
New Insights into Bile Acids Related Signaling Pathways in the Onset of Colorectal Cancer

 ,
,  , ,
, , 
Abstract
:1. Introduction
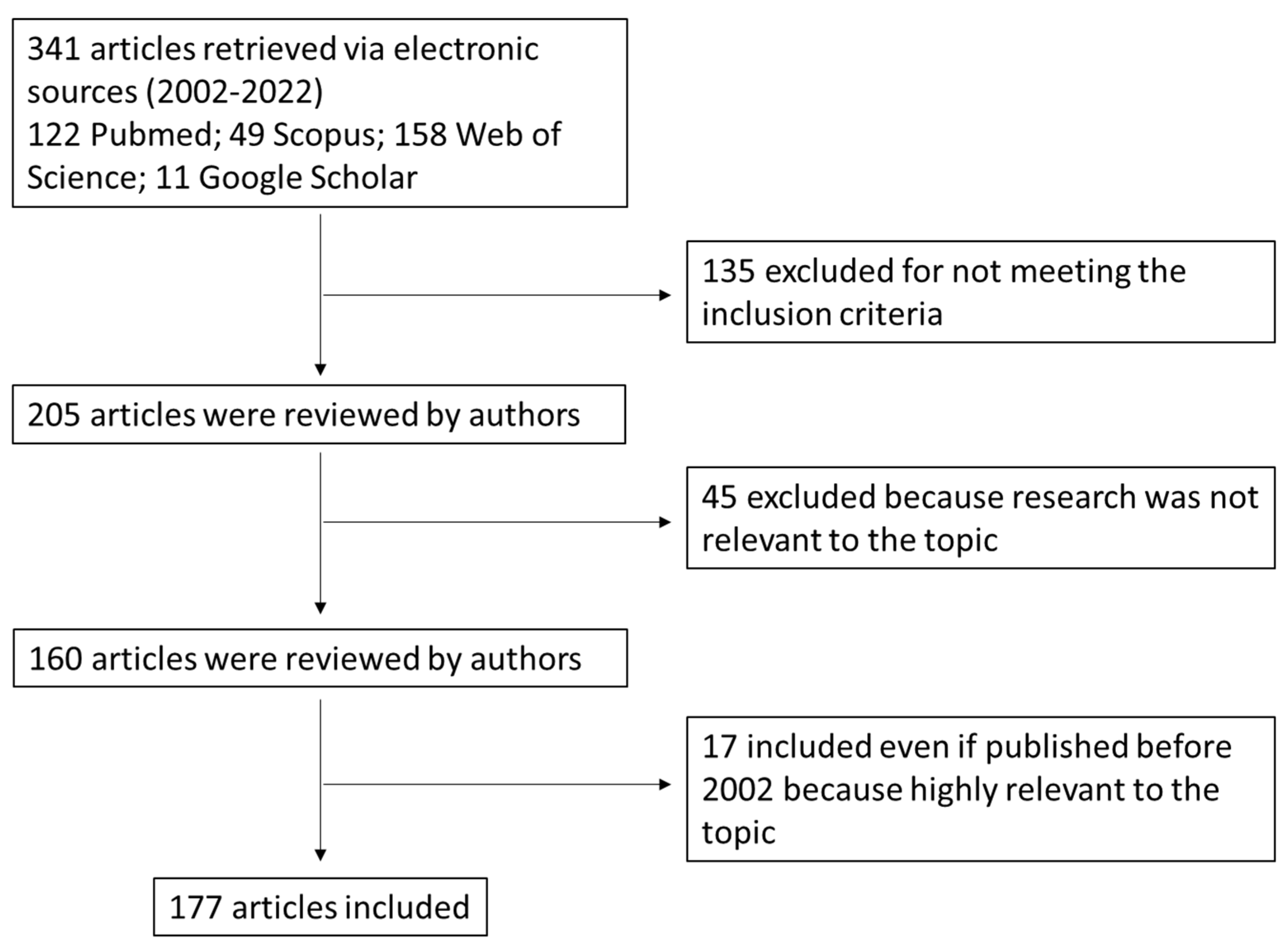
2. Search Strategy (Methods)
- Discussing BAs synthesis and metabolism, physicochemical properties and BA receptors in the gut;
- Including findings from human and animal studies (if relevant) with the support of preclinical data related to the role of BAs in CRC onset;
- Published in peer-reviewed journals;
- Available in full-text;
- Written in English.
- Published within the time frame of 2002–2022. Papers published before 2002 were included if they were relevant in the field.
3. Bile Acids: An Overview
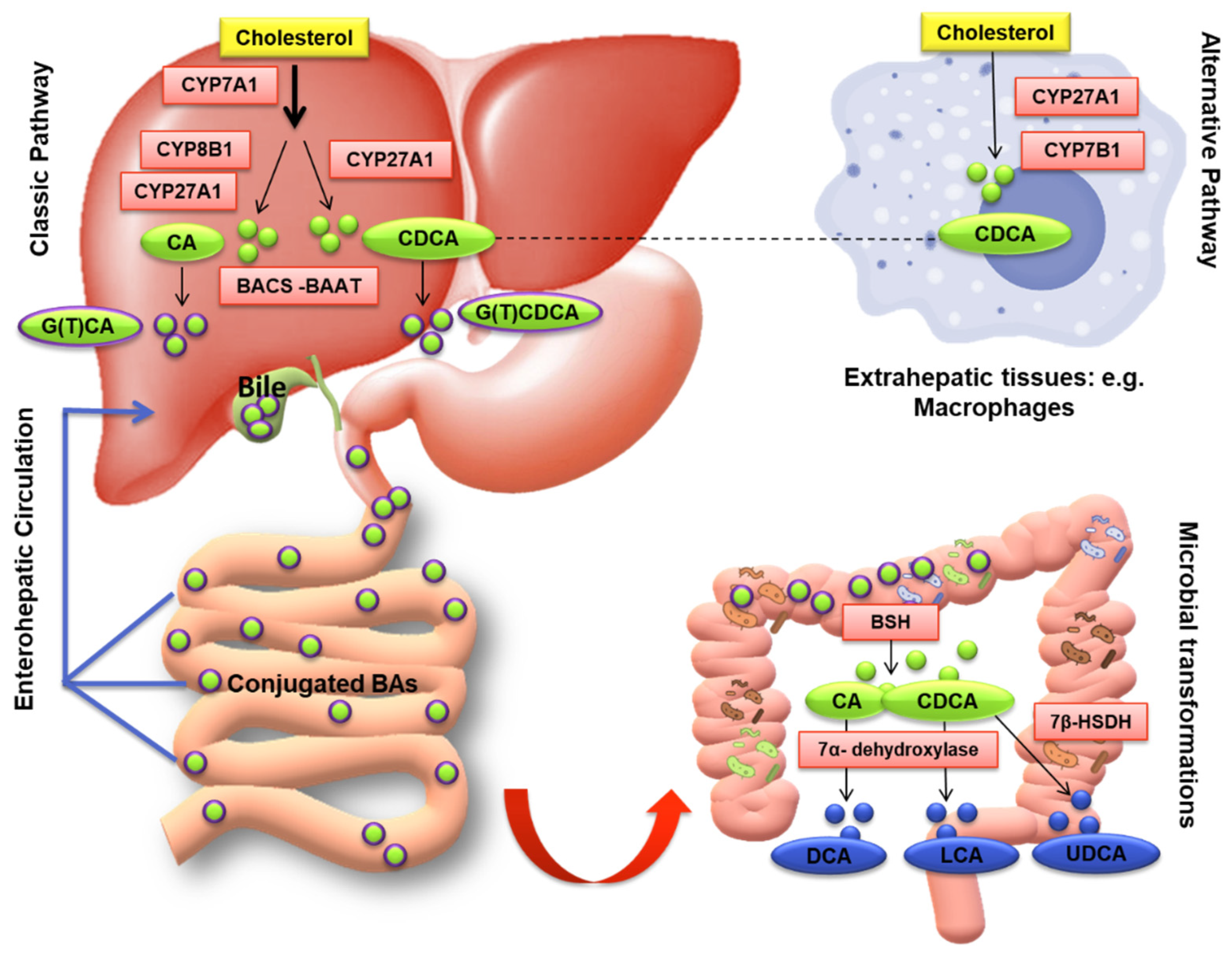
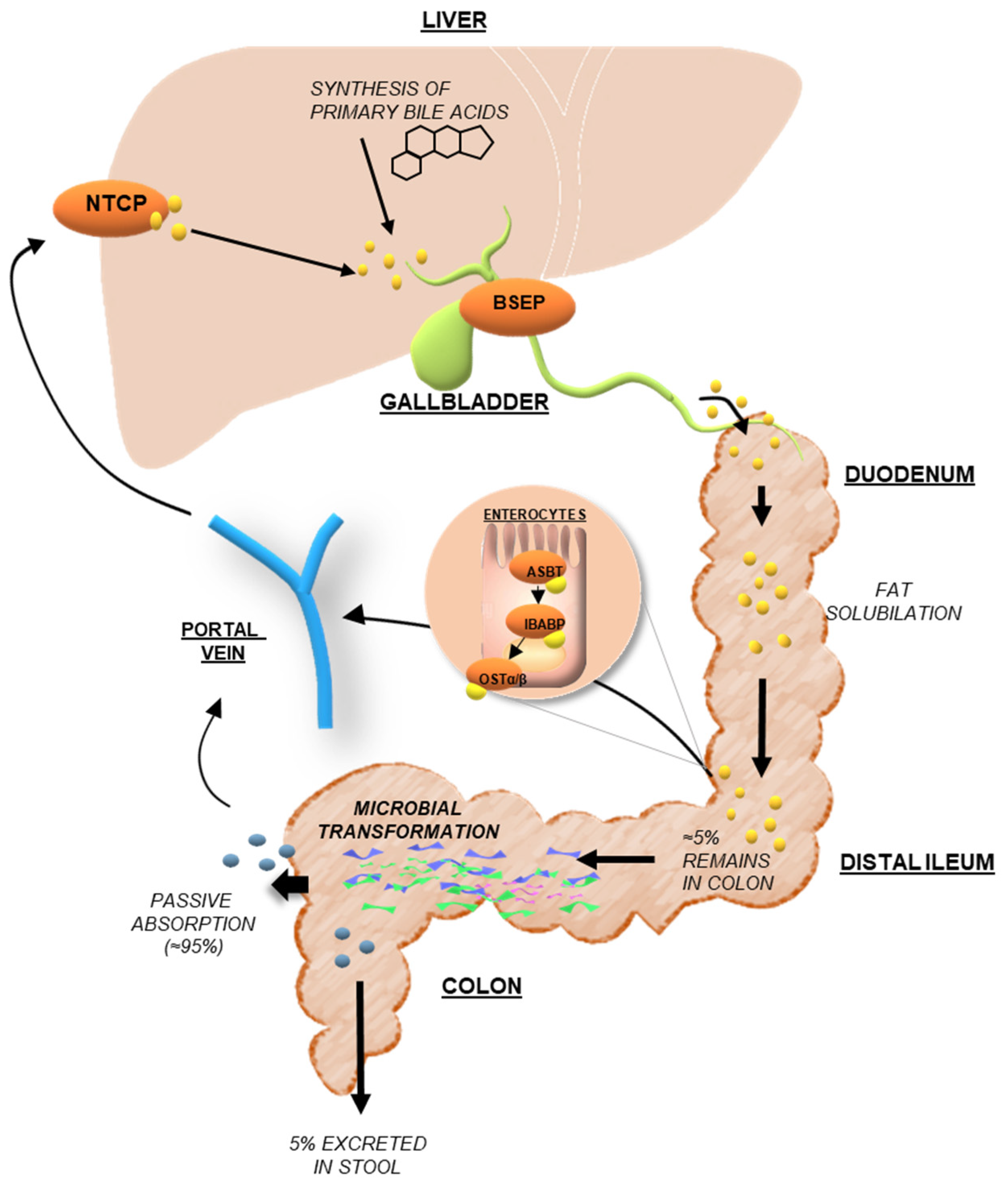
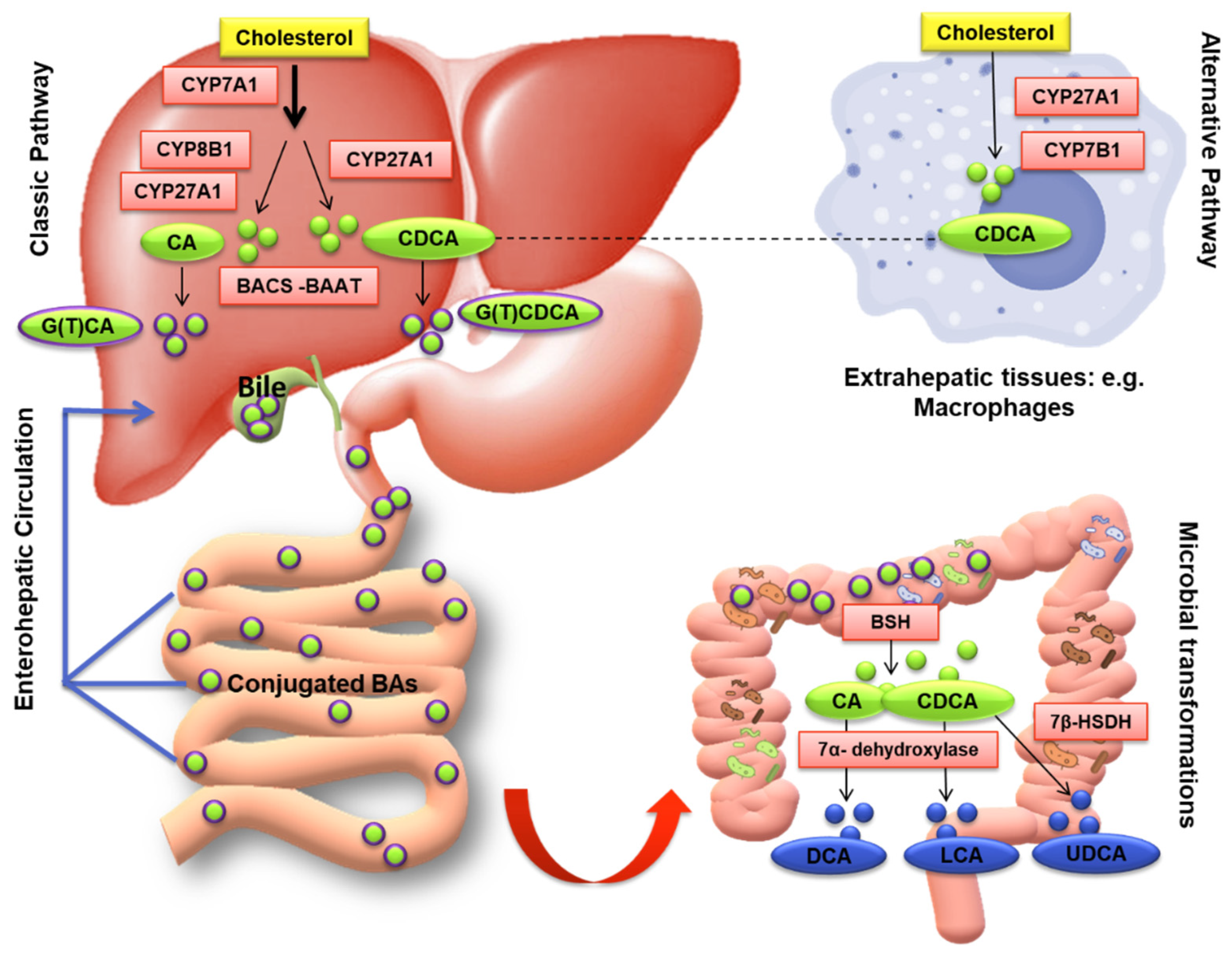
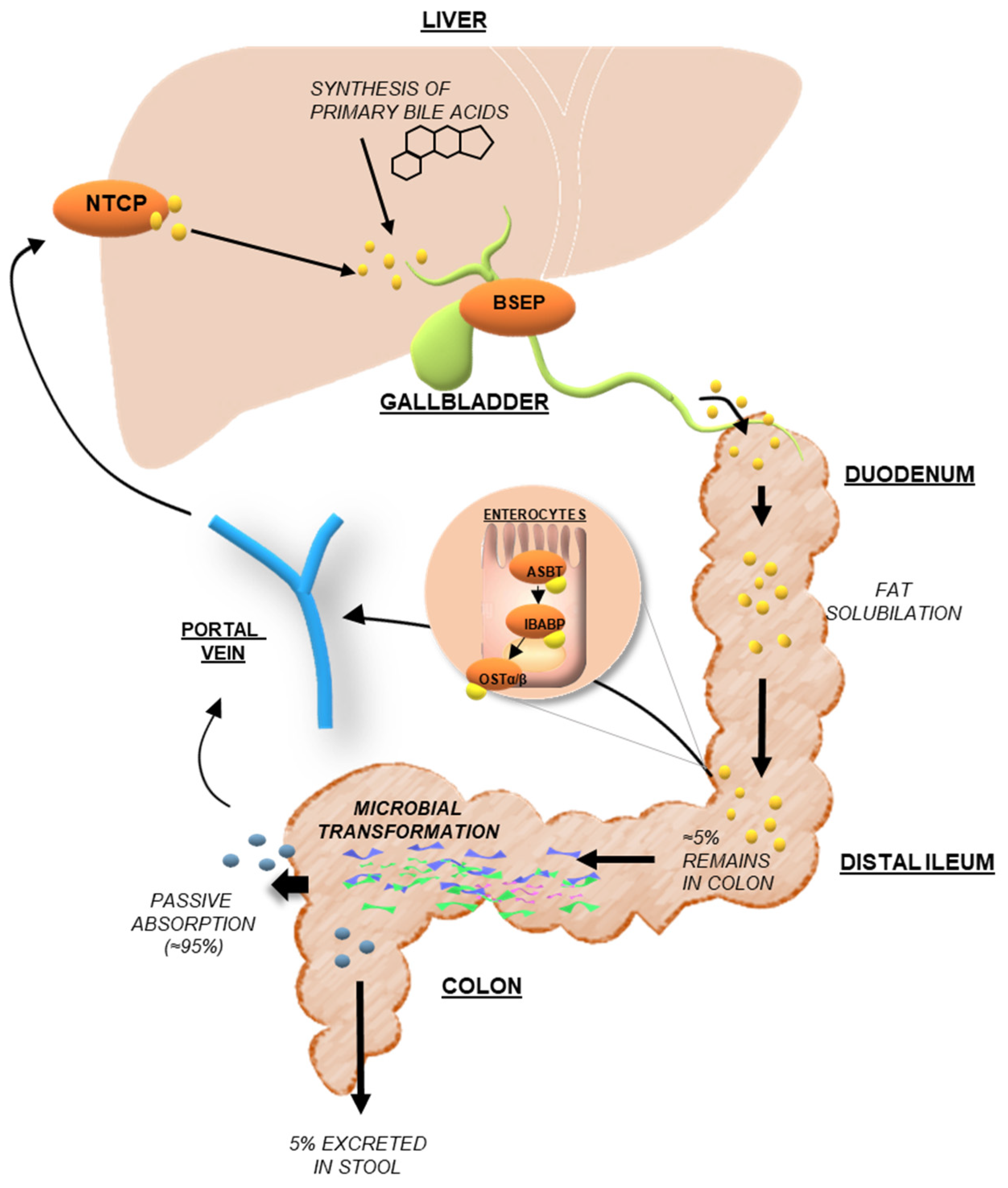
