

Disruption of Glutamate Homeostasis in the Brain of Rat Offspring Induced by Prenatal and Early Postnatal Exposure to Maternal High-Sugar Diet
 , , and
, , and
Abstract
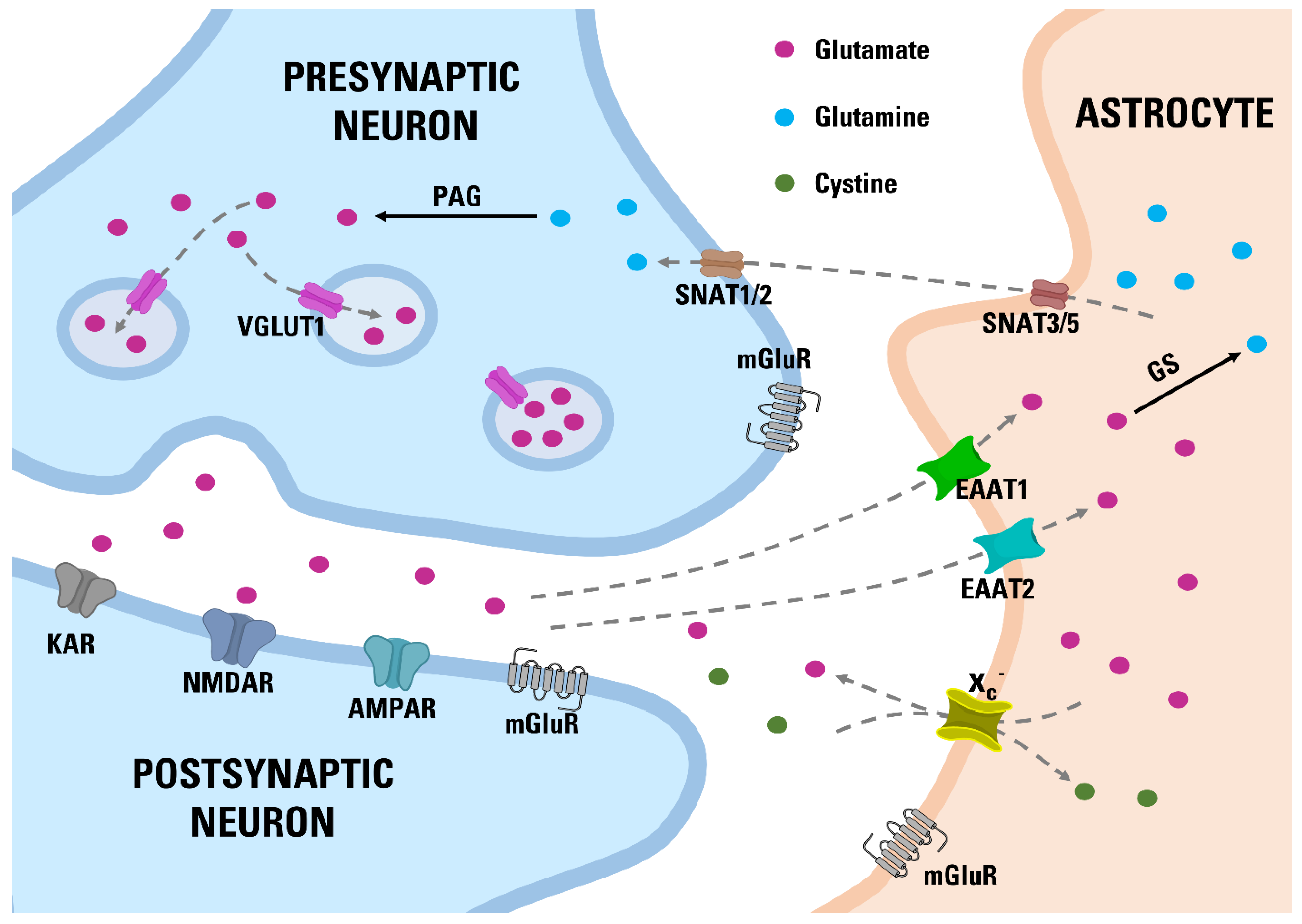
:1. Introduction
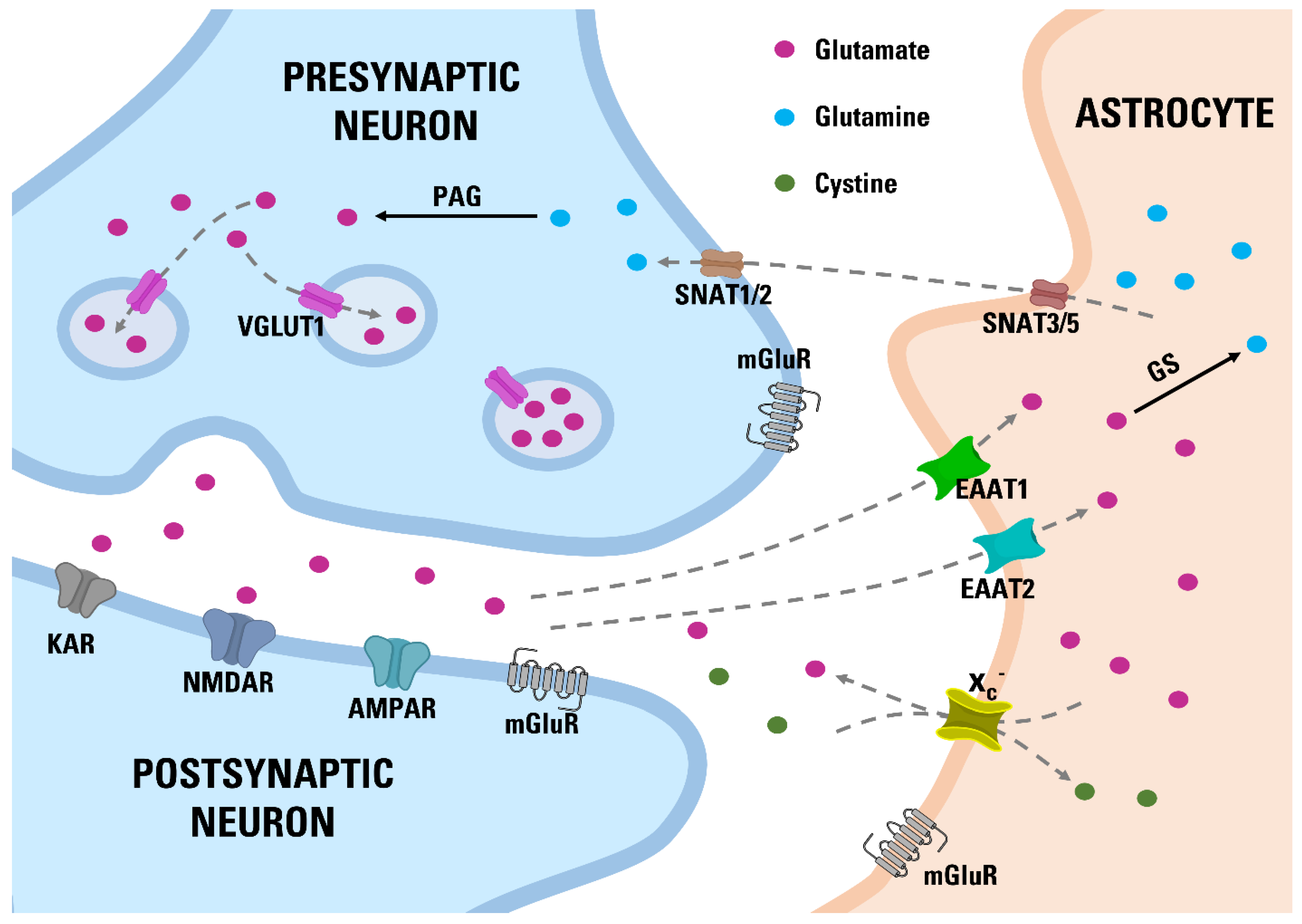
2. Materials and Methods
2.1. Animals
2.2. Diets
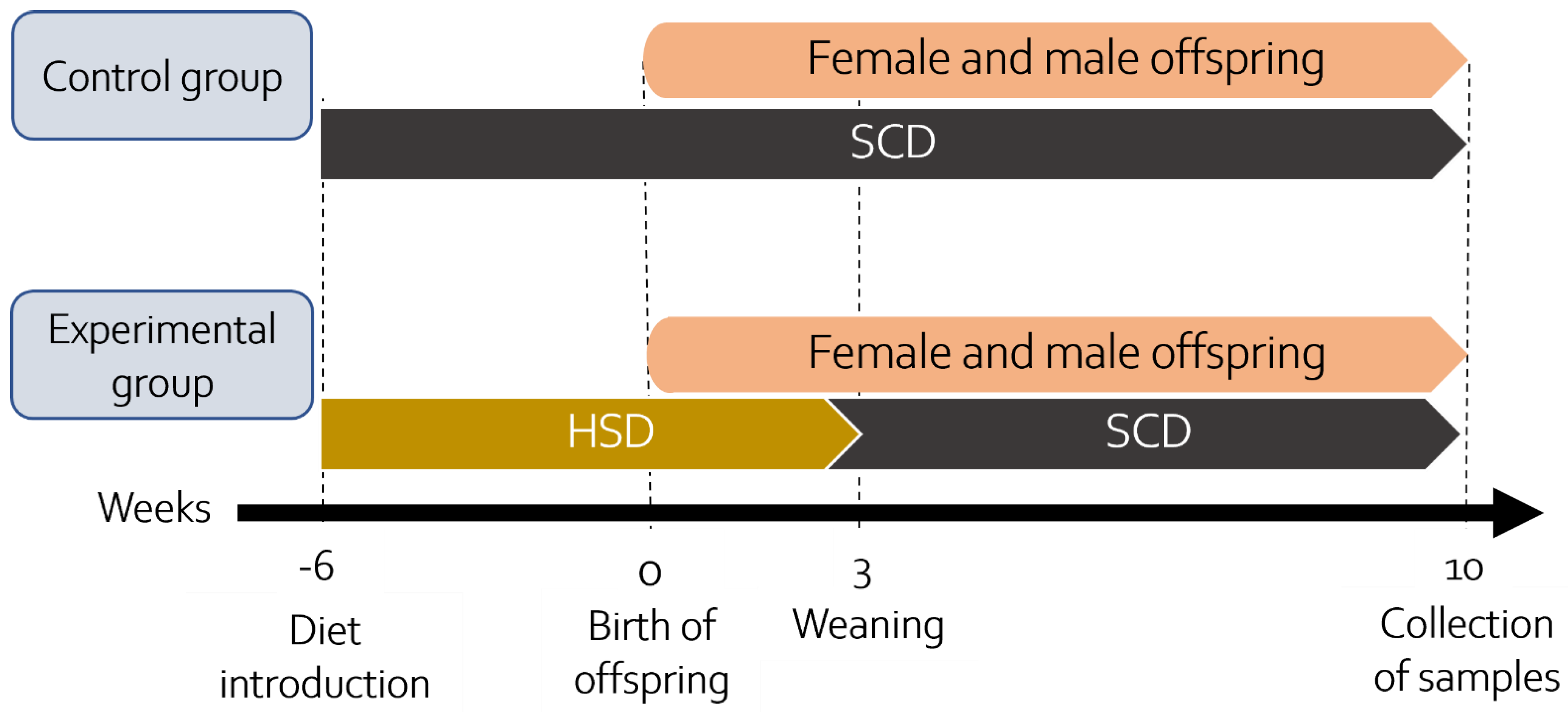
2.3. Experimental Design
2.4. Tissue Sampling
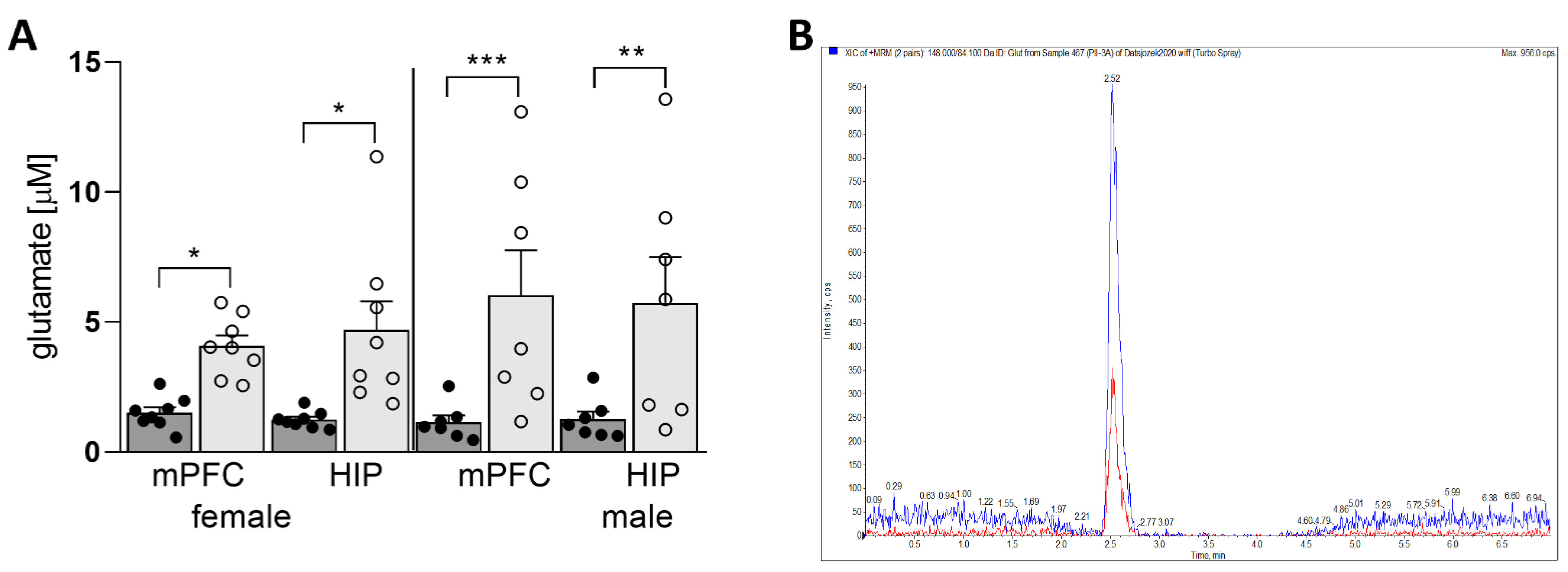
2.5. Microdialysis
2.6. LC/MS/MS
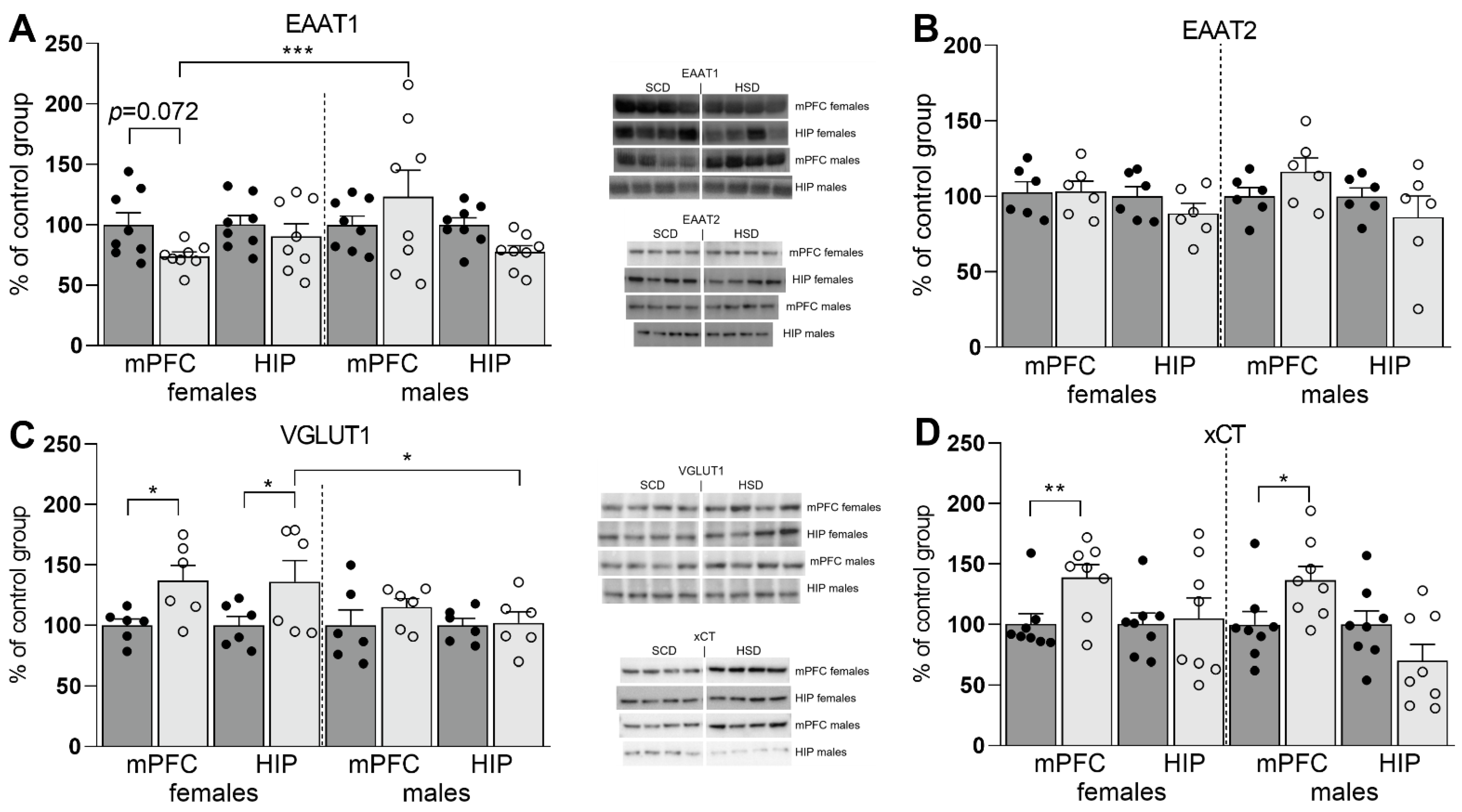
2.7. Western Blot
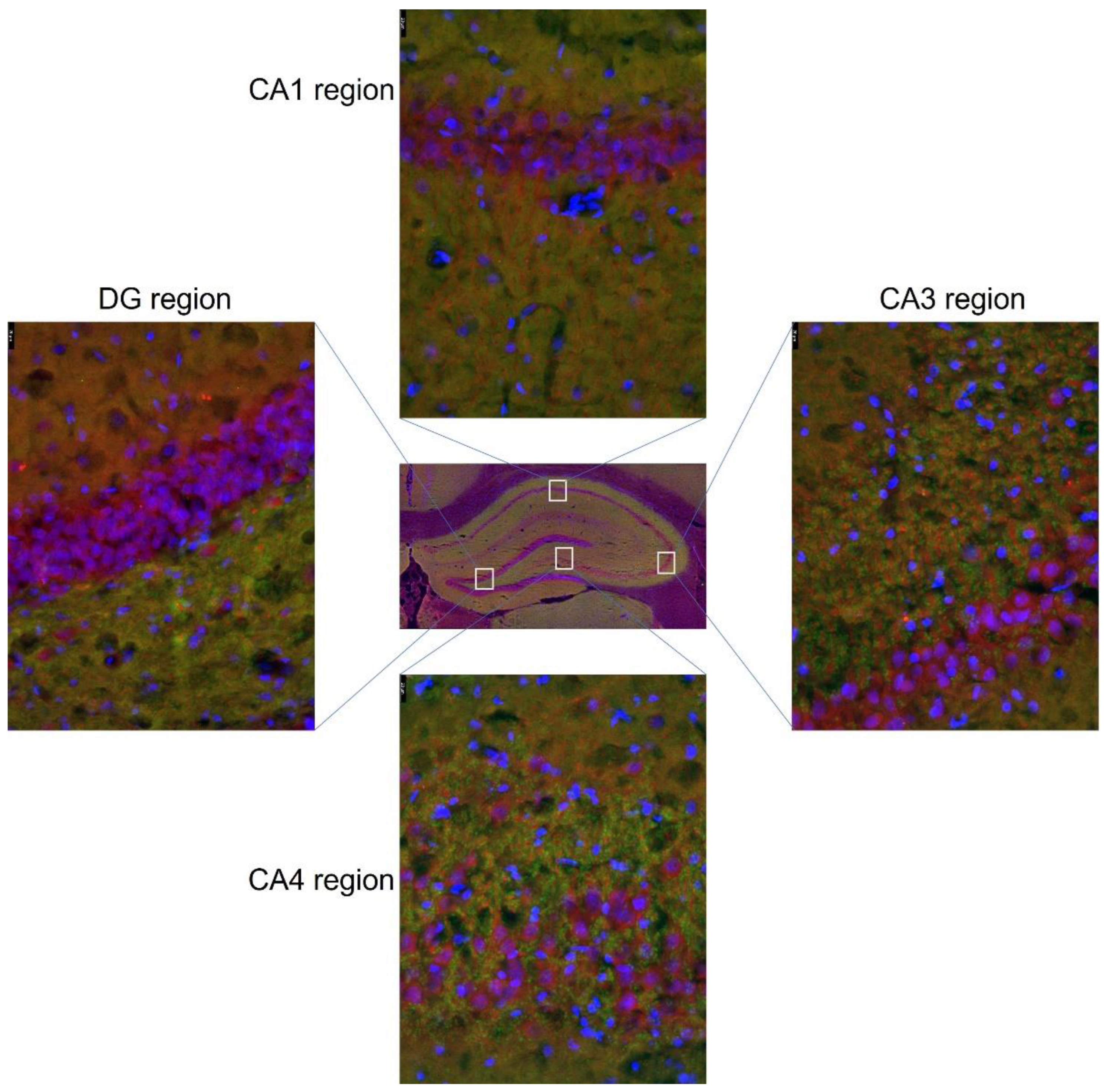
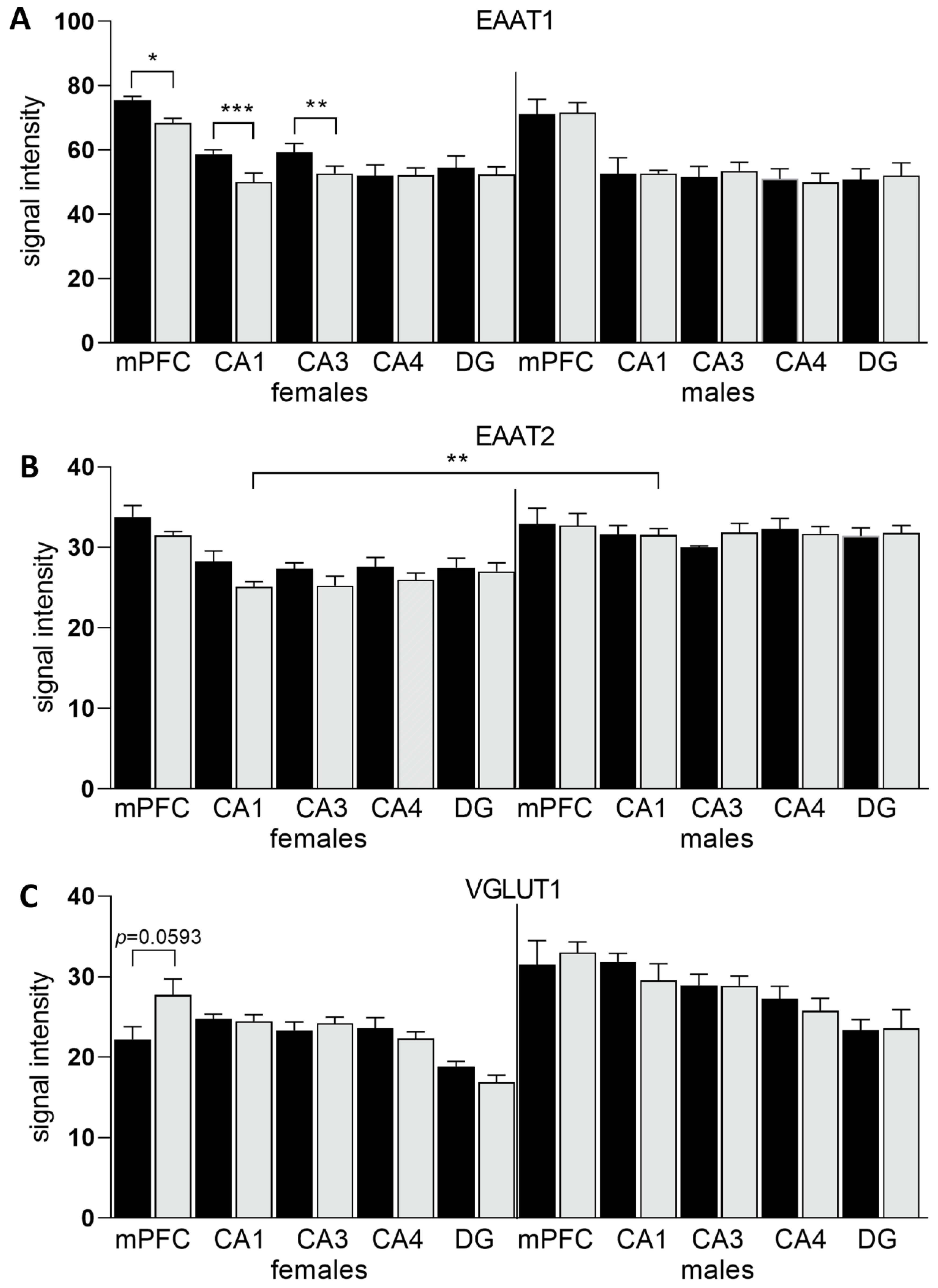
2.8. Immunofluorescence Assays
2.9. Statistical Analysis
3. Results
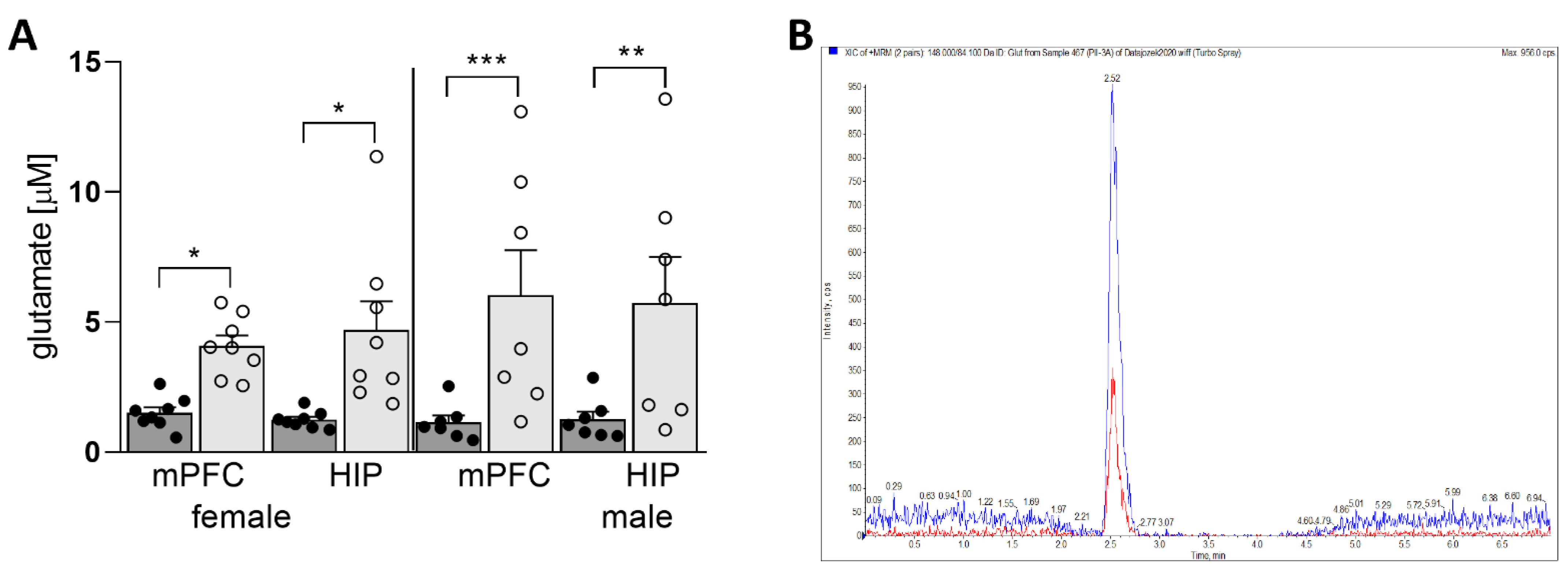
3.1. Maternal HSD Increases Extracellular Glu Level in Both Female and Male Offspring
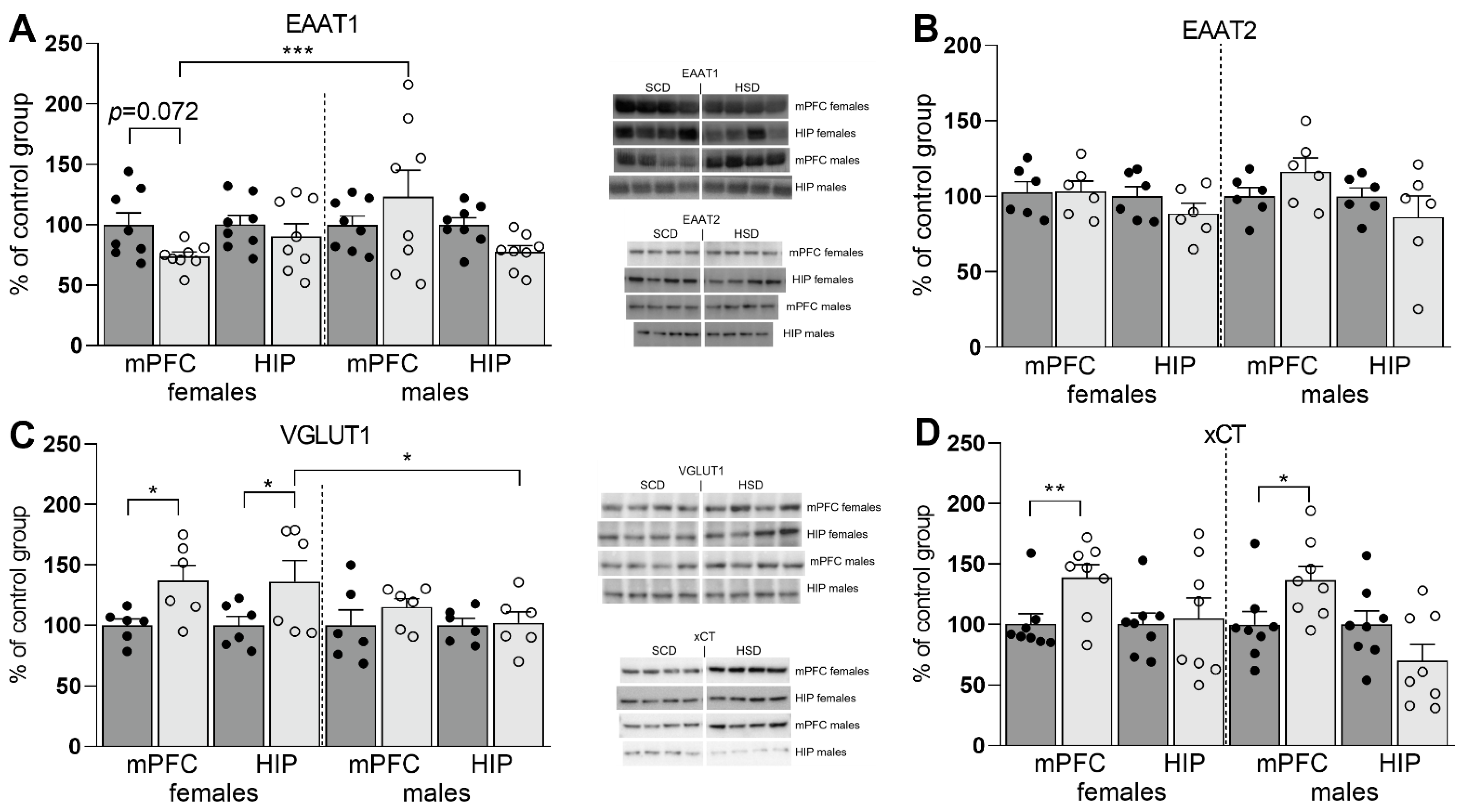
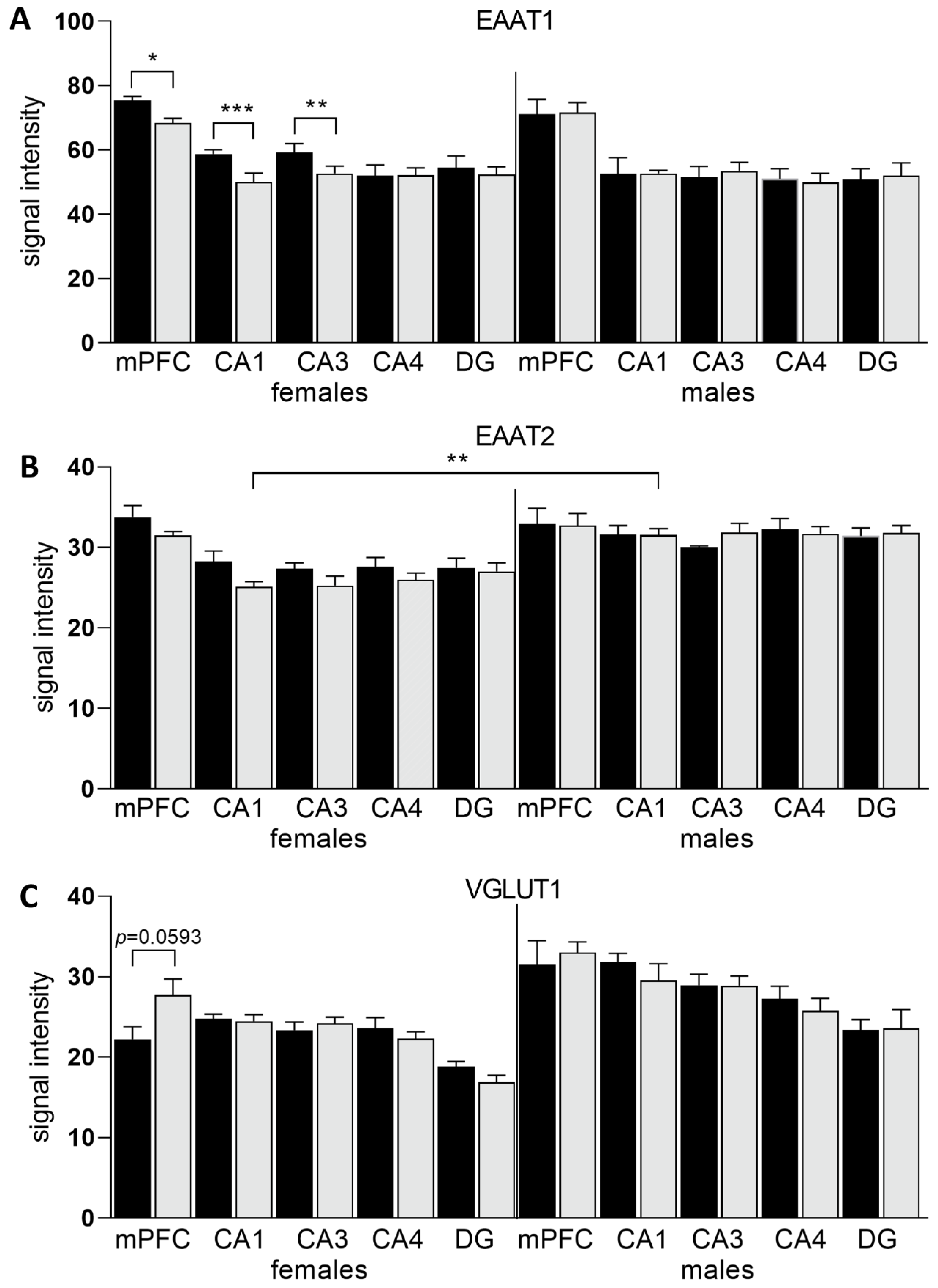
3.2. Expression of Glu Transporter Proteins (Western Blot)
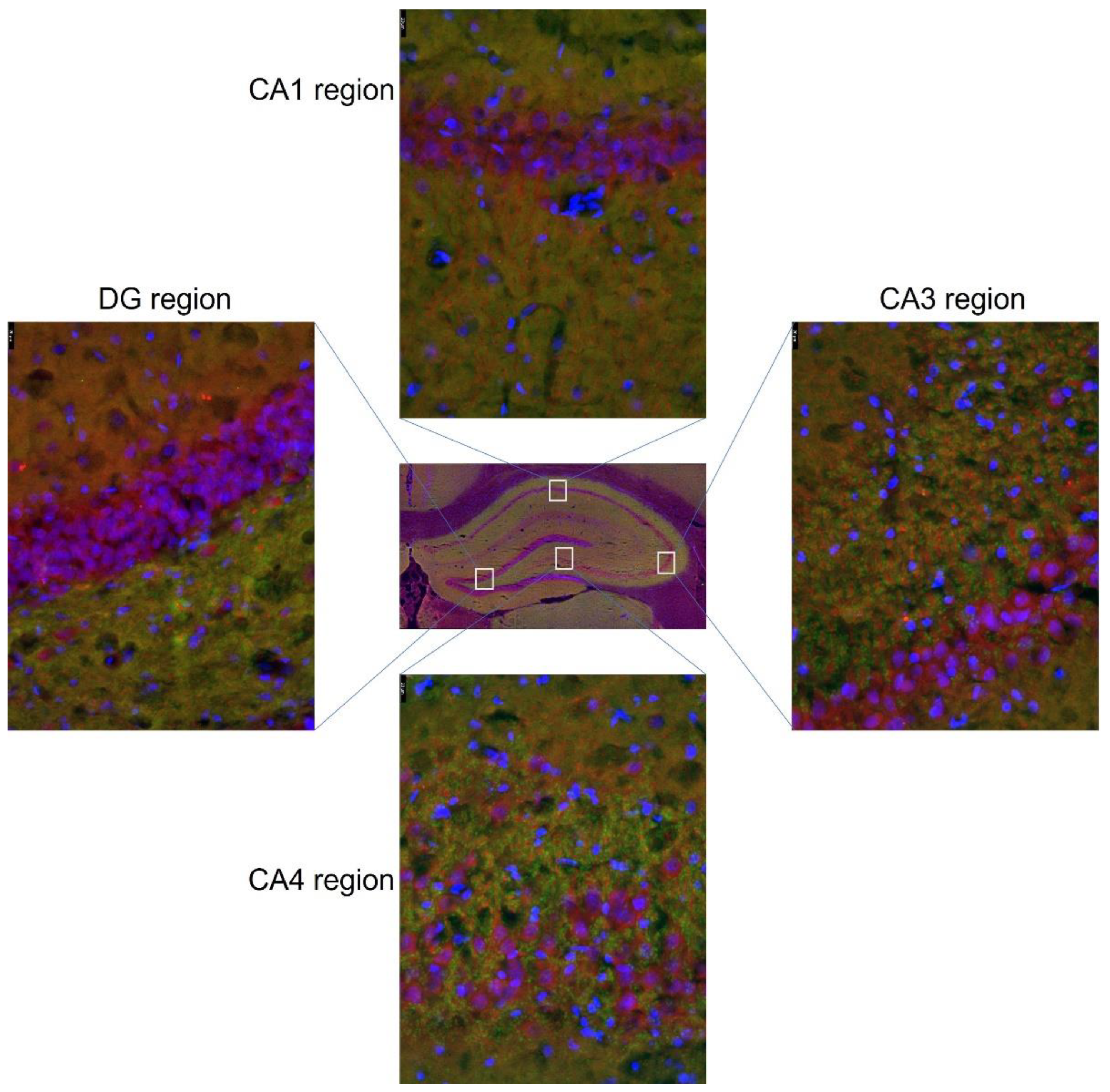
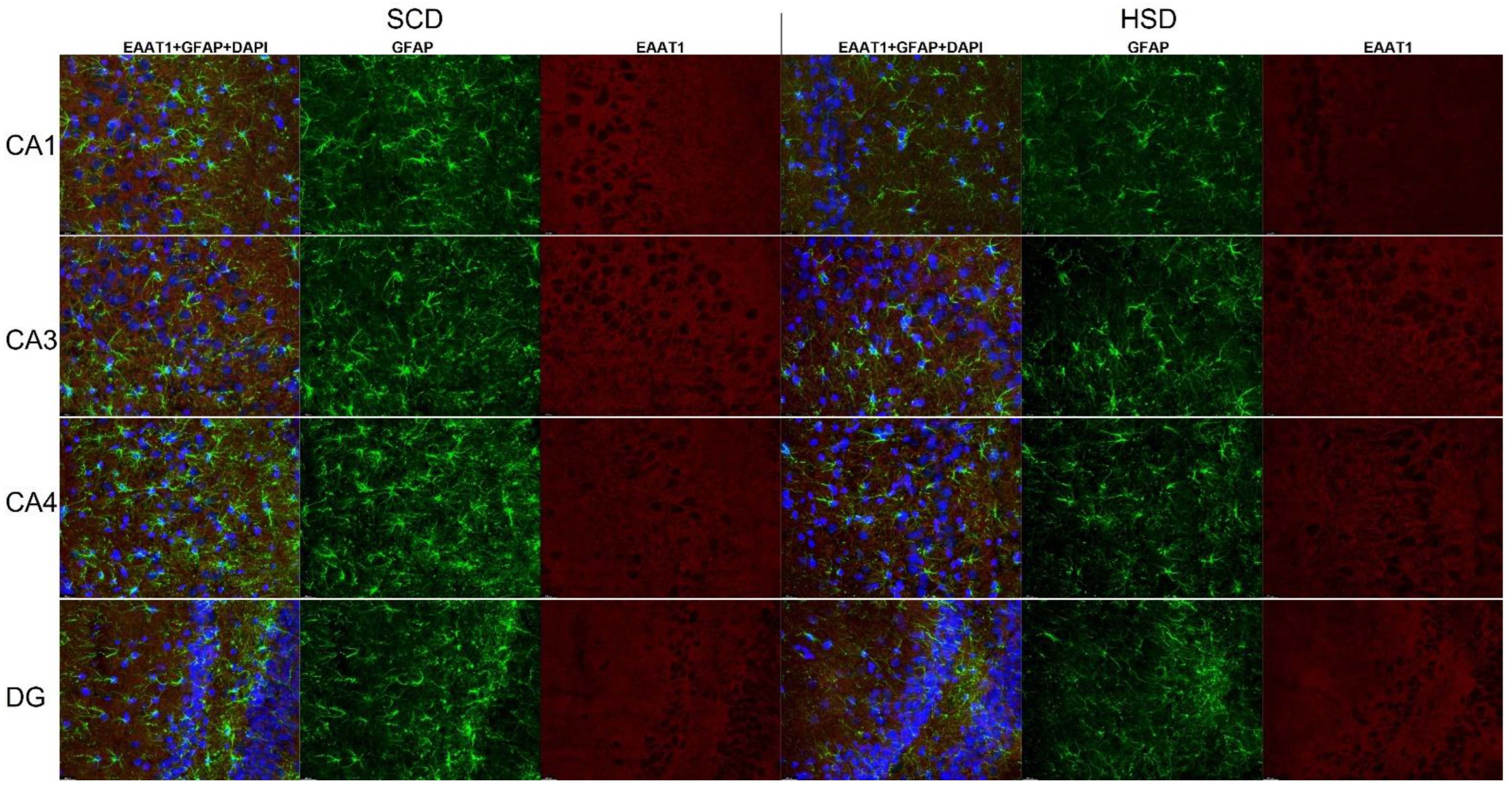
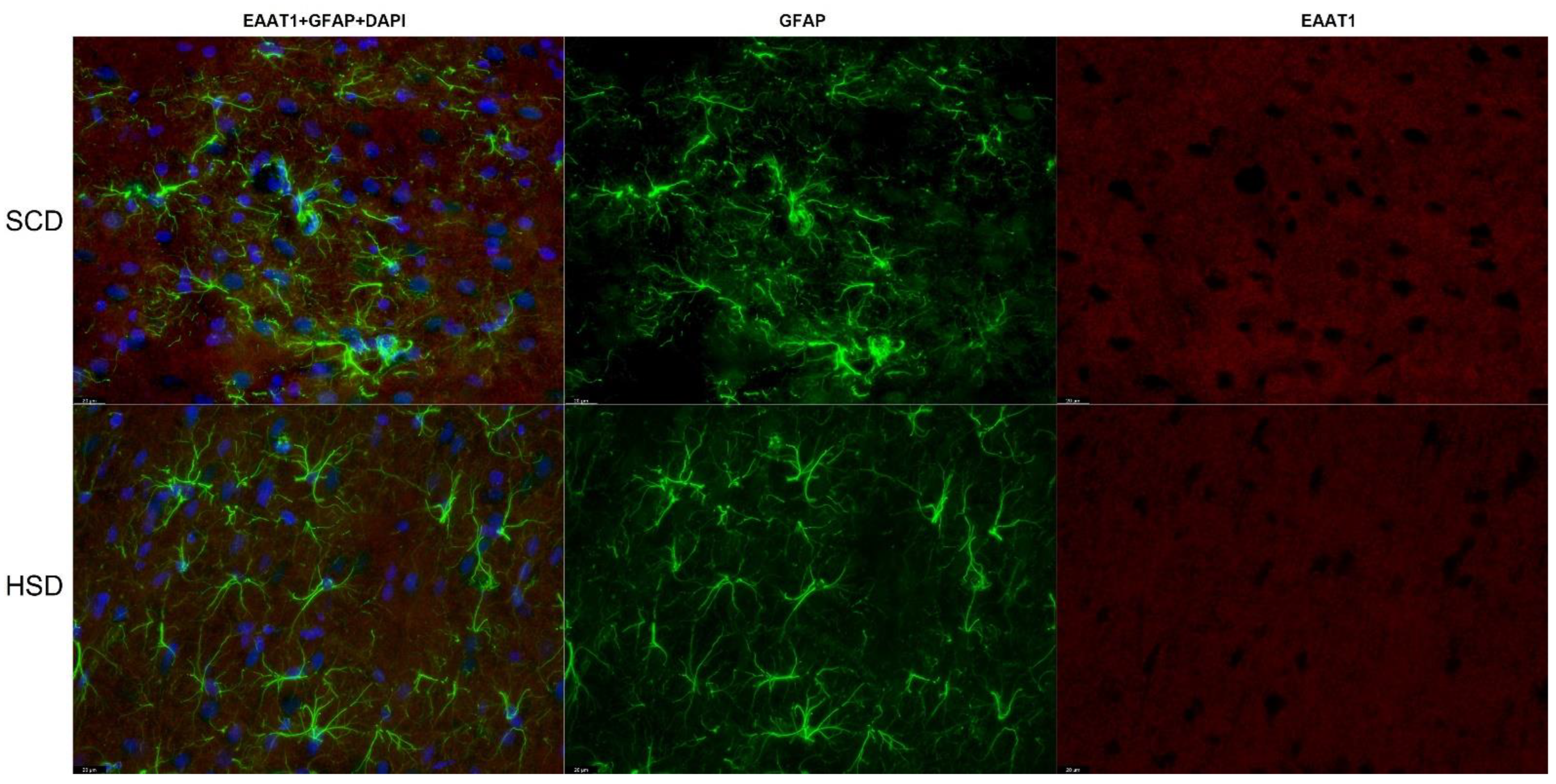
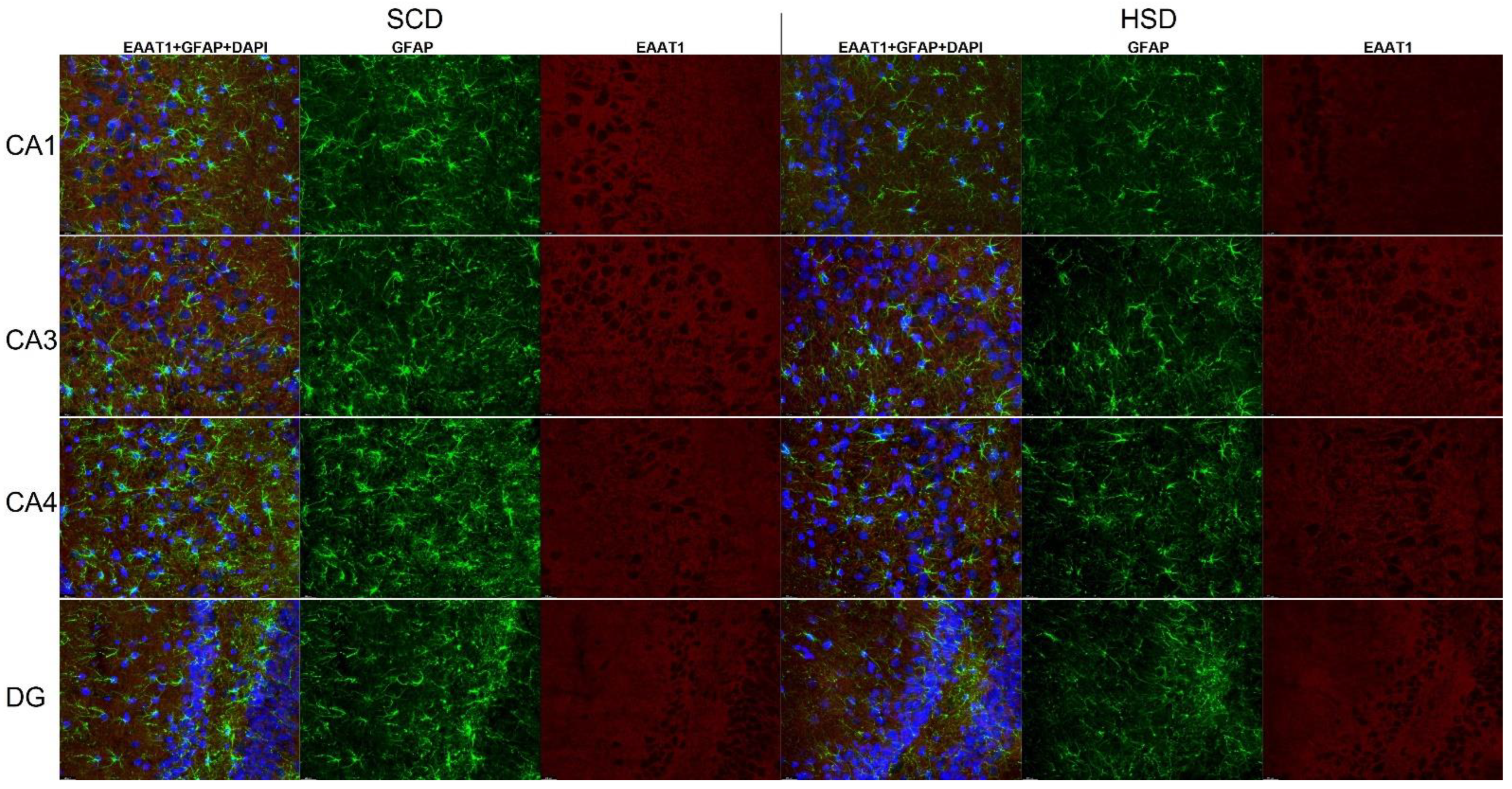
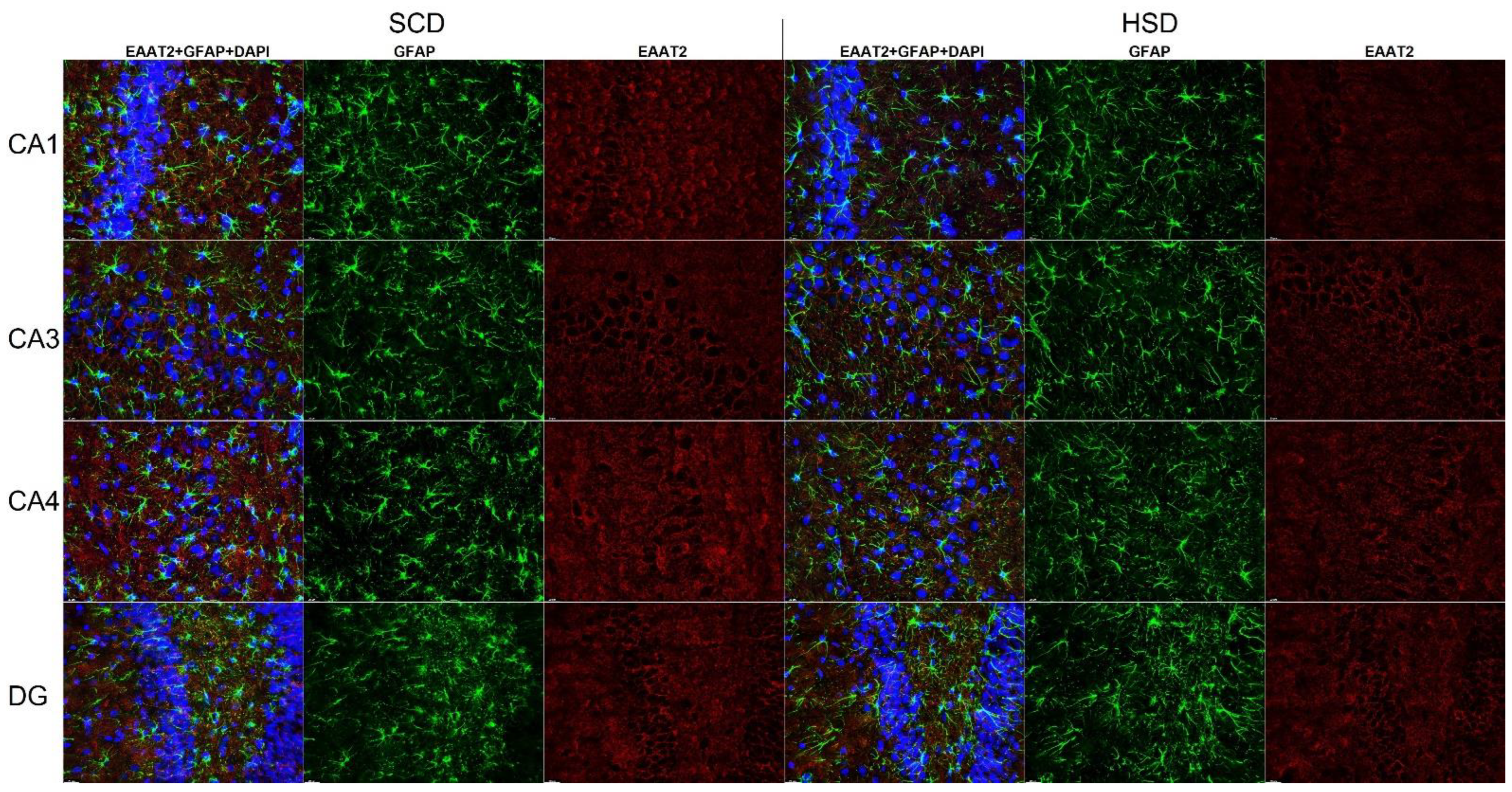
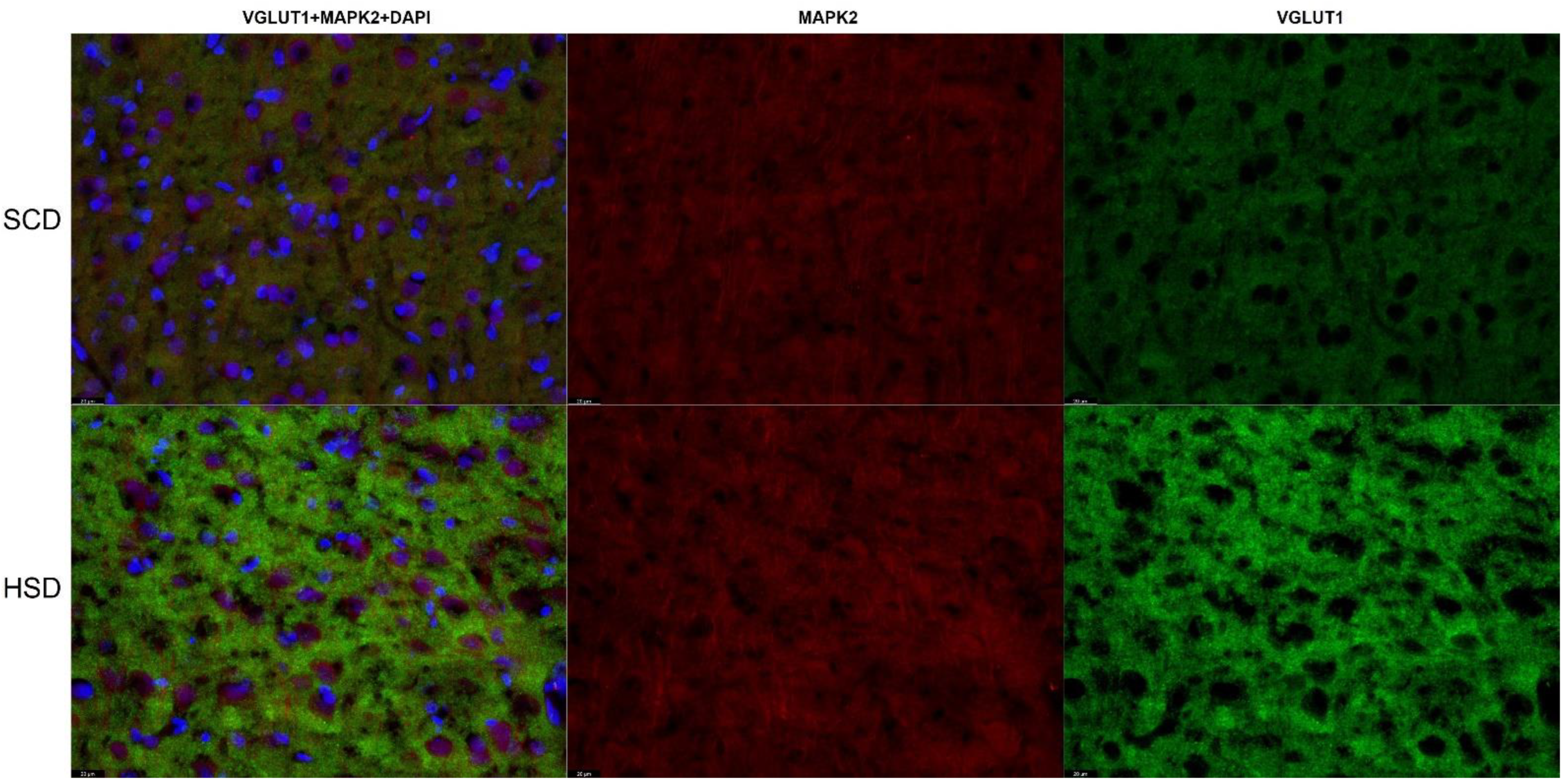
3.3. Expression of Glu Transporter Proteins (Immunofluorescence Staining)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, C.; Xu, X.; Yan, Y. Estimated global overweight and obesity burden in pregnant women based on panel data model. PLoS ONE 2018, 13, e0202183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffey, K.J.; Popkin, B.M. Shifts in patterns and consumption of beverages between 1965 and 2002. Obesity 2007, 15, 2739–2747. [Google Scholar] [CrossRef] [PubMed]
