
Mild Choline Deficiency and MTHFD1 Synthetase Deficiency Interact to Increase Incidence of Developmental Delays and Defects in Mice

 , , and
, , and
Abstract
:
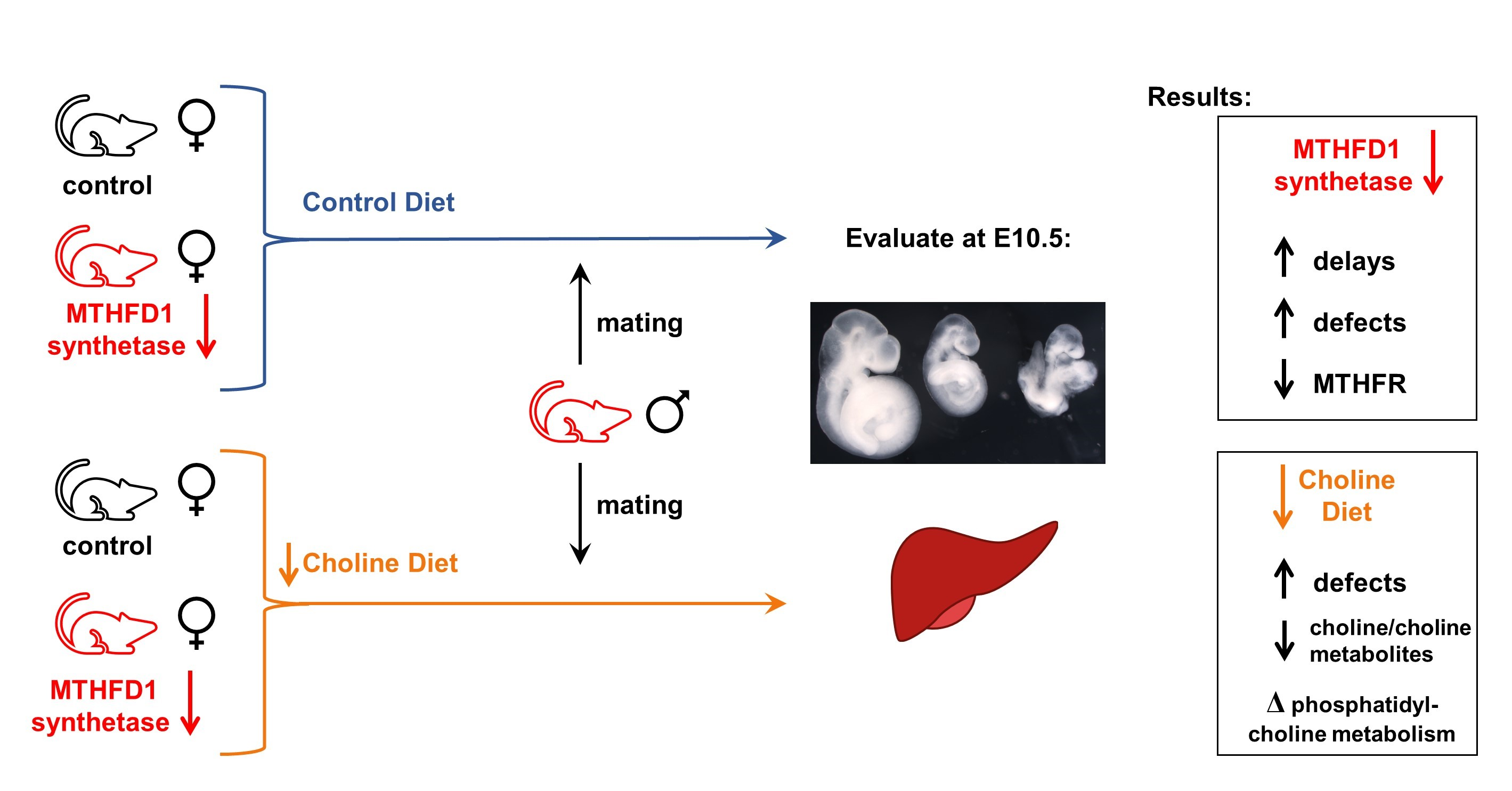{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mice and Diets
2.2. Timed Matings, Tissue and Embryo Collection
2.3. Measurement of Plasma and Liver Folates
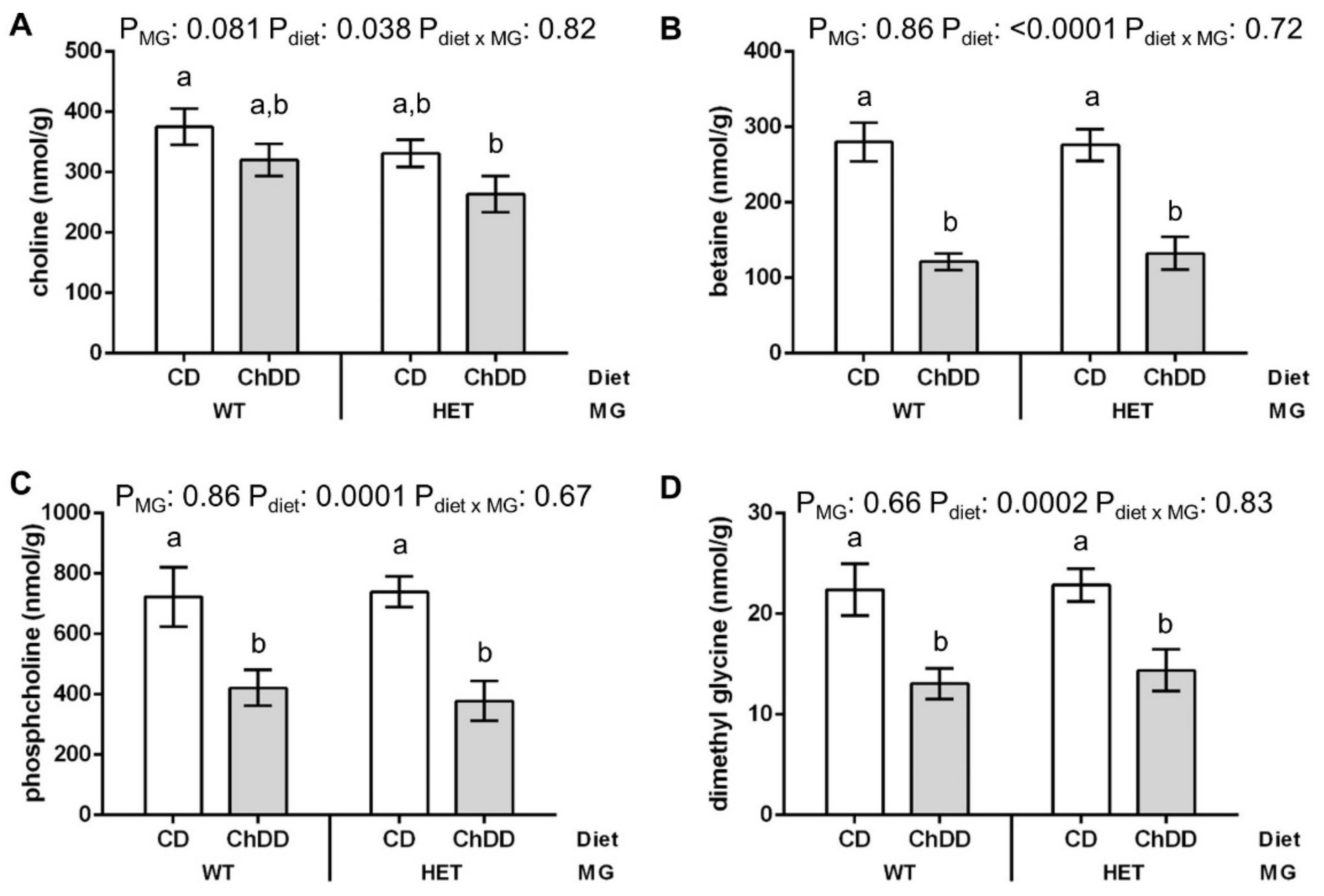
2.4. Choline and Methylation Metabolite Measurement in Maternal Liver
2.5. Measurement of PtdCho:PtdE Ratio in Liver
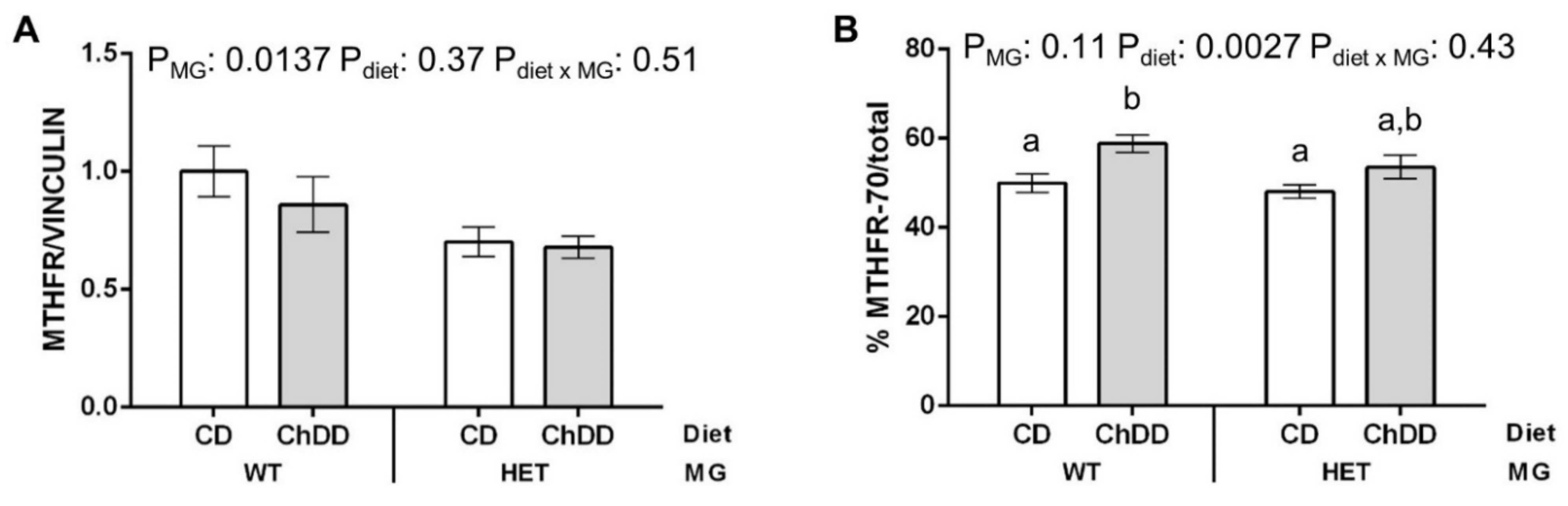
2.6. Immunoblotting
2.7. Statistical Analysis
3. Results
3.1. Maternal Food Consumption and Weight Gain
3.2. Maternal Body and Organ Weights at E10.5
3.3. Fertility
3.4. Embryonic Genotype and Sex
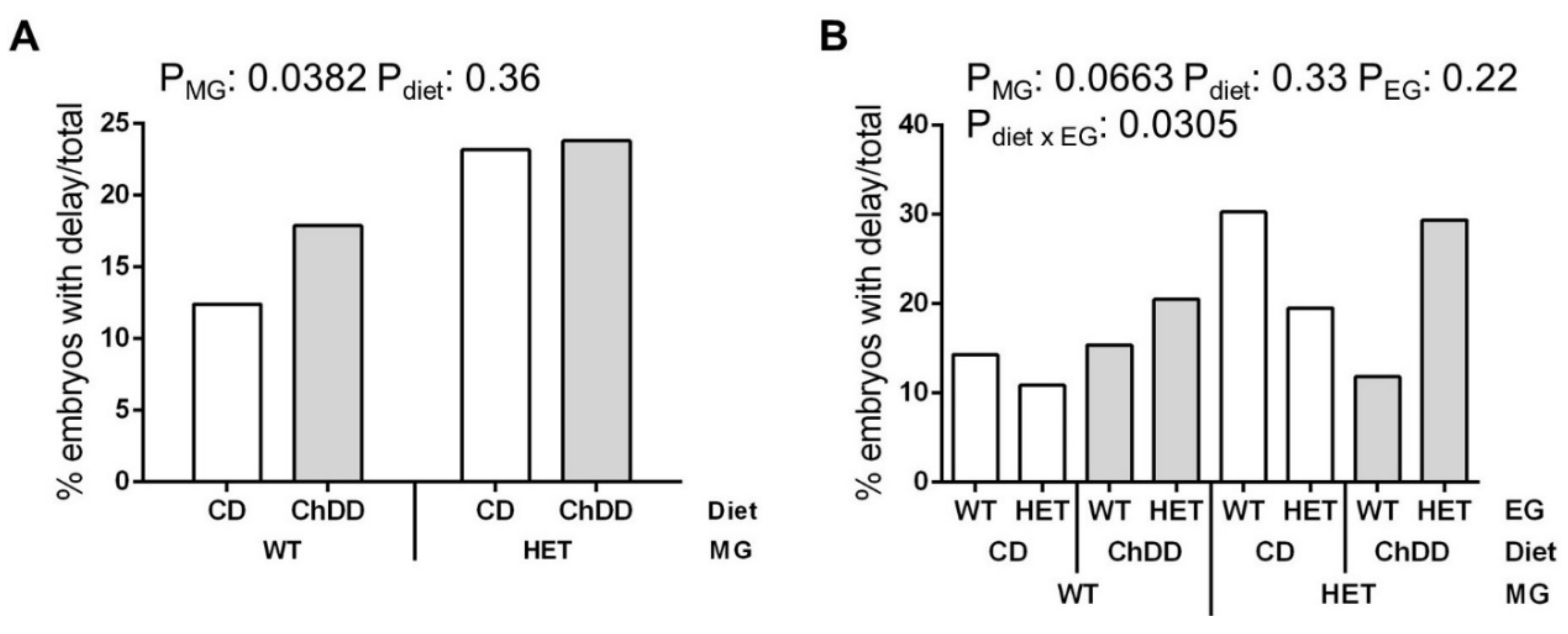
3.5. Embryo Characteristics at E10.5
3.5.1. Developmental Delay
3.5.2. Defects
3.6. Maternal E10.5 Liver and Plasma Metabolites
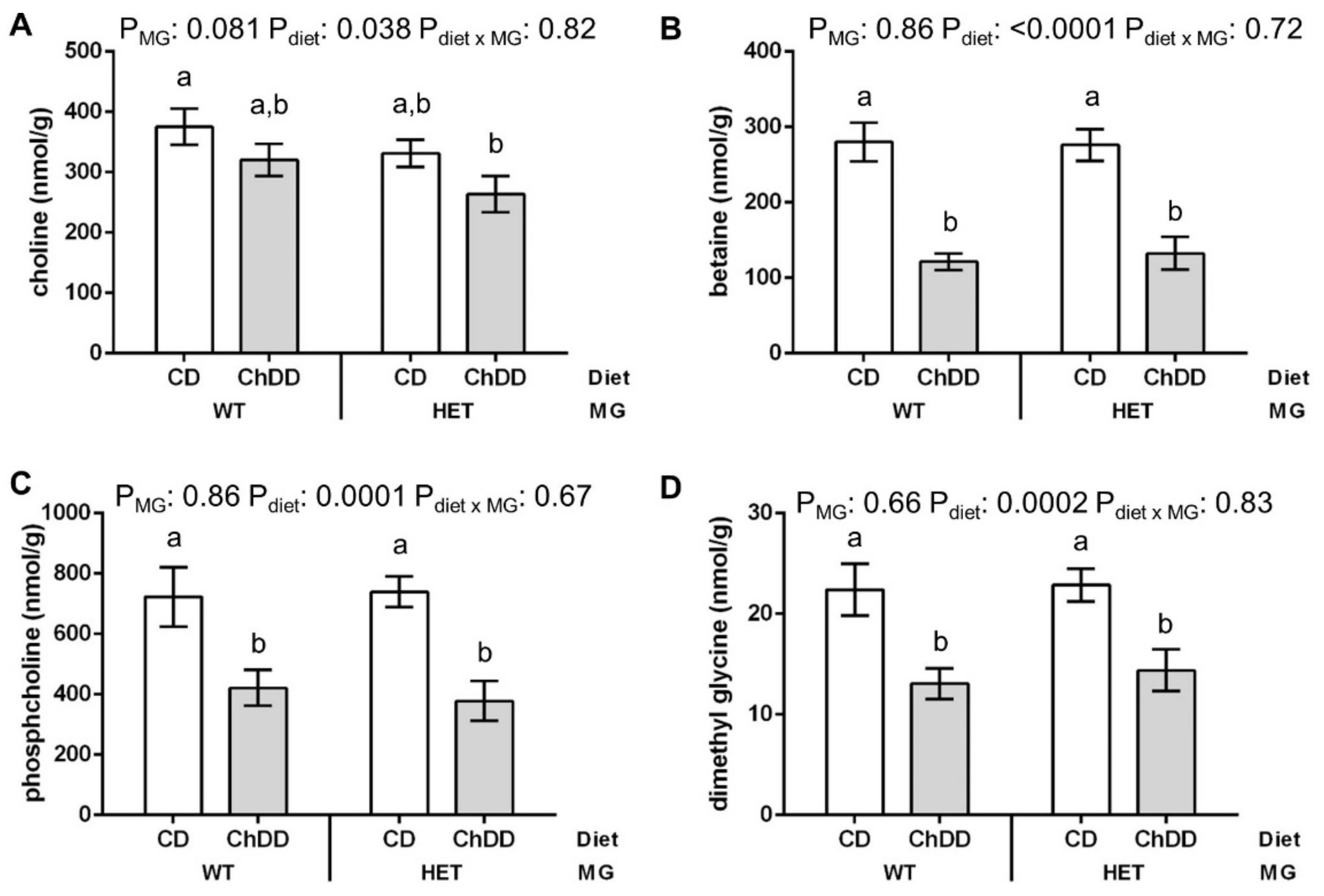
3.6.1. Choline and Methyl Metabolites in Maternal Liver