3.1. Bile Acid Synthesis and Metabolism
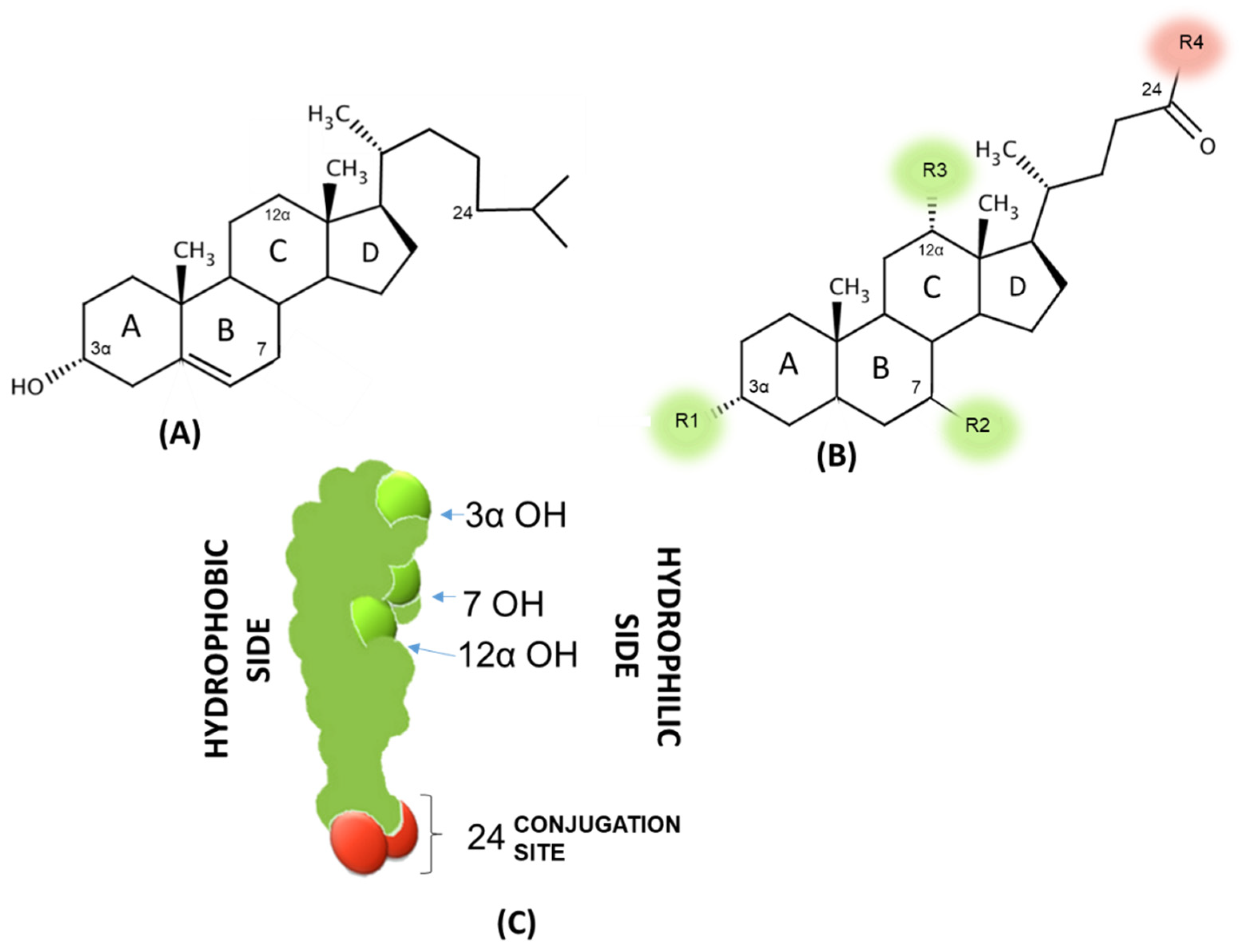
3.2. Physicochemical Properties of Bile Acids
4. Dysregulated Signaling Pathways in Colorectal Cancer: The Emerging Role of Bile Acids
4.1. The Molecular Characteristics of Colorectal Cancer: Implications of the Major Signaling Pathways
4.2. Involvement of Bile Acids in Colorectal Carcinogenesis
4.3. Role of Bile Acid Receptors—Derived Signaling in Colorectal Cancer
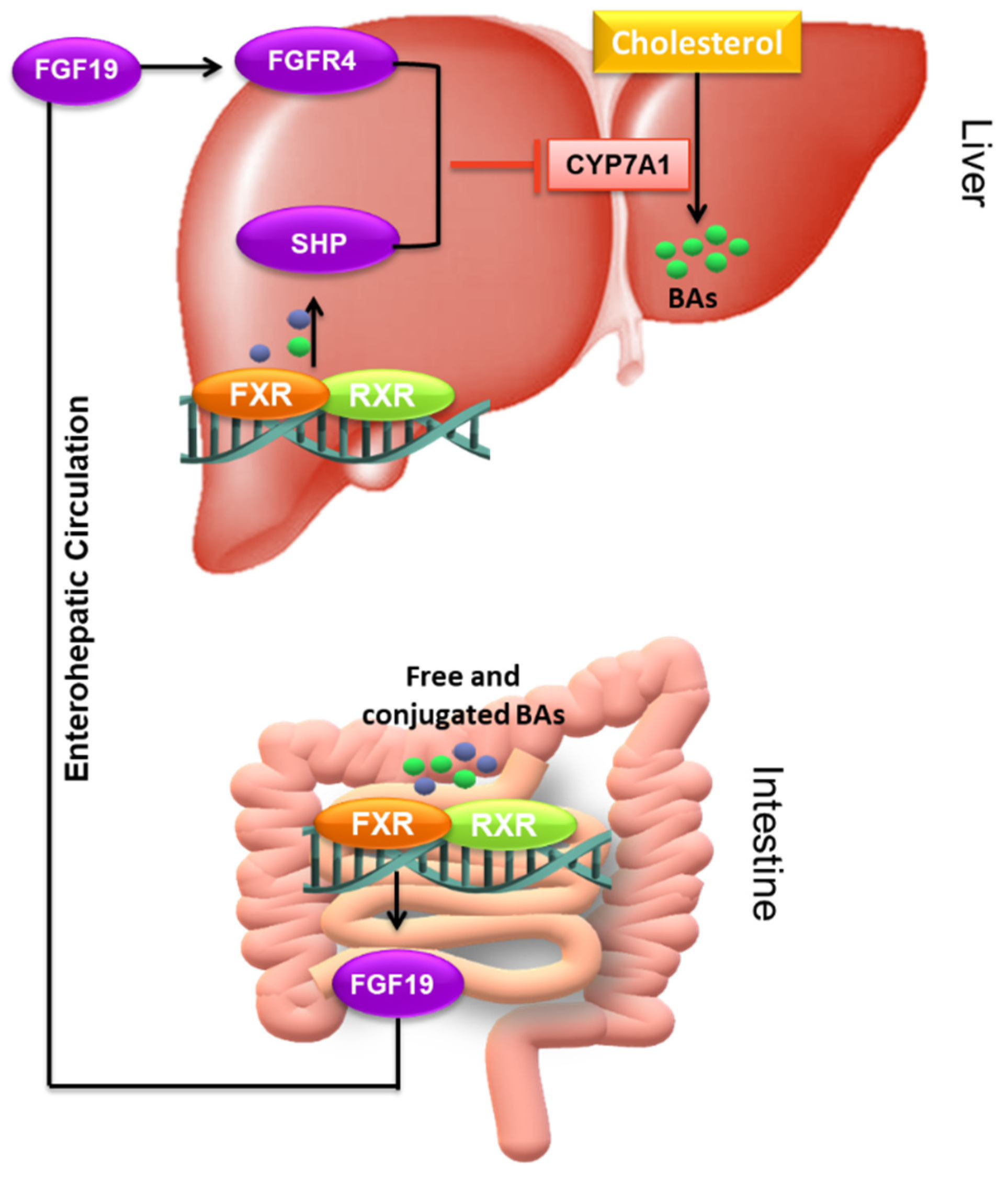
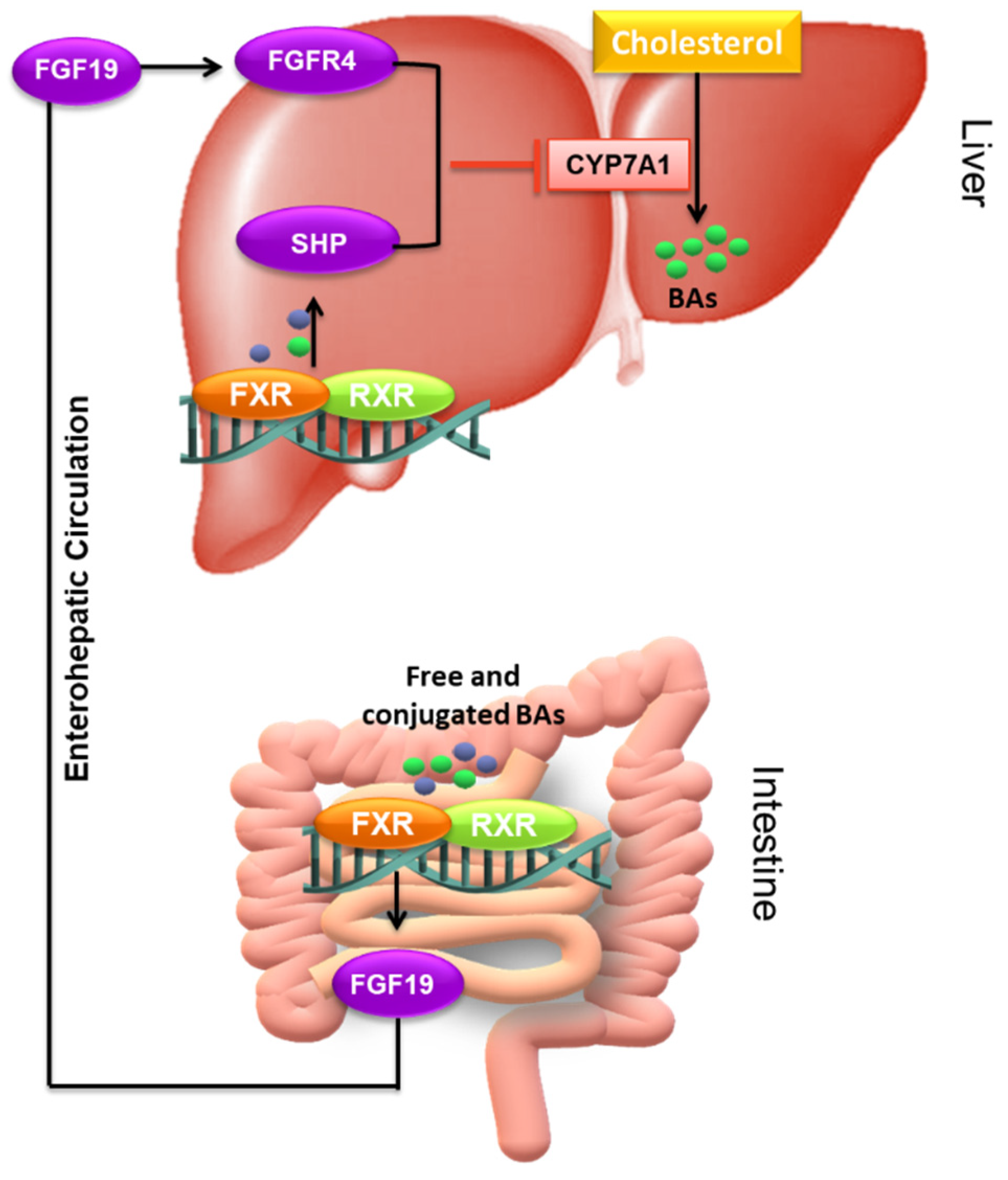
4.3.1. The Nuclear Receptors: FXR, VDR, and SXR/PXR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Bile Acid | BAs Receptor | References |
|---|---|---|---|
| NF-κB signaling | DCA | Not specified | [92] |
| CA | Not specified | [96] | |
| CDCA | TGR5 | [153] | |
| Not specified | FXR | [90,119,124] | |
| Not specified | TGR5 | [149,154] | |
| EGFR axis | DCA | Not specified | [93] |
| MAPK signaling | DCA | Not specified | [94] |
| CA | Not specified | [96] | |
| Not specified | FXR | [105,106] | |
| Not specified | Not specified | [92] | |
| K-Ras signaling | Not specified | FXR | [110] |
| Wnt/β-catenin signaling | OCA | FXR | [112] |
| DCA | FXR | [114] | |
| Not specified | FXR | [108,111,113,115] | |
| JAK2/STAT3 signaling | OCA | FXR | [112] |
| Not specified | Not specified | [138] | |
| CDCA | TGR5 | [153] | |
| Not specified | TGR5 | [154] | |
| PI3K signaling | Not specified | FXR | [123] |
4.3.2. The Membrane Receptors: TGR5 and S1PR2
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Cancer Today-Global Cancer Observatory-IARC. Available online: https://gco.iarc.fr/today/data/factsheets/cancers/10_8_9-Colorectum-fact-sheet.pdf (accessed on 3 May 2022).