- Spagnuolo, M.S.; Iossa, S.; Cigliano, L. Sweet but bitter: Focus on fructose impact on brain function in rodent models. Nutrients 2021, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Mandy, M.; Nyirenda, M. Developmental Origins of Health and Disease: The relevance to developing nations. Int. Health 2018, 10, 66–70. [Google Scholar] [CrossRef] [Green Version]
- Goyal, D.; Limesand, S.W.; Goyal, R. Epigenetic responses and the developmental origins of health and disease. J. Endocrinol. 2019, 242, T105–T119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godfrey, K.M.; Reynolds, R.; Prescott, S.L.; Nyirenda, M.; Jaddoe, V.W.V.; Eriksson, J.G.; Broekman, B.F.P. Broekman, Influence of maternal obesity on the long-term health of offspring. Lancet Diabetes Endocrinol. 2017, 5, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Mina, T.H.; Lahti, M.; Drake, A.J.; Räikkönen, K.; Minnis, H.; Denison, F.C.; Norman, J.E.; Reynolds, R.M. Prenatal exposure to very severe maternal obesity is associated with adverse neuropsychiatric outcomes in children. Psychol. Med. 2017, 47, 353–362. [Google Scholar] [CrossRef]
- Sullivan, E.L.; Nousen, E.K.; Chamlou, K.A. Maternal high fat diet consumption during the perinatal period programs offspring behavior. Physiol. Behav. 2014, 123, 236–242. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, A. Maternal pre-pregnancy obesity and risk for inattention and negative emotionality in children. J. Child Psychol. Psychiatry Allied Discip. 2010, 51, 134–143. [Google Scholar] [CrossRef]