3.6.2. Maternal Plasma and Liver Folate
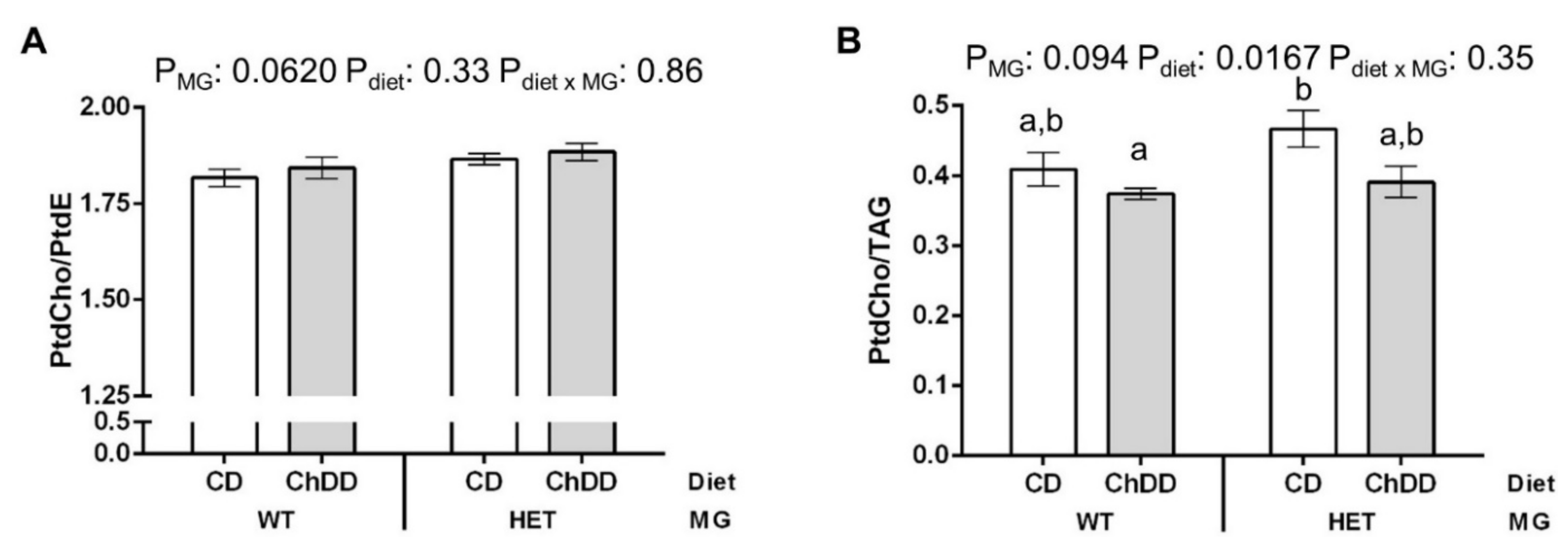
3.6.3. Phosphatidylcholine:Phosphatidylethanolamine in Maternal Liver
3.7. Immunoblotting
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Bailey, L.B.; Stover, P.J.; McNulty, H.; Fenech, M.F.; Gregory, J.F.; Mills, J.L.; Pfeiffer, C.M.; Fazili, Z.; Zhang, M.; Ueland, P.M.; et al. Biomarkers of nutrition for development—Folate review. J. Nutr. 2015, 145, 1636S–1680S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, T.C.; Blusztajn, J.K.; Caudill, M.A.; Klatt, K.C.; Zeisel, S.H. Choline: The neurocognitive essential nutrient of interest to obstetricians and gynecologists. J. Diet. Suppl. 2020, 17, 733–752. [Google Scholar] [CrossRef] [PubMed]
- Korsmo, H.W.; Jiang, X.; Caudill, M.A. Choline: Exploring the Growing Science on Its Benefits for Moms and Babies. Nutrients 2019, 11, 1823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, K.E.; Rohlicek, C.V.; Andelfinger, G.U.; Michaud, J.; Bigras, J.L.; Richter, A.; Mackenzie, R.E.; Rozen, R. The