- DE Rosa, M.; Pace, U.; Rega, D.; Costabile, V.; Duraturo, F.; Izzo, P.; Delrio, P. Genetics, diagnosis and management of colorectal cancer (Review). Oncol. Rep. 2015, 34, 1087–1096. [Google Scholar] [CrossRef] [Green Version]
- Fodde, R. The APC gene in colorectal cancer. Eur. J. Cancer 2002, 38, 867–871. [Google Scholar] [CrossRef]
- Oh, H.-H.; Joo, Y.-E. Novel biomarkers for the diagnosis and prognosis of colorectal cancer. Intest. Res. 2020, 18, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.G.; Karlitz, J.J.; Yen, T.; Lieu, C.H.; Boland, C.R. The rising tide of early-onset colorectal cancer: A comprehensive review of epidemiology, clinical features, biology, risk factors, prevention, and early detection. Lancet Gastroenterol. Hepatol. 2022, 7, 262–274. [Google Scholar] [CrossRef]
- Saif, M.W.; Chu, E. Biology of Colorectal Cancer. Cancer J. 2010, 16, 196–201. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas (TCGA) Research Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Doll, R.; Peto, R. The Causes of Cancer: Quantitative Estimates of Avoidable Risks of Cancer in the United States Today. JNCI J. Natl. Cancer Inst. 1981, 66, 1192–1308. [Google Scholar] [CrossRef]
- Feagins, L.A.; Souza, R.F.; Spechler, S.J. Carcinogenesis in IBD: Potential targets for the prevention of colorectal cancer. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 297–305. [Google Scholar] [CrossRef]
- Yang, J.; Yu, J. The association of diet, gut microbiota and colorectal cancer: What we eat may imply what we get. Protein Cell 2018, 9, 474–487. [Google Scholar] [CrossRef] [Green Version]
- Aldini, R.; Montagnani, M.; Roda, A.; Hrelia, S.; Biagi, P.; Roda, E. Intestinal absorption of bile acids in the rabbit: Different transport rates in jejunum and ileum. Gastroenterology 1996, 110, 459–468. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Ung, T.T.; Kim, N.H.; Jung, Y.D. Role of bile acids in colon carcinogenesis. World J. Clin. Cases 2018, 6, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Ocvirk, S.; O’Keefe, S.J. Dietary fat, bile acid metabolism and colorectal cancer. Semin. Cancer Biol. 2021, 73, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Salic, K.; Kleemann, R.; Wilkins-Port, C.; McNulty, J.; Verschuren, L.; Palmer, M. Apical sodium-dependent bile acid transporter inhibition with volixibat improves metabolic aspects and components of non-alcoholic steatohepatitis in Ldlr-/- Leiden mice. PLoS ONE 2019, 14, e0218459. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.W. The Enzymes, Regulation, and Genetics of Bile Acid Synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Hylemon, P.B. Bile acids are nutrient signaling hormones. Steroids 2014, 86, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Monte, M.J.; Marin, J.J.; Antelo, A.; Vazquez-Tato, J. Bile acids: Chemistry, physiology, and pathophysiology. World J. Gastroenterol. 2009, 15, 804–816. [Google Scholar] [CrossRef]
- Chiang, J.Y.L. Bile Acid Metabolism and Signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef] [Green Version]
- Alan, F.H. The enterohepatic circulation of bile acids in mammals: Form and functions. Front. Biosci. 2009, 14, 2584–2598. [Google Scholar] [CrossRef] [Green Version]
- Cardenia, V.; Rodriguez-Estrada, M.T.; Lorenzini, A.; Bandini, E.; Angeloni, C.; Hrelia, S.; Malaguti, M. Effect of broccoli extract enriched diet on liver cholesterol oxidation in rats subjected to exhaustive exercise. J. Steroid Biochem. Mol. Biol. 2017, 169, 137–144. [Google Scholar] [CrossRef]
- Vaz, F.M.; Ferdinandusse, S. Bile acid analysis in human disorders of bile acid biosynthesis. Mol. Asp. Med. 2017, 56, 10–24. [Google Scholar] [CrossRef]
- Roda, A.; Hofmann, A.F.; Mysels, K.J. The influence of bile salt structure on self-association in aqueous solutions. J. Biol. Chem. 1983, 258, 6362–6370. [Google Scholar] [CrossRef]
- Roda, A.; Minutello, A.; Angellotti, M.; Fini, A. Bile acid structure-activity relationship: Evaluation of bile acid lipophilicity using 1-octanol/water partition coefficient and reverse phase HPLC. J. Lipid Res. 1990, 31, 1433–1443. [Google Scholar] [CrossRef]
- Roda, A.; Pellicciari, R.; Gioiello, A.; Neri, F.; Camborata, C.; Passeri, D.; De Franco, F.; Spinozzi, S.; Colliva, C.; Adorini, L.; et al. Semisynthetic Bile Acid FXR and TGR5 Agonists: Physicochemical Properties, Pharmacokinetics, and Metabolism in the Rat. J. Pharmacol. Exp. Ther. 2014, 350, 56–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roda, A.; Grigolo, B.; Roda, E.; Simoni, P.; Pellicciari, R.; Natalini, B.; Fini, A.; Labate, A.M.M. Quantitative Relationship between Bile Acid Structure and Biliary Lipid Secretion in Rats. J. Pharm. Sci. 1988, 77, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Schneider, K.M.; Albers, S.; Trautwein, C. Role of bile acids in the gut-liver axis. J. Hepatol. 2018, 68, 1083–1085. [Google Scholar] [CrossRef]
- Heidi Eigenrauch Karpen, S.J.K. Bile Acid Metabolism During Development. In Fetal and Neonatal Physiology, 5th ed.; Polin, R.A., Abman, S.H., Rowitch, D., Benitz, W.E., Eds.; Elsevier: Philadelphia, PA, USA, 2017; Volume 2, pp. 913–929. ISBN 978-032-371-284-2. [Google Scholar]
- Roda, A.; Cappelleri, G.; Aldini, R.; Roda, E.; Barbara, L. Quantitative aspects of the interaction of bile acids with human serum albumin. J. Lipid Res. 1982, 23, 490–495. [Google Scholar] [CrossRef]
- Hofmann, A.F. The Continuing Importance of Bile Acids in Liver and Intestinal Disease. Arch. Intern. Med. 1999, 159, 2647–2658. [Google Scholar] [CrossRef]
- Chen, M.-J.; Liu, C.; Wan, Y.; Yang, L.; Jiang, S.; Qian, D.-W.; Duan, J.-A. Enterohepatic circulation of bile acids and their emerging roles on glucolipid metabolism. Steroids 2021, 165, 108757. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [Green Version]
- Durník, R.; Šindlerová, L.; Babica, P.; Jurček, O. Bile Acids Transporters of Enterohepatic Circulation for Targeted Drug Delivery. Molecules 2022, 27, 2961. [Google Scholar] [CrossRef]
- Gérard, P. Metabolism of Cholesterol and Bile Acids by the Gut Microbiota. Pathogens 2013, 3, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez, B. Bile acid–microbiota crosstalk in gastrointestinal inflammation and carcinogenesis: A role for bifidobacteria and lactobacilli? Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porru, E.; Scicchitano, D.; Interino, N.; Tavella, T.; Candela, M.; Roda, A.; Fiori, J. Analysis of fecal bile acids and metabolites by high resolution mass spectrometry in farm animals and correlation with microbiota. Sci. Rep. 2022, 12, 2866. [Google Scholar] [CrossRef]
- Funabashi, M.; Grove, T.L.; Wang, M.; Varma, Y.; McFadden, M.E.; Brown, L.C.; Guo, C.; Higginbottom, S.; Almo, S.C.; Fischbach, M.A. A metabolic pathway for bile acid dehydroxylation by the gut microbiome. Nature 2020, 582, 566–570. [Google Scholar] [CrossRef]
- Fini, A.; Roda, A. Chemical properties of bile acids. IV. Acidity constants of glycine-conjugated bile acids. J. Lipid Res. 1987, 28, 755–759. [Google Scholar] [CrossRef]
- Franco, P.; Porru, E.; Fiori, J.; Gioiello, A.; Cerra, B.; Roda, G.; Caliceti, C.; Simoni, P.; Roda, A. Identification and quantification of oxo-bile acids in human faeces with liquid chromatography–mass spectrometry: A potent tool for human gut acidic sterolbiome studies. J. Chromatogr. A 2019, 1585, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Wahlström, A.; Sayin, S.I.; Marschall, H.-U.; Bäckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridlon, J.M.; Bajaj, J.S. The human gut sterolbiome: Bile acid-microbiome endocrine aspects and therapeutics. Acta Pharm. Sin. B 2015, 5, 99–105. [Google Scholar] [CrossRef] [Green Version]
- Ajouz, H.; Mukherji, D.; Shamseddine, A. Secondary bile acids: An underrecognized cause of colon cancer. World J. Surg. Oncol. 2014, 12, 164. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, H.; Masuno, H.; Kawasaki, H.; Yoshihara, A.; Numoto, N.; Ito, N.; Ishida, H.; Yamamoto, K.; Hirata, N.; Kanda, Y.; et al. Lithocholic Acid Derivatives as Potent Vitamin D Receptor Agonists. J. Med. Chem. 2021, 64, 516–526. [Google Scholar] [CrossRef]
- Pols, T.W.H.; Puchner, T.; Korkmaz, H.I.; Vos, M.; Soeters, M.R.; De Vries, C.J.M. Lithocholic acid controls adaptive immune responses by inhibition of Th1 activation through the Vitamin D receptor. PLoS ONE 2017, 12, e0176715. [Google Scholar] [CrossRef] [PubMed]