- Sarker, G.; Litwan, K.; Kastli, R.; Peleg-Raibstein, D. Maternal overnutrition during critical developmental periods leads to different health adversities in the offspring: Relevance of obesity, addiction and schizophrenia. Sci. Rep. 2019, 9, 17322. [Google Scholar] [CrossRef] [Green Version]
- Tozuka, Y.; Wada, E.; Wada, K. Diet-induced obesity in female mice leads to peroxidized lipid accumulations and impairment of hippocampal neurogenesis during the early life of their offspring. FASEB J. 2009, 23, 1920–1934. [Google Scholar] [CrossRef]
- Mendes-da-Silva, C.; Lemes, S.F.; da Silva Baliani, T.; Versutti, M.D.; Torsoni, M.A. Increased expression of Hes5 protein in Notch signaling pathway in the hippocampus of mice offspring of dams fed a high-fat diet during pregnancy and suckling. Int. J. Dev. Neurosci. 2015, 40, 35–42. [Google Scholar] [CrossRef]
- Tozuka, Y.; Kumon, M.; Wada, E.; Onodera, M.; Mochizuki, H.; Wada, K. Maternal obesity impairs hippocampal BDNF production and spatial learning performance in young mouse offspring. Neurochem. Int. 2010, 57, 235–247. [Google Scholar] [CrossRef]
- Winther, G.; Elfving, B.; Müller, H.K.; Lund, S.; Wegener, G. Maternal High-fat Diet Programs Offspring Emotional Behavior in Adulthood. Neuroscience 2018, 388, 87–101. [Google Scholar] [CrossRef]
- Sullivan, E.; Grayson, B.; Takahashi, D.; Robertson, N.; Maier, A.; Bethea, C.L.; Smith, M.S.; Coleman, K.; Grove, K.L. Chronic consumption of a high-fat diet during pregnancy causes perturbations in the serotonergic system and increased anxiety-like behavior in nonhuman primate offspring. J. Neurosci. 