MTHFD1 p.Arg653Gln variant alters enzyme function and increases risk for congenital heart defects. Hum. Mutat. 2009, 30, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Parle-McDermott, A.; Kirke, P.N.; Mills, J.L.; Molloy, A.M.; Cox, C.; O’Leary, V.B.; Pangilinan, F.; Conley, M.; Cleary, L.; Brody, L.C.; et al. Confirmation of the R653Q polymorphism of the trifunctional C1-synthase enzyme as a maternal risk for neural tube defects in the Irish population. Eur. J. Hum. Genet. 2006, 14, 768–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parle-McDermott, A.; Pangilinan, F.; Mills, J.L.; Signore, C.C.; Molloy, A.M.; Cotter, A.; Conley, M.; Cox, C.; Kirke, P.N.; Scott, J.M.; et al. A polymorphism in the MTHFD1 gene increases a mother’s risk of having an unexplained second trimester pregnancy loss. Mol. Hum. Reprod. 2005, 11, 477–480. [Google Scholar] [CrossRef] [PubMed]
- Furness, D.L.; Fenech, M.F.; Khong, Y.T.; Romero, R.; Dekker, G.A. One-carbon metabolism enzyme polymorphisms and uteroplacental insufficiency. Am. J. Obstet. Gynecol. 2008, 199, 276.e1–276.e8. [Google Scholar] [CrossRef]
- Kohlmeier, M.; da Costa, K.A.; Fischer, L.M.; Zeisel, S.H. Genetic variation of folate-mediated one-carbon transfer pathway predicts susceptibility to choline deficiency in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 16025–16030. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, A.; Nash-Barboza, S.; Hinkis, S.; Caudill, M.A. Genetic variants in phosphatidylethanolamine N-methyltransferase and methylenetetrahydrofolate dehydrogenase influence biomarkers of choline metabolism when folate intake is restricted. J. Am. Diet. Assoc. 2009, 109, 313–318. [Google Scholar] [CrossRef] [Green Version]
- Ganz, A.B.; Shields, K.; Fomin, V.G.; Lopez, Y.S.; Mohan, S.; Lovesky, J.; Chuang, J.C.; Ganti, A.; Carrier, B.; Yan, J.; et al. Genetic impairments in folate enzymes increase dependence on dietary choline for phosphatidylcholine production at the expense of betaine synthesis. FASEB J. 2016, 30, 3321–3333. [Google Scholar] [CrossRef] [Green Version]