- Stellaard, F.; Lütjohann, D. Dynamics of the enterohepatic circulation of bile acids in healthy humans. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 321, G55–G66. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, M. Bile acid detergency: Permeability, inflammation, and effects of sulfation. Am. J. Physiol. Gastrointest. Liver Physiol. 2022, 322, G480–G488. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.F. Detoxification of Lithocholic Acid, A Toxic Bile Acid: Relevance to Drug Hepatotoxicity. Drug Metab. Rev. 2004, 36, 703–722. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, G.; Perino, A.; Yildiz, E.; El Alam, G.; Sleiman, M.B.; Gioiello, A.; Pellicciari, R.; Schoonjans, K. Bile Acids Signal via TGR5 to Activate Intestinal Stem Cells and Epithelial Regeneration. Gastroenterology 2020, 159, 956–968.e958. [Google Scholar] [CrossRef]
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef]
- Brenner, H.; Kloor, M.; Pox, C.P. Colorectal cancer. Lancet 2014, 383, 1490–1502. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Elinav, E.; Nowarski, R.; Thaiss, C.A.; Hu, B.; Jin, C.; Flavell, R.A. Inflammation-induced cancer: Crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 2013, 13, 759–771. [Google Scholar] [CrossRef]
- Buhrmann, C.; Brockmueller, A.; Harsha, C.; Kunnumakkara, A.B.; Kubatka, P.; Aggarwal, B.B.; Shakibaei, M. Evidence That Tumor Microenvironment Initiates Epithelial-To-Mesenchymal Transition and Calebin A can Suppress it in Colorectal Cancer Cells. Front. Pharmacol. 2021, 12, 699842. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Vogelstein, B. Lessons from Hereditary Colorectal Cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Fearon, E.R. Molecular Genetics of Colorectal Cancer. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 479–507. [Google Scholar] [CrossRef] [PubMed]
- Tape, C.J. The Heterocellular Emergence of Colorectal Cancer. Trends Cancer 2017, 3, 79–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paterson, C.; Clevers, H.; Bozic, I. Mathematical model of colorectal cancer initiation. Proc. Natl. Acad. Sci. USA 2020, 117, 20681–20688. [Google Scholar] [CrossRef] [PubMed]
- van Neerven, S.M.; de Groot, N.E.; Nijman, L.E.; Scicluna, B.P.; van Driel, M.S.; Lecca, M.C.; Warmerdam, D.O.; Kakkar, V.; Moreno, L.F.; Braga, F.A.V.; et al. Apc-mutant cells act as supercompetitors in intestinal tumour initiation. Nature 2021, 594, 436–441. [Google Scholar] [CrossRef]
- Flanagan, D.J.; Pentinmikko, N.; Luopajärvi, K.; Willis, N.J.; Gilroy, K.; Raven, A.P.; Mcgarry, L.; Englund, J.I.; Webb, A.T.; Scharaw, S.; et al. NOTUM from Apc-mutant cells biases clonal competition to initiate cancer. Nature 2021, 594, 430–435. [Google Scholar] [CrossRef]
- Pentinmikko, N.; Iqbal, S.; Mana, M.; Andersson, S.; Cognetta, A.B., III.; Suciu, R.M.; Roper, J.; Luopajärvi, K.; Markelin, E.; Gopalakrishnan, S.; et al. Notum produced by Paneth cells attenuates regeneration of aged intestinal epithelium. Nat. Cell Biol. 2019, 571, 398–402. [Google Scholar] [CrossRef]
- Chia, S.B.; DeGregori, J. Cancer stem cells in the gut have a bad influence on neighbouring cells. Nature 2021, 594, 340–341. [Google Scholar] [CrossRef]
- Malinowsky, K.; Nitsche, U.; Janssen, K.-P.; Bader, F.G.; Späth, C.; Drecoll, E.; Keller, G.; Höfler, H.; Slotta-Huspenina, J.; Becker, K.-F. Activation of the PI3K/AKT pathway correlates with prognosis in stage II colon cancer. Br. J. Cancer 2014, 110, 2081–2089. [Google Scholar] [CrossRef] [Green Version]
- Koveitypour, Z.; Panahi, F.; Vakilian, M.; Peymani, M.; Forootan, F.S.; Esfahani, M.H.N.; Ghaedi, K. Signaling pathways involved in colorectal cancer progression. Cell Biosci. 2019, 9, 97. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, P.; Köhne, C.-H.; Qvortrup, C. The changing face of treatment for metastatic colorectal cancer. Expert Rev. Anticancer Ther. 2019, 19, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High Frequency of Mutations of the PIK3CA Gene in Human Cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.-F.; Yu, B.-H.; Li, D.-L.; Ke, H.-L.; Guo, X.-Z.; Xiao, X.-Y. PI3K expression and PIK3CA mutations are related to colorectal cancer metastases. World J. Gastroenterol. 2012, 18, 3745–3751. [Google Scholar] [CrossRef]
- Bowen, K.A.; Doan, H.Q.; Zhou, B.P.; Wang, Q.; Zhou, Y.; Rychahou, P.G.; Evers, B.M. PTEN loss induces epithelial--mesenchymal transition in human colon cancer cells. Anticancer Res. 2009, 29, 4439–4449. [Google Scholar] [PubMed]
- Wilson, P.M.; LaBonte, M.J.; Lenz, H.-J. Molecular Markers in the Treatment of Metastatic Colorectal Cancer. Cancer J. 2010, 16, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Bai, D.; Ueno, L.; Vogt, P.K. Akt-mediated regulation of NFκB and the essentialness of NFκB for the oncogenicity of PI3K and Akt. Int. J. Cancer 2009, 125, 2863–2870. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Liu, Z.; Wang, L.; Zhang, X. NF-κB Signaling Pathway, Inflammation and Colorectal Cancer. Cell. Mol. Immunol. 2009, 6, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Nolan, G.P.; Ghosh, S.; Liou, H.-C.; Tempst, P.; Baltimore, D. DNA binding and IκB inhibition of the cloned p65 subunit of NF-κB, a rel-related polypeptide. Cell 1991, 64, 961–969. [Google Scholar] [CrossRef]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Siveen, K.S.; Prabhu, K.; Krishnankutty, R.; Kuttikrishnan, S.; Tsakou, M.; Alali, F.; Dermime, S.; Mohammad, R.M.; Uddin, S. Vascular Endothelial Growth Factor (VEGF) Signaling in Tumour Vascularization: Potential and Challenges. Curr. Vasc. Pharmacol. 2017, 15, 339–351. [Google Scholar] [CrossRef]
- Zhu, D.; Yang, Z.; Miao, X.; Yang, W.; Jiang, M.; Chen, Y. Association of Serum Vascular Endothelial Growth Factor (VEGF) with Colorectal Cancer: A Systemic Review and Meta-Analysis. J. Cancer Sci. Clin. Ther. 2020, 4, 15–31. [Google Scholar] [CrossRef] [Green Version]
- Hansen, T.; Qvortrup, C.; Pfeiffer, P. Angiogenesis Inhibitors for Colorectal Cancer. A Review of the Clinical Data. Cancers 2021, 13, 1031. [Google Scholar] [CrossRef]
- Caliceti, C.; Zambonin, L.; Rizzo, B.; Fiorentini, D.; Sega, F.V.D.; Hrelia, S.; Prata, C. Role of plasma membrane caveolae/lipid rafts in VEGF-induced redox signaling in human leukemia cells. BioMed Res. Int. 2014, 2014, 857504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortini, F.; Sega, F.V.D.; Caliceti, C.; Lambertini, E.; Pannuti, A.; Peiffer, D.S.; Balla, C.; Rizzo, P. Estrogen-mediated protection against coronary heart disease: The role of the Notch pathway. J. Steroid Biochem. Mol. Biol. 2019, 189, 87–100. [Google Scholar] [CrossRef]
- Kuhnert, F.; Kirshner, J.R.; Thurston, G. Dll4-Notch signaling as a therapeutic target in tumor angiogenesis. Vasc. Cell 2011, 3, 20. [Google Scholar] [CrossRef] [Green Version]
- Massard, C.; Azaro, A.; Soria, J.-C.; Lassen, U.; Le Tourneau, C.; Sarker, D.; Smith, C.; Ohnmacht, U.; Oakley, G.; Patel, B.; et al. First-in-human study of LY3039478, an oral Notch signaling inhibitor in advanced or metastatic cancer. Ann. Oncol. 2018, 29, 1911–1917. [Google Scholar] [CrossRef]
- Yeom, D.-H.; Lee, Y.-S.; Ryu, I.; Lee, S.; Sung, B.; Lee, H.-B.; Kim, D.; Ahn, J.-H.; Ha, E.; Choi, Y.-S.; et al. ABL001, a Bispecific Antibody Targeting VEGF and DLL4, with Chemotherapy, Synergistically Inhibits Tumor Progression in Xenograft Models. Int. J. Mol. Sci. 2020, 22, 241. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.; Sharma, A.K.; Damodaran, C. A Review on Notch Signaling and Colorectal Cancer. Cells 2020, 9, 1549. [Google Scholar] [CrossRef] [PubMed]
- Veenendaal, L.M.; Kranenburg, O.; Smakman, N.; Klomp, A.; Rinkes, I.H.M.B.; van Diest, P.J. Differential Notch and TGFβ Signaling in Primary Colorectal Tumors and Their Corresponding Metastases. Anal. Cell. Pathol. 2008, 30, 1–11. [Google Scholar] [CrossRef]
- Rajendran, D.; Subramaniyan, B.; Ganeshan, M. Role of Notch Signaling in Colorectal Cancer. In Role of Transcription Factors in Gastrointestinal Malignancies, 5th ed.; Rajendran, D.T., Subramaniyan, B., Ganeshan, M., Eds.; Springer: Singapore, 2017; Volume 1, pp. 307–314. ISBN 978-981-106-727-3. [Google Scholar]