2010, 30, 3826–3830. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.; Valleau, J.C.; Barling, A.N.; Franco, J.G.; Decapo, M.; Bagley, J.L.; Sullivan, E.L. Exposure to a high-fat diet during early development programs behavior and impairs the central serotonergic system in juvenile non-human primates. Front. Endocrinol. 2017, 8, 21. [Google Scholar] [CrossRef] [Green Version]
- Rivera, H.M.; Kievit, P.; Kirigiti, M.A.; Bauman, L.A.; Baquero, K.; Blundell, P.; Dean, T.A.; Valleau, J.C.; Takahashi, D.L.; Frazee, T.; et al. Maternal high-fat diet and obesity impact palatable food intake and dopamine signaling in nonhuman primate offspring. In Obesity; Blackwell Publishing Inc.: Malden, MA, USA, 2015; pp. 2157–2164. [Google Scholar] [CrossRef] [Green Version]
- Pilc, A.; Chaki, S.; Nowak, G.; Witkin, J.M. Mood disorders: Regulation by metabotropic glutamate receptors. Biochem. Pharmacol. 2008, 75, 997–1006. [Google Scholar] [CrossRef]
- Pomierny-Chamioło, L.; Rup, K.; Pomierny, B.; Niedzielska, E.; Kalivas, P.W.; Filip, M. Metabotropic glutamatergic receptors and their ligands in drug addiction. Pharmacol. Ther. 2014, 142, 281–305. [Google Scholar] [CrossRef]
- Mizera, J.; Kazek, G.; Niedzielska-Andres, E.; Pomierny-Chamiolo, L. Maternal high-sugar diet results in NMDA receptors abnormalities and cognitive impairment in rat offspring. FASEB J. 2021, 35, e21547. [Google Scholar] [CrossRef]
- Zhou, Y.; Danbolt, N.C. Glutamate as a neurotransmitter in the healthy brain. J. Neural Transm. 2014, 121, 799–817. [Google Scholar] [CrossRef] [Green Version]