- Ilozumba, M.N.; Cheng, T.-Y.D.; Neuhouser, M.L.; Miller, J.W.; Beresford, S.A.A.; Duggan, D.J.; Toriola, A.T.; Song, X.; Zheng, Y.; Bailey, L.B.; et al. Associations between plasma choline metabolites and genetic polymorphisms in one-carbon metabolism in postmenopausal women: The women’s health initiative observational study. J. Nutr. 2020, 150, 2874–2881. [Google Scholar] [CrossRef]
- Yan, J.; Winter, L.B.; Burns-Whitmore, B.; Vermeylen, F.; Caudill, M.A. Plasma choline metabolites associate with metabolic stress among young overweight men in a genotype-specific manner. Nutr. Diabetes 2012, 2, e49. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.I.; Miller, J.W.; da Costa, K.A.; Nadeau, M.; Smith, D.; Selhub, J.; Zeisel, S.H.; Mason, J.B. Severe folate deficiency causes secondary depletion of choline and phosphocholine in rat liver. J. Nutr. 1994, 124, 2197–2203. [Google Scholar] [CrossRef]
- Jacob, R.A.; Jenden, D.J.; Allman-Farinelli, M.A.; Swendseid, M.E. Folate nutriture alters choline status of women and men fed low choline diets. J. Nutr. 1999, 129, 712–717. [Google Scholar] [CrossRef] [Green Version]
- Selhub, J.; Seyoum, E.; Pomfret, E.A.; Zeisel, S.H. Effects of choline deficiency and methotrexate treatment upon liver folate content and distribution. Cancer Res. 1991, 51, 16–21. [Google Scholar]
- Christensen, K.E.; Wu, Q.; Wang, X.; Deng, L.; Caudill, M.A.; Rozen, R. Steatosis in mice is associated with gender, folate intake, and expression of genes of one-carbon metabolism. J. Nutr. 2010, 140, 1736–1741. [Google Scholar] [CrossRef] [Green Version]
- Derbyshire, E.; Obeid, R. Choline, neurological development and brain function: A systematic review focusing on the first 1000 days. Nutrients 2020, 12, 1731. [Google Scholar] [CrossRef]