- Fazio, C.; Piazzi, G.; Vitaglione, P.; Fogliano, V.; Munarini, A.; Prossomariti, A.; Milazzo, M.; D’Angelo, L.; Napolitano, M.; Chieco, P.; et al. Inflammation increases NOTCH1 activity via MMP9 and is counteracted by Eicosapentaenoic Acid-free fatty acid in colon cancer cells. Sci. Rep. 2016, 6, 20670. [Google Scholar] [CrossRef] [Green Version]
- Raskov, H.; Burcharth, J.; Pommergaard, H.-C. Linking Gut Microbiota to Colorectal Cancer. J. Cancer 2017, 8, 3378–3395. [Google Scholar] [CrossRef] [PubMed]
- Rokavec, M.; Öner, M.G.; Li, H.; Jackstadt, R.; Jiang, L.; Lodygin, D.; Kaller, M.; Horst, D.; Ziegler, P.K.; Schwitalla, S.; et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J. Clin. Investig. 2014, 124, 1853–1867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bingham, S.A.; Day, N.E.; Luben, R.; Ferrari, P.; Slimani, N.; Norat, T.; Clavel-Chapelon, F.; Kesse, E.; Nieters, A.; Boeing, H.; et al. Dietary fibre in food and protection against colorectal cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC): An observational study. Lancet 2003, 361, 1496–1501. [Google Scholar] [CrossRef]
- Wynder, E.L.; Reddy, B.S. Metabolic epidemiology of colorectal cancer. Cancer 1974, 34, 801–806. [Google Scholar] [CrossRef]
- Li, J.; Zhang, A.-H.; Wu, F.-F.; Wang, X.-J. Alterations in the Gut Microbiota and Their Metabolites in Colorectal Cancer: Recent Progress and Future Prospects. Front. Oncol. 2022, 12, 841552. [Google Scholar] [CrossRef]
- Slattery, M.L.; Boucher, K.; Caan, B.; Potter, J.; Ma, K.-N. Eating Patterns and Risk of Colon Cancer. Am. J. Epidemiol. 1998, 148, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Archambault, A.N.; Jeon, J.; Lin, Y.; Thomas, M.; Harrison, T.A.; Bishop, D.T.; Brenner, H.; Casey, G.; Chan, A.T.; Chang-Claude, J.; et al. Risk Stratification for Early-Onset Colorectal Cancer Using a Combination of Genetic and Environmental Risk Scores: An International Multi-Center Study. JNCI J. Natl. Cancer Inst. 2022, 114, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Mizutani, S.; Shiroma, H.; Shiba, S.; Nakajima, T.; Sakamoto, T.; Watanabe, H.; Masuda, K.; Nishimoto, Y.; Kubo, M.; et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat. Med. 2019, 25, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Ocvirk, S.; O’Keefe, S.J. Influence of Bile Acids on Colorectal Cancer Risk: Potential Mechanisms Mediated by Diet-Gut Microbiota Interactions. Curr. Nutr. Rep. 2017, 6, 315–322. [Google Scholar] [CrossRef]
- Kühn, T.; Stepien, M.; López-Nogueroles, M.; Damms-Machado, A.; Sookthai, D.; Johnson, T.; Roca, M.; Hüsing, A.; Maldonado, S.G.; Cross, A.J.; et al. Prediagnostic Plasma Bile Acid Levels and Colon Cancer Risk: A Prospective Study. JNCI J. Natl. Cancer Inst. 2020, 112, 516–524. [Google Scholar] [CrossRef]
- Perez, M.-J.; Briz, O. Bile-acid-induced cell injury and protection. World J. Gastroenterol. 2009, 15, 1677–1689. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Wang, M.; Gu, W.; Chen, L. Intestine-specific FXR agonists as potential therapeutic agents for colorectal cancer. Biochem. Pharmacol. 2021, 186, 114430. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.W.; Kennaway, E.L.; Kennaway, N.M. Production of Tumours in Mice by Deoxycholic Acid. Nature 1940, 145, 627. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Wang, D.Q.-H.; Molina-Molina, E.; Baccetto, R.L.; Calamita, G.; Palmieri, V.O.; Portincasa, P. Bile Acids and Cancer: Direct and Environmental-Dependent Effects. Ann. Hepatol. 2017, 16, S87–S105. [Google Scholar] [CrossRef]
- Dong, W.; Liu, L.; Dou, Y.; Xu, M.; Liu, T.; Wang, S.; Zhang, Y.; Deng, B.; Wang, B.; Cao, H. Deoxycholic acid activates epidermal growth factor receptor and promotes intestinal carcinogenesis byADAM17-dependent ligand release. J. Cell. Mol. Med. 2018, 22, 4263–4273. [Google Scholar] [CrossRef]
- Zeng, H.; Botnen, J.H.; Briske-Anderson, M. Deoxycholic Acid and Selenium Metabolite Methylselenol Exert Common and Distinct Effects on Cell Cycle, Apoptosis, and MAP Kinase Pathway in HCT116 Human Colon Cancer Cells. Nutr. Cancer 2010, 62, 85–92. [Google Scholar] [CrossRef]
- Xie, G.; Wang, X.; Huang, F.; Zhao, A.; Chen, W.; Yan, J.; Zhang, Y.; Lei, S.; Ge, K.; Zheng, X.; et al. Dysregulated hepatic bile acids collaboratively promote liver carcinogenesis. Int. J. Cancer 2016, 139, 1764–1775. [Google Scholar] [CrossRef]
- Li, S.; Ung, T.T.; Nguyen, T.T.; Sah, D.K.; Park, S.Y.; Jung, Y.D. Cholic Acid Stimulates MMP-9 in Human Colon Cancer Cells via Activation of MAPK, AP-1, and NF-κB Activity. Int. J. Mol. Sci. 2020, 21, 3420. [Google Scholar] [CrossRef]
- Bernstein, C.; Holubec, H.; Bhattacharyya, A.K.; Nguyen, H.; Payne, C.M.; Zaitlin, B.; Bernstein, H. Carcinogenicity of deoxycholate, a secondary bile acid. Arch. Toxicol. 2011, 85, 863–871. [Google Scholar] [CrossRef] [Green Version]
- Ticho, A.; Malhotra, P.; Dudeja, P.K.; Gill, R.K.; Alrefai, W.A. Bile acid receptors and gastrointestinal functions. Liver Res. 2019, 3, 31–39. [Google Scholar] [CrossRef]
- Tang, Q.; Evans, R.M. Colon cancer checks in when bile acids check out: The bile acid–nuclear receptor axis in colon cancer. Essays Biochem. 2021, 65, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.R.; Mangelsdorf, D.J. Nuclear receptors of the enteric tract: Guarding the frontier. Nutr. Rev. 2008, 66, S88–S97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dent, P.; Fang, Y.; Gupta, S.; Studer, E.; Mitchell, C.; Spiegel, S.; Hylemon, P.B. Conjugated bile acids promote ERK1/2 and AKT activation via a pertussis toxin-sensitive mechanism in murine and human hepatocytes. Hepatology 2005, 42, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Degirolamo, C.; Modica, S.; Palasciano, G.; Moschetta, A. Bile acids and colon cancer: Solving the puzzle with nuclear receptors. Trends Mol. Med. 2011, 17, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a Nuclear Receptor for Bile Acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Mangelsdorf, D.J. Bile Acids as Hormones: The FXR-FGF15/19 Pathway. Dig. Dis. 2015, 33, 327–331. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.-J.; Wang, L. Bile acid-activated receptors: A review on FXR and other nuclear receptors. In Bile Acids and Their Receptors. Handbook of Experimental Pharmacology, 1st ed.; Fiorucci, S., Distrutti, E., Eds.; Springer: Cham, Switzerland, 2019; Volume 256, pp. 51–72. ISBN 978-3-030-22004-4. [Google Scholar]
- Wang, L.X.; Frey, M.R.; Kohli, R. The Role of FGF19 and MALRD1 in Enterohepatic Bile Acid Signaling. Front. Endocrinol. 2021, 12, 799648. [Google Scholar] [CrossRef]
- Gadaleta, R.M.; Garcia-Irigoyen, O.; Moschetta, A. Bile acids and colon cancer: Is FXR the solution of the conundrum? Mol. Asp. Med. 2017, 56, 66–74. [Google Scholar] [CrossRef]
- Fu, T.; Coulter, S.; Yoshihara, E.; Oh, T.G.; Fang, S.; Cayabyab, F.; Zhu, Q.; Zhang, T.; Leblanc, M.; Liu, S.; et al. FXR Regulates Intestinal Cancer Stem Cell Proliferation. Cell 2019, 176, 1098–1112.e1018. [Google Scholar] [CrossRef] [Green Version]
- De Gottardi, A.; Touri, F.; Maurer, C.A.; Perez, A.; Maurhofer, O.; Ventre, G.; Bentzen, C.L.; Niesor, E.J.; Dufour, J.-F. The Bile Acid Nuclear Receptor FXR and the Bile Acid Binding Protein IBABP Are Differently Expressed in Colon Cancer. Am. J. Dig. Dis. 2004, 49, 982–989. [Google Scholar] [CrossRef]
- Bailey, A.M.; Zhan, L.; Maru, D.; Shureiqi, I.; Pickering, C.; Kiriakova, G.; Izzo, J.; He, N.; Wei, C.; Baladandayuthapani, V.; et al. FXR silencing in human colon cancer by DNA methylation and KRAS signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G48–G58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Li, S.; Guo, J.; Xu, Z.; Zheng, J.; Sun, X. Farnesoid X receptor antagonizes Wnt/β-catenin signaling in colorectal tumorigenesis. Cell Death Dis. 2020, 11, 640. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xu, Z.; Guo, J.; Zheng, J.; Sun, X.; Yu, J. Farnesoid X receptor activation induces antitumour activity in colorectal cancer by suppressing JAK2/STAT3 signalling via transactivation of SOCS3 gene. J. Cell. Mol. Med. 2020, 24, 14549–14560. [Google Scholar] [CrossRef] [PubMed]