- Murrough, J.W.; Abdallah, C.; Mathew, S.J. Targeting glutamate signalling in depression: Progress and prospects. Nat. Rev. Drug Discov. 2017, 16, 472–486. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.R.; Willnow, T.E. Excitatory amino acid transporters in physiology and disorders of the central nervous system. Int. J. Mol. Sci. 2019, 20, 5671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fremeau, R.T.; Burman, J.; Qureshi, T.; Tran, C.H.; Proctor, J.; Johnson, J.; Zhang, H.; Sulzer, D.; Copenhagen, D.R.; Storm-Mathisen, J.; et al. The identification of vesicular glutamate transporter 3 suggests novel modes of signaling by glutamate. Proc. Natl. Acad. Sci. USA 2002, 99, 14488–14493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Li, J.; Li, M.; Yang, X.; Qi, Z.; Xu, B.; Liu, W.; Xu, Z.; Deng, Y. Research progress on the role of type I vesicular glutamate transporter (VGLUT1) in nervous system diseases. Cell Biosci. 2020, 10, 26. [Google Scholar] [CrossRef] [Green Version]
- Kashani, A.; Betancur, C.; Giros, B.; Hirsch, E.; El Mestikawy, S. Altered expression of vesicular glutamate transporters VGLUT1 and VGLUT2 in Parkinson disease. Neurobiol. Aging 2007, 28, 568–578. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.-R.; Yang, Y.; Zhou, W.-X.; Zhang, Y.-X. Expression of VGLUTs contributes to degeneration and acquisition of learning and memory. Neurobiol. Learn. Mem. 2011, 95, 361–375. [Google Scholar] [CrossRef]
- Balschun, D.; Moechars, D.; Callaerts-Vegh, Z.; Vermaercke, B.; Van Acker, N.; Andries, L.; D’Hooge, R. Vesicular glutamate transporter VGLUT1 has a role in hippocampal long-term potentiation and spatial reversal learning. Cereb. Cortex 2010, 20, 684–693. [Google Scholar] [CrossRef] [Green Version]
