doi.org/10.3390/nu14142982
Kim S, Park S, Choi TG, Kim SS. Role of Short Chain Fatty Acids in Epilepsy and Potential Benefits of Probiotics and Prebiotics: Targeting “Health” of Epileptic Patients. Nutrients. 2022; 14(14):2982. https://doi.org/10.3390/nu14142982
Chicago/Turabian StyleKim, Soomin, Siyeon Park, Tae Gyu Choi, and Sung Soo Kim. 2022. "Role of Short Chain Fatty Acids in Epilepsy and Potential Benefits of Probiotics and Prebiotics: Targeting “Health” of Epileptic Patients" Nutrients 14, no. 14: 2982. https://doi.org/10.3390/nu14142982
APA StyleKim, S., Park, S., Choi, T. G., & Kim, S. S. (2022). Role of Short Chain Fatty Acids in Epilepsy and Potential Benefits of Probiotics and Prebiotics: Targeting “Health” of Epileptic Patients. Nutrients, 14(14), 2982. https://doi.org/10.3390/nu14142982