- Shaw, G.M.; Carmichael, S.L.; Yang, W.; Selvin, S.; Schaffer, D.M. Periconceptional dietary intake of choline and betaine and neural tube defects in offspring. Am. J. Epidemiol. 2004, 160, 102–109. [Google Scholar] [CrossRef] [Green Version]
- Shaw, G.M.; Finnell, R.H.; Blom, H.J.; Carmichael, S.L.; Vollset, S.E.; Yang, W.; Ueland, P.M. Choline and risk of neural tube defects in a folate-fortified population. Epidemiology 2009, 20, 714–719. [Google Scholar] [CrossRef] [Green Version]
- Fisher, M.C.; Zeisel, S.H.; Mar, M.H.; Sadler, T.W. Inhibitors of choline uptake and metabolism cause developmental abnormalities in neurulating mouse embryos. Teratology 2001, 64, 114–122. [Google Scholar] [CrossRef]
- Chan, J.; Deng, L.; Mikael, L.G.; Yan, J.; Pickell, L.; Wu, Q.; Caudill, M.A.; Rozen, R. Low dietary choline and low dietary riboflavin during pregnancy influence reproductive outcomes and heart development in mice. Am. J. Clin. Nutr. 2010, 91, 1035–1043. [Google Scholar] [CrossRef] [Green Version]
- Lewis, E.D.; Subhan, F.B.; Bell, R.C.; McCargar, L.J.; Curtis, J.M.; Jacobs, R.L