- Kirvell, S.L.; Esiri, M.; Francis, P.T. Down-regulation of vesicular glutamate transporters precedes cell loss and pathology in Alzheimer’s disease. J. Neurochem. 2006, 98, 939–950. [Google Scholar] [CrossRef]
- Tordera, R.M.; Totterdell, S.; Wojcik, S.M.; Brose, N.; Elizalde, N.; Lasheras, B.; Del Rio, J. Enhanced anxiety, depressive-like behaviour and impaired recognition memory in mice with reduced expression of the vesicular glutamate transporter 1 (VGLUT1). Eur. J. Neurosci. 2007, 25, 281–290. [Google Scholar] [CrossRef] [Green Version]
- McKenna, M.C.; Ferreira, G.C. Ferreira, Enzyme Complexes Important for the Glutamate-Glutamine Cycle. Adv. Neurobiol. 2016, 13, 59–98. [Google Scholar] [CrossRef]
- Lander, S.S.; Chornyy, S.; Safory, H.; Gross, A.; Wolosker, H.; Gaisler-Salomon, I.; Sima, L.S.; Sergiy, C.; Hazem, S.; Amit, G.; et al. Glutamate dehydrogenase deficiency disrupts glutamate homeostasis in hippocampus and prefrontal cortex and impairs recognition memory. Genes. Brain Behav. 2020, 19, e12636. [Google Scholar] [CrossRef]
- Ottestad-Hansen, S.; Hu, Q.X.; Follin-Arbelet, V.V.; Bentea, E.; Sato, H.; Massie, A.; Zhou, Y.; Danbolt, N.C. The cystine-glutamate exchanger (xCT, Slc7a11) is expressed in significant concentrations in a subpopulation of astrocytes in the mouse brain. Glia 2018, 66, 951–970. [Google Scholar] [CrossRef] [Green Version]