- Maran, R.R.; Thomas, A.; Roth, M.; Sheng, Z.; Esterly, N.; Pinson, D.; Gao, X.; Zhang, Y.; Ganapathy, V.; Gonzalez, F.J.; et al. Farnesoid X Receptor Deficiency in Mice Leads to Increased Intestinal Epithelial Cell Proliferation and Tumor Development. J. Pharmacol. Exp. Ther. 2009, 328, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Farhana, L.; Nangia-Makker, P.; Arbit, E.; Shango, K.; Sarkar, S.; Mahmud, H.; Hadden, T.; Yu, Y.; Majumdar, A.P.N. Bile acid: A potential inducer of colon cancer stem cells. Stem Cell Res. Ther. 2016, 7, 181. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.; Chen, X.; Wang, C.; Li, W.; Li, J. Effects and mechanism of the bile acid (farnesoid X) receptor on the Wnt/β-catenin signaling pathway in colon cancer. Oncol. Lett. 2020, 20, 337–345. [Google Scholar] [CrossRef] [Green Version]
- Modica, S.; Murzilli, S.; Salvatore, L.; Schmidt, D.R.; Moschetta, A. Nuclear Bile Acid Receptor FXR Protects against Intestinal Tumorigenesis. Cancer Res. 2008, 68, 9589–9594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lax, S.; Schauer, G.; Prein, K.; Kapitan, M.; Silbert, D.; Berghold, A.; Berger, A.; Trauner, M. Expression of the nuclear bile acid receptor/farnesoid X receptor is reduced in human colon carcinoma compared to nonneoplastic mucosa independent from site and may be associated with adverse prognosis. Int. J. Cancer 2012, 130, 2232–2239. [Google Scholar] [CrossRef]
- Selmin, O.I.; Fang, C.; Lyon, A.M.; Doetschman, T.; Thompson, P.A.; Martinez, J.D.; Smith, J.W.; Lance, P.M.; Romagnolo, D.F. Inactivation of Adenomatous Polyposis Coli Reduces Bile Acid/Farnesoid X Receptor Expression through Fxr gene CpG Methylation in Mouse Colon Tumors and Human Colon Cancer Cells. J. Nutr. 2016, 146, 236–242. [Google Scholar] [CrossRef]
- Gadaleta, R.M.; Oldenburg, B.; Willemsen, E.C.; Spit, M.; Murzilli, S.; Salvatore, L.; Klomp, L.W.; Siersema, P.D.; van Erpecum, K.J.; van Mil, S.W. Activation of bile salt nuclear receptor FXR is repressed by pro-inflammatory cytokines activating NF-κB signaling in the intestine. Biochim. Biophys. Acta Mol. Basis Dis. 2011, 1812, 851–858. [Google Scholar] [CrossRef]
- Zhao, D.; Cai, C.; Chen, Q.; Jin, S.; Yang, B.; Li, N. High-Fat Diet Promotes DSS-Induced Ulcerative Colitis by Downregulated FXR Expression through the TGFB Pathway. BioMed Res. Int. 2020, 2020, 3516128. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular Basis of Colorectal Cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136.e123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girisa, S.; Henamayee, S.; Parama, D.; Rana, V.; Dutta, U.; Kunnumakkara, A.B. Targeting Farnesoid X receptor (FXR) for developing novel therapeutics against cancer. Mol. Biomed. 2021, 2, 21. [Google Scholar] [CrossRef]
- Cao, L.; Che, Y.; Meng, T.; Deng, S.; Zhang, J.; Zhao, M.; Xu, W.; Wang, D.; Pu, Z.; Wang, G.; et al. Repression of intestinal transporters and FXR-FGF15 signaling explains bile acids dysregulation in experimental colitis-associated colon cancer. Oncotarget 2017, 8, 63665–63679. [Google Scholar] [CrossRef] [Green Version]
- Peng, Z.; Chen, J.; Drachenberg, C.B.; Raufman, J.-P.; Xie, G. Farnesoid X receptor represses matrix metalloproteinase 7 expression, revealing this regulatory axis as a promising therapeutic target in colon cancer. J. Biol. Chem. 2019, 294, 8529–8542. [Google Scholar] [CrossRef]
- Inagaki, T.; Moschetta, A.; Lee, Y.-K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.J.; et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, H.; Bernstein, C.; Payne, C.M.; Dvorak, K. Bile acids as endogenous etiologic agents in gastrointestinal cancer. World J. Gastroenterol. 2009, 15, 3329–3340. [Google Scholar] [CrossRef]
- Makishima, M.; Lu, T.T.; Xie, W.; Whitfield, G.K.; Domoto, H.; Evans, R.M.; Haussler, M.R.; Mangelsdorf, D.J. Vitamin D Receptor As an Intestinal Bile Acid Sensor. Science 2002, 296, 1313–1316. [Google Scholar] [CrossRef] [Green Version]
- Dou, R.; Ng, K.; Giovannucci, E.L.; Manson, J.E.; Qian, Z.R.; Ogino, S. Vitamin D and colorectal cancer: Molecular, epidemiological and clinical evidence. Br. J. Nutr. 2016, 115, 1643–1660. [Google Scholar] [CrossRef] [Green Version]
- Carlberg, C.; Muñoz, A. An update on vitamin D signaling and cancer. Semin. Cancer Biol. 2022, 79, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Pálmer, H.G.; Larriba, M.J.; García, J.M.; Ordóñez-Morán, P.; Peña, C.; Peiró, S.; Puig, I.; Rodríguez, R.; De La Fuente, R.; Bernad, A.; et al. The transcription factor SNAIL represses vitamin D receptor expression and responsiveness in human colon cancer. Nat. Med. 2004, 10, 917–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larriba, M.J.; Bonilla, F.; Muñoz, A. The transcription factors Snail1 and Snail2 repress vitamin D receptor during colon cancer progression. J. Steroid Biochem. Mol. Biol. 2010, 121, 106–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larriba, M.J.; Munoz, A. SNAIL vs vitamin D receptor expression in colon cancer: Therapeutics implications. Br. J. Cancer 2005, 92, 985–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urdaneta, V.; Casadesús, J. Interactions between Bacteria and Bile Salts in the Gastrointestinal and Hepatobiliary Tracts. Front. Med. 2017, 4, 163. [Google Scholar] [CrossRef]
- Ferrer-Mayorga, G.; López, G.G.; Barbáchano, A.; Fernández-Barral, A.; Peña, C.; Pisano, D.G.; Cantero, R.; Rojo, F.; Muñoz, A.; Larriba, M.J. Vitamin D receptor expression and associated gene signature in tumour stromal fibroblasts predict clinical outcome in colorectal cancer. Gut 2017, 66, 1449–1462. [Google Scholar] [CrossRef]
- Xu, C.; Li, C.Y.-T.; Kong, A.-N.T. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch. Pharmacal Res. 2005, 28, 249–268. [Google Scholar] [CrossRef]
- Scripture, C.D.; Sparreboom, A.; Figg, W.D. Modulation of cytochrome P450 activity: Implications for cancer therapy. Lancet Oncol. 2005, 6, 780–789. [Google Scholar] [CrossRef]
- Wu, S.; Zhang, Y.; Lu, R.; Xia, Y.; Zhou, D.; Sun, J. Intestinal vitamin D receptor protects mice from dysbiosis via modulating JAK/STAT pathway in tumorigenesis. FASEB J. 2015, 29, 999.4. [Google Scholar] [CrossRef]
- He, Y.; The SUNLIGHT Consortium; Timofeeva, M.; Farrington, S.M.; Vaughan-Shaw, P.; Svinti, V.; Walker, M.; Zgaga, L.; Meng, X.; Li, X.; et al. Exploring causality in the association between circulating 25-hydroxyvitamin D and colorectal cancer risk: A large Mendelian randomisation study. BMC Med. 2018, 16, 142. [Google Scholar] [CrossRef]
- Irving, A.A.; Halberg, R.B.; Albrecht, D.M.; Plum, L.A.; Krentz, K.J.; Clipson, L.; Drinkwater, N.; Amos-Landgraf, J.M.; Dove, W.F.; DeLuca, H.F. Supplementation by vitamin D compounds does not affect colonic tumor development in vitamin D sufficient murine models. Arch. Biochem. Biophys. 2011, 515, 64–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irving, A.A.; Plum, L.A.; Blaser, W.J.; Ford, M.R.; Weng, C.; Clipson, L.; DeLuca, H.F.; Dove, W.F. Cholecalciferol or 25-hydroxycholecalciferol neither prevents nor treats adenomas in a rat model of familial colon cancer. J. Nutr. 2015, 145, 291–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irving, A.A.; Waters, B.J.; Seeman, J.R.; Plum, L.A.; DeLuca, H.F. Vitamin D receptor absence does not enhance intestinal tumorigenesis in Apc Pirc/+ rats. Biol. Open 2022, 11, bio059290. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Yan, J.; Niu, Y. PXR: A center of transcriptional regulation in cancer. Acta Pharm. Sin. B 2020, 10, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Synold, T.; Dussault, I.; Forman, B.M. The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat. Med. 2001, 7, 584–590. [Google Scholar] [CrossRef]
- Basseville, A.; Preisser, L.; Trécesson, S.D.C.; Boisdron-Celle, M.; Gamelin, E.; Coqueret, O.; Morel, A. Irinotecan induces steroid and xenobiotic receptor (SXR) signaling to detoxification pathway in colon cancer cells. Mol. Cancer 2011, 10, 80. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Verma, S.; Blumberg, B. The steroid and xenobiotic receptor (SXR), beyond xenobiotic metabolism. Nucl. Recept. Signal. 2009, 7, e001. [Google Scholar] [CrossRef] [Green Version]