- Fournier, M.; Monin, A.; Ferrari, C.; Baumann, P.S.; Conus, P.; Do, K. Implication of the glutamate-cystine antiporter xCT in schizophrenia cases linked to impaired GSH synthesis. NPJ Schizophr. 2017, 3, 31. [Google Scholar] [CrossRef]
- Massie, A.; Boillée, S.; Hewett, S.; Knackstedt, L.; Lewerenz, J. Main path and byways: Non-vesicular glutamate release by system Xc− as an important modifier of glutamatergic neurotransmission. J. Neurochem. 2015, 135, 1062–1079. [Google Scholar] [CrossRef] [Green Version]
- Shih, A.Y.; Erb, H.; Sun, X.; Toda, S.; Kalivas, P.W.; Murphy, T.H. Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J. Neurosci. 2006, 26, 10514–10523. [Google Scholar] [CrossRef]
- Baker, D.A.; Xi, Z.-X.; Shen, H.; Swanson, C.J.; Kalivas, P.W. The origin and neuronal function of in vivo nonsynaptic glutamate. J. Neurosci. 2002, 22, 9134–9141. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.; McFarland, K.; Lake, R.W.; Shen, H.; Tang, X.-C.; Toda, S.; Kalivas, P.W. Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat. Neurosci. 2003, 6, 743–749. [Google Scholar] [CrossRef]
- De Groot, J.; Sontheimer, H. Glutamate and the biology of gliomas. Glia 2011, 59, 1181–1189. [Google Scholar] [CrossRef] [Green Version]
- Augustin, H.; Grosjean, Y.; Chen, K.; Sheng, Q.; Featherstone, D.E. Nonvesicular release of glutamate by glial xCT transporters suppresses glutamate receptor clustering in vivo. J. Neurosci. 2007, 27, 111–123. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, P. The Laboratory Rat: Relating Its Age with Human’s. Int. J. Prev. Med. 2013, 4, 624–630. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3733029/ (accessed on 6 May 2022).
- Sit, D. Women and bipolar disorder across the life span. J. Am. Med. Womens. Assoc. 2004, 59, 91–100. [Google Scholar]
- Roca, M.; Vives, M.; López-Navarro, E.; García-Campayo, J.; Gili, M. Cognitive impairments and depression: A critical review. Actas Esp. Psiquiatr. 2015, 43, 187–193. [Google Scholar] [PubMed]
- Chan, V. Schizophrenia and Psychosis: Diagnosis, Current Research Trends, and Model Treatment Approaches with Implications for Transitional Age Youth. Child Adolesc. Psychiatr. Clin. N. Am. 2017, 26, 341–366. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Li, Q.; DU, H.-Z.; Shen, C.-X.; Li, A.-P.; Pei, X.-P.; DU, C.-H.; Qin, X.-M. Determination of five neurotransmitters in the rat brain for the study of the hypnotic effects of Ziziphi Spinosae Semen aqueous extract on insomnia rat model by UPLC-MS/MS. Chin. J. Nat. Med. 2019, 17, 551–560. [Google Scholar] [CrossRef]
- Mesci, P.; Zaïdi, S.; Lobsiger, C.S.; Millecamps, S.; Escartin, C.; Seilhean, D.; Sato, H.; Mallat, M.; Boillée, S. System Xc− is a mediator of microglial function and its deletion slows symptoms in amyotrophic lateral sclerosis mice. Brain 2015, 138, 53–68. [Google Scholar] [CrossRef] [Green Version]
- Van Liefferinge, J.; Bentea, E.; Demuyser, T.; Albertini, G.; Follin-Arbelet, V.; Holmseth, S.; Merckx, E.; Sato, H.; Aerts, J.L.; Smolders, I.; et al. Comparative analysis of antibodies to xCT (Slc7a11): Forewarned is forearmed. J. Comp. Neurol. 2016, 524, 1015–1032. [Google Scholar] [CrossRef]
- Hinzman, J.M.; Wilson, J.A.; Mazzeo, A.T.; Bullock, M.R.; Hartings, J. Excitotoxicity and Metabolic Crisis Are Associated with Spreading Depolarizations in Severe Traumatic Brain Injury Patients. J. Neurotrauma 2016, 33, 1775–1783. [Google Scholar] [CrossRef]
- Bridges, R.; Lutgen, V.; Lobner, D.; Baker, D.A. Thinking outside the cleft to understand synaptic activity: Contribution of the cystine-glutamate antiporter (System Xc−) to normal and pathological glutamatergic signaling. Pharmacol. Rev. 2012, 64, 780–802. [Google Scholar] [CrossRef] [Green Version]
- Rivera, P.; Tovar, R.; Ramírez-López, M.T.; Navarro, J.A.; Vargas, A.; Suárez, J.; de Fonseca, F.R. Sex-Specific Anxiety and Prefrontal Cortex Glutamatergic Dysregulation Are Long-Term Consequences of Pre- and Postnatal Exposure to Hypercaloric Diet in a Rat Model. Nutrients 2020, 12, 1829. [Google Scholar] [CrossRef]
- Hascup, E.; Broderick, S.O.; Russell, M.K.; Fang, Y.; Bartke, A.; Boger, H.A.; Hascup, K.N. Diet-induced insulin resistance elevates hippocampal glutamate as well as VGLUT1 and GFAP expression in AβPP/PS1 mice. J. Neurochem. 2019, 148, 219–237. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Perdigon, M.; Tordera, R.M.; Gil-Bea, F.J.; Gerenu, G.; Ramirez, M.J.; Solas, M. Down-regulation of glutamatergic terminals (VGLUT1) driven by Aβ in Alzheimer’s disease. Hippocampus 2016, 26, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Minelli, A.; Edwards, R.H.; Manzoni, T.; Conti, F. Postnatal development of the glutamate vesicular transporter VGLUT1 in rat cerebral cortex. Brain Res. Dev. 2003, 140, 309–314. [Google Scholar] [CrossRef]
- Sousa, F.J.; Correia, R.G.; Cruz, A.F.; Martins, J.M.; Rodrigues, M.S.; Gomes, C.A.; Ambrósio, A.F.; Baptista, F.I. Sex differences in offspring neurodevelopment, cognitive performance and microglia morphology associated with maternal diabetes: Putative targets for insulin therapy. Brain Behav. Immun. Health 2020, 5, 100075. [Google Scholar] [CrossRef] [PubMed]
- Lewerenz, J.; Hewett, S.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system Xc− in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef] [Green Version]
- Batandier, C.; Poyot, T.; Marissal-Arvy, N.; Couturier, K.; Canini, F.; Roussel, A.; Hininger-Favier, I. Acute emotional stress and high fat/high fructose diet modulate brain oxidative damage through NrF2 and uric acid in rats. Nutr. Res. 2020, 79, 23–34. [Google Scholar] [CrossRef]
- Hsu, C.-N.; Lin, Y.-J.; Yu, H.-R.; Lin, I.-C.; Sheen, J.-M.; Huang, L.-T.; Tain, Y.-L. Protection of Male Rat Offspring against Hypertension Programmed by Prenatal Dexamethasone Administration and Postnatal High-Fat Diet with the Nrf2 Activator Dimethyl Fumarate during Pregnancy. Int. J. Mol. Sci. 2019, 20, 3957. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Saad, S.; Tan, Y.; Pollock, C.; Chen, H. Maternal high-fat diet induces metabolic stress response disorders in offspring hypothalamus. J. Mol. Endocrinol. 2017, 59, 81–92. [Google Scholar] [CrossRef]
- Liu, W.-C.; Wu, C.-W.; Fu, M.-H.; Tain, Y.-L.; Liang, C.-K.; Hung, C.-Y.; Chen, I.-C.; Lee, Y.-C.; Wu, C.-Y.; Wu, K.L. Maternal high fructose-induced hippocampal neuroinflammation in the adult female offspring via PPARγ-NF-κB signaling. J. Nutr. Biochem. 2020, 81, 108378. [Google Scholar] [CrossRef]
- Tarry-Adkins, J.L.; Martin-Gronert, M.S.; Chen, J.; Cripps, R.L.; Ozanne, S.E. Maternal diet influences DNA damage, aortic telomere length, oxidative stress, and antioxidant defense capacity in rats. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2008, 22, 2037–2044. [Google Scholar] [CrossRef]
- Kjaergaard, M.; Nilsson, C.; Nielsen, M.O.; Grove, K.; Raun, K. Hypothalamic oxidative stress and inflammation, and peripheral glucose homeostasis in Sprague-Dawley rat offspring exposed to maternal and postnatal chocolate and soft drink. Nutr. Diabetes 2018, 8, 44. [Google Scholar] [CrossRef]
- Ballesteros-Guzmán, A.K.; Carrasco-Legleu, C.E.; Levario-Carrillo, M.; Chávez-Corral, D.V.; Sánchez-Ramírez, B.; Mariñelarena-Carrillo, E.O.; Guerrero-Salgado, F.; Reza-López, S.A. Maternal Dietary Intake, and Oxidative Stress Biomarkers in the Fetomaternal Unit. BioMed Res. Int. 2019, 2019, 5070453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, A.L.-Y.; Díaz-López, K.M.; Vilchis-Gil, J.; Diaz-Garcia, H.; Gomez-Lopez, J.; Medina-Bravo, P.; Granados-Riveron, J.T.; Gallardo, J.M.; Klünder-Klünder, M.; Sánchez-Urbina, R. Diet and Maternal Obesity Are Associated with Increased Oxidative Stress in Newborns: A Cross-Sectional Study. Nutrients 2022, 14, 746. [Google Scholar] [CrossRef] [PubMed]
- Bordeleau, M.; de Cossío, L.F.; Chakravarty, M.M.; Tremblay, M. From Maternal Diet to Neurodevelopmental Disorders: A Story of Neuroinflammation. Front. Cell. Neurosci. 2020, 14, 612705. [Google Scholar] [CrossRef] [PubMed]
- Jamar, G.; Ribeiro, D.A.; Pisani, L.P. High-fat or high-sugar diets as trigger inflammation in the microbiota-gut-brain axis. Crit. Rev. Food Sci. Nutr. 2021, 61, 836–854. [Google Scholar] [CrossRef] [PubMed]
- Tsien, J.Z.; Huerta, P.; Tonegawa, S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell 1996, 87, 1327–1338. [Google Scholar] [CrossRef] [Green Version]
- Lau, B.K.; Murphy-Royal, C.; Kaur, M.; Qiao, M.; Bains, J.S.; Gordon, G.R.; Borgland, S.L. Obesity-induced astrocyte dysfunction impairs heterosynaptic plasticity in the orbitofrontal cortex. Cell Rep. 2021, 36, 109563. [Google Scholar] [CrossRef]
- Poblete-Naredo, I.; Angulo, C.; Hernández-Kelly, L.; López-Bayghen, E.; Aguilera, J.; Ortega, A. Insulin-dependent regulation of GLAST/EAAT1 in Bergmann glial cells. Neurosci. Lett. 2009, 451, 134–138. [Google Scholar] [CrossRef]
- Ward, M.M.; Jobling, A.I.; Kalloniatis, M.; Fletcher, E.L. Glutamate uptake in retinal glial cells during diabetes. Diabetologia 2005, 48, 351–360. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Dilution | Manufacturer | Cat. Number |
|---|---|---|---|
| Western blot—primary antibodies | |||
| EAAT1 | 1:1000 | Abcam (Cambridge, UK) | ab416 |
| EAAT2 | 1:500 | Abcam (Cambridge, UK) | ab178401 |
| xCT | 1:1000 | Abcam (Cambridge, UK) | ab175186 |
| VGLUT1 | 1:1000 | Abcam (Cambridge, UK) | ab77822 |
| Western blot—secondary antibodies | |||
| Goat anti-rabbit IgG | 1:5000 | Invitrogen (Waltham, MA, USA) | A27036 |
| Immunofluorescence assays—primary antibodies | |||
| EAAT1 | 1:300 | Abcam (Cambridge, UK) | ab416 |
| EAAT2 | 1:600 | Abcam (Cambridge, UK) | ab41621 |
| VGLUT1 | 1:1000 | Merck Sigma Aldrich (Darmstadt, Germany) | AMAb91041 |
| GFAP | 1:1000 | Abcam (Cambridge, UK) | ab4674 |
| MAP2 | 1:1000 | Abcam (Cambridge, UK) | ab5392 |
| Immunofluorescence assays—secondary antibodies | |||
| Goat anti-chicken AF488 | 1:300 | Abcam (Cambridge, UK) | ab150173 |
| Goat anti-rabbit AF594 | 1:300 | Abcam (Cambridge, UK) | ab150080 |
| Goat anti-chicken AF594 | 1:400 | Thermo Fischer Scientific (Waltham, MA, USA) | A32759 |
| Goat anti-mouse AF488 | 1:400 | Thermo Fischer Scientific (Waltham, MA, USA) | A32723 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mizera, J.; Pomierny, B.; Sadakierska-Chudy, A.; Bystrowska, B.; Pomierny-Chamiolo, L. Disruption of Glutamate Homeostasis in the Brain of Rat Offspring Induced by Prenatal and Early Postnatal Exposure to Maternal High-Sugar Diet. Nutrients 2022, 14, 2184. https://doi.org/10.3390/nu14112184
Mizera J, Pomierny B, Sadakierska-Chudy A, Bystrowska B, Pomierny-Chamiolo L. Disruption of Glutamate Homeostasis in the Brain of Rat Offspring Induced by Prenatal and Early Postnatal Exposure to Maternal High-Sugar Diet. Nutrients. 2022; 14(11):2184. https://doi.org/10.3390/nu14112184
Chicago/Turabian StyleMizera, Jozef, Bartosz Pomierny, Anna Sadakierska-Chudy, Beata Bystrowska, and Lucyna Pomierny-Chamiolo. 2022. "Disruption of Glutamate Homeostasis in the Brain of Rat Offspring Induced by Prenatal and Early Postnatal Exposure to Maternal High-Sugar Diet" Nutrients 14, no. 11: 2184. https://doi.org/10.3390/nu14112184
APA StyleMizera, J., Pomierny, B., Sadakierska-Chudy, A., Bystrowska, B., & Pomierny-Chamiolo, L. (2022). Disruption of Glutamate Homeostasis in the Brain of Rat Offspring Induced by Prenatal and Early Postnatal Exposure to Maternal High-Sugar Diet. Nutrients, 14(11), 2184. https://doi.org/10.3390/nu14112184