- Planque, C.; Rajabi, F.; Grillet, F.; Finetti, P.; Bertucci, F.; Gironella, M.; Lozano, J.J.; Beucher, B.; Giraud, J.; Garambois, V.; et al. Pregnane X-receptor promotes stem cell-mediated colon cancer relapse. Oncotarget 2016, 7, 56558–56573. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Liu, M.; Zhai, Y.; Xie, W. The Antiapoptotic Role of Pregnane X Receptor in Human Colon Cancer Cells. Mol. Endocrinol. 2008, 22, 868–880. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, N.; Ke, S.; Eagleton, N.; Xie, Y.; Chen, G.; Laffins, B.; Yao, H.; Zhou, B.; Tian, Y. Pregnane X receptor suppresses proliferation and tumourigenicity of colon cancer cells. Br. J. Cancer 2010, 102, 1753–1761. [Google Scholar] [CrossRef]
- Guo, C.; Chen, W.-D.; Wang, Y.-D. TGR5, Not Only a Metabolic Regulator. Front. Physiol. 2016, 7, 646. [Google Scholar] [CrossRef] [PubMed]
- Duboc, H.; Taché, Y.; Hofmann, A.F. The bile acid TGR5 membrane receptor: From basic research to clinical application. Dig. Liver Dis. 2014, 46, 302–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepanov, V.; Stankov, K.; Mikov, M. The bile acid membrane receptor TGR5: A novel pharmacological target in metabolic, inflammatory and neoplastic disorders. J. Recept. Signal Transduct. Res. 2013, 33, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Miyamoto, Y.; Nakamura, T.; Tamai, Y.; Okada, H.; Sugiyama, E.; Nakamura, T.; Itadani, H.; Tanaka, K. Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 2002, 298, 714–719. [Google Scholar] [CrossRef]
- van Nierop, F.S.; Scheltema, M.J.; Eggink, H.M.; Pols, T.W.; Sonne, D.P.; Knop, F.K.; Soeters, M.R. Clinical relevance of the bile acid receptor TGR5 in metabolism. Lancet Diabetes Endocrinol. 2017, 5, 224–233. [Google Scholar] [CrossRef]
- Cipriani, S.; Mencarelli, A.; Chini, M.G.; Distrutti, E.; Renga, B.; Bifulco, G.; Baldelli, F.; Donini, A.; Fiorucci, S. The Bile Acid Receptor GPBAR-1 (TGR5) Modulates Integrity of Intestinal Barrier and Immune Response to Experimental Colitis. PLoS ONE 2011, 6, e25637. [Google Scholar] [CrossRef]
- Wang, Y.-D.; Chen, W.-D.; Yu, D.; Forman, B.M.; Huang, W. The G-Protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light-chain enhancer of activated B cells (NF-κB) in mice. Hepatology 2011, 54, 1421–1432. [Google Scholar] [CrossRef] [Green Version]
- Pols, T.W.; Nomura, M.; Harach, T.; Sasso, G.L.; Oosterveer, M.H.; Thomas, C.; Rizzo, G.; Gioiello, A.; Adorini, L.; Pellicciari, R.; et al. TGR5 Activation Inhibits Atherosclerosis by Reducing Macrophage Inflammation and Lipid Loading. Cell Metab. 2011, 14, 747–757. [Google Scholar] [CrossRef] [Green Version]
- Režen, T.; Rozman, D.; Kovács, T.; Kovács, P.; Sipos, A.; Bai, P.; Mikó, E. The role of bile acids in carcinogenesis. Cell. Mol. Life Sci. 2022, 79, 243. [Google Scholar] [CrossRef]
- Ward, J.B.J.; Lajczak, N.K.; Kelly, O.B.; O’Dwyer, A.M.; Giddam, A.K.; Gabhann, J.N.; Franco, P.; Tambuwala, M.M.; Jefferies, C.A.; Keely, S.; et al. Ursodeoxycholic acid and lithocholic acid exert anti-inflammatory actions in the colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G550–G558. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Qi, H.; Yu, Y.; Zhang, Q.; Su, J.; Yu, D.; Huang, W.; Chen, W.-D.; Wang, Y.-D. The G-Protein-Coupled Bile Acid Receptor Gpbar1 (TGR5) Inhibits Gastric Inflammation Through Antagonizing NF-κB Signaling Pathway. Front. Pharmacol. 2015, 6, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.-D.; Yu, N.; Forman, B.M.; Huang, W.; Wang, Y.-D. Deficiency of G-protein-coupled bile acid receptor Gpbar1 (TGR5) enhances chemically induced liver carcinogenesis. Hepatology 2012, 57, 656–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalikonda, G.; Lee, H.; Sheik, A.; Huh, Y.S. Targeting key transcriptional factor STAT3 in colorectal cancer. Mol. Cell. Biochem. 2021, 476, 3219–3228. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xu, H.; Zhang, C.; Tang, Q.; Bi, F. Ursodeoxycholic acid suppresses the malignant progression of colorectal cancer through TGR5-YAP axis. Cell Death Discov. 2021, 7, 207. [Google Scholar] [CrossRef]
- Zhou, H.; Chen, T.; Huang, Z.; Liu, R.; Yang, J.; Hylemon, P.B. Sphingosine-1 phosphate promotes intestinal epithelial cell proliferation via S1PR2. Front. Biosci. 2017, 22, 596–608. [Google Scholar] [CrossRef]
- Petti, L.; Rizzo, G.; Rubbino, F.; Elangovan, S.; Colombo, P.; Restelli, S.; Piontini, A.; Arena, V.; Carvello, M.; Romano, B.; et al. Unveiling role of sphingosine-1-phosphate receptor 2 as a brake of epithelial stem cell proliferation and a tumor suppressor in colorectal cancer. J. Exp. Clin. Cancer Res. 2020, 39, 253. [Google Scholar] [CrossRef]
- Zhao, S.; Gong, Z.; Du, X.; Tian, C.; Wang, L.; Zhou, J.; Xu, C.; Chen, Y.; Cai, W.; Wu, J. Deoxycholic Acid-Mediated Sphingosine-1-Phosphate Receptor 2 Signaling Exacerbates DSS-Induced Colitis through Promoting Cathepsin B Release. J. Immunol. Res. 2018, 2018, 2481418. [Google Scholar] [CrossRef] [Green Version]
- Shida, D.; Inoue, S.; Yoshida, Y.; Kodaka, A.; Tsuji, T.; Tsuiji, M. Sphingosine kinase 1 is upregulated with lysophosphatidic acid receptor 2 in human colorectal cancer. World J. Gastroenterol. 2016, 22, 2503–2511. [Google Scholar] [CrossRef]
- Uranbileg, B.; Nishikawa, T.; Ikeda, H.; Kurano, M.; Sato, M.; Saigusa, D.; Aoki, J.; Watanabe, T.; Yatomi, Y. Evidence Suggests Sphingosine 1-Phosphate Might Be Actively Generated, Degraded, and Transported to Extracellular Spaces with Increased S1P2 and S1P3 Expression in Colon Cancer. Clin. Color. Cancer 2018, 17, e171–e182. [Google Scholar] [CrossRef]
- Fiorucci, S.; Distrutti, E. The Pharmacology of Bile Acids and Their Receptors. In Handbook of Experimental Pharmacology, 1st ed.; Fiorucci, S., Distrutti, E., Eds.; Springer: Cham, Switzerland, 2019; Volume 256, pp. 3–18. ISBN 978-3-030-22004-4. [Google Scholar]
- Fiorucci, S.; Carino, A.; Baldoni, M.; Santucci, L.; Costanzi, E.; Graziosi, L.; Distrutti, E.; Biagioli, M. Bile acid signaling in inflammatory bowel diseases. Dig. Dis. Sci. 2021, 66, 674–693. [Google Scholar] [CrossRef]
- Nagahashi, M.; Yuza, K.; Hirose, Y.; Nakajima, M.; Ramanathan, R.; Hait, N.C.; Hylemon, P.B.; Zhou, H.; Takabe, K.; Wakai, T. The roles of bile acids and sphingosine-1-phosphate signaling in the hepatobiliary diseases. J. Lipid Res. 2016, 57, 1636–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duineveld, L.A.M.; Van Asselt, K.M.; Bemelman, W.A.; Smits, A.B.; Tanis, P.; Van Weert, H.C.P.M.; Wind, J. Symptomatic and Asymptomatic Colon Cancer Recurrence: A Multicenter Cohort Study. Ann. Fam. Med. 2016, 14, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
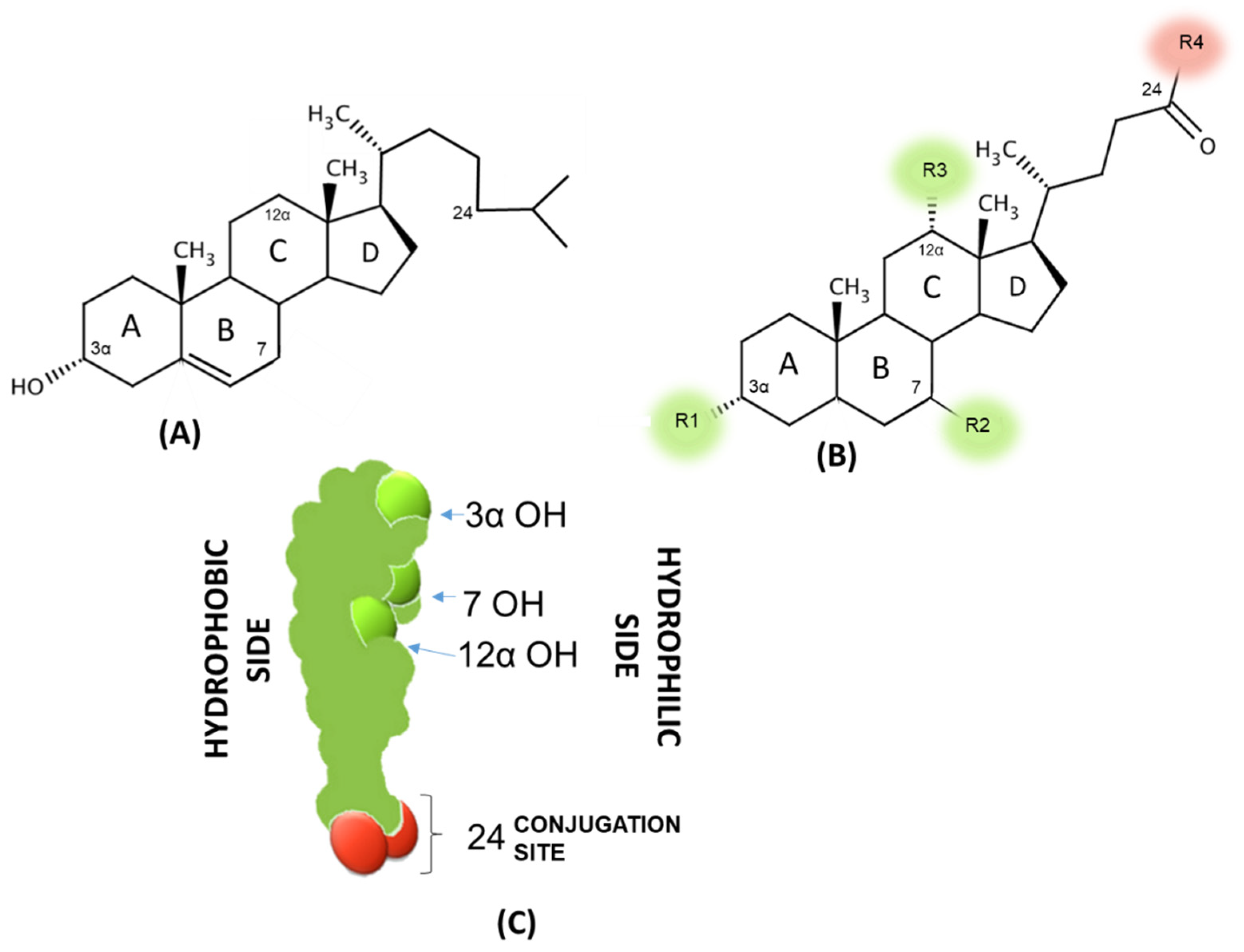
| R1 | R2 | R3 | R4 | LogPA- | CMC (mM in 0.15 M Na+) | |
|---|---|---|---|---|---|---|
| CA | OH | OH (α) | OH | OH | 1.1 | 9 |
| CDCA | OH | OH (α) | H | OH | 2.3 | 3.2 |
| UDCA | OH | OH (β) | H | OH | 2.2 | 10 |
| DCA | OH | H (α) | OH | OH | 2.7 | 3 |
| LCA | OH | H (α) | H | OH | 5.5 (est) | -* |
| G-CA | OH | OH (α) | OH | Glycine | −0.4 | 8 |
| T-CA | OH | OH (α) | OH | Taurine | −0.5 | 4 |
| G-CDCA | OH | OH (α) | H | Glycine | 0.5 | 2 |
| T-CDCA | OH | OH (α) | H | Taurine | 0.9 | 3 |
| G-UDCA | OH | OH (β) | H | Glycine | 0.2 | 4 |
| T-UDCA | OH | OH (β) | H | Taurine | 1.1 | 6 |
| G-DCA | OH | H (α) | OH | Glycine | 0.8 | 2 |
| T-DCA | OH | H (α) | OH | Taurine | 0.7 | 2.5 |
| G-LCA | OH | H (α) | H | Glycine | 3.5 est) | -* |
| T-LCA | OH | H (α) | H | Taurine | 3 (est) | -* |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caliceti, C.; Punzo, A.; Silla, A.; Simoni, P.; Roda, G.; Hrelia, S. New Insights into Bile Acids Related Signaling Pathways in the Onset of Colorectal Cancer. Nutrients 2022, 14, 2964. https://doi.org/10.3390/nu14142964
Caliceti C, Punzo A, Silla A, Simoni P, Roda G, Hrelia S. New Insights into Bile Acids Related Signaling Pathways in the Onset of Colorectal Cancer. Nutrients. 2022; 14(14):2964. https://doi.org/10.3390/nu14142964
Chicago/Turabian StyleCaliceti, Cristiana, Angela Punzo, Alessia Silla, Patrizia Simoni, Giulia Roda, and Silvana Hrelia. 2022. "New Insights into Bile Acids Related Signaling Pathways in the Onset of Colorectal Cancer" Nutrients 14, no. 14: 2964. https://doi.org/10.3390/nu14142964
APA StyleCaliceti, C., Punzo, A., Silla, A., Simoni, P., Roda, G., & Hrelia, S. (2022). New Insights into Bile Acids Related Signaling Pathways in the Onset of Colorectal Cancer. Nutrients, 14(14), 2964. https://doi.org/10.3390/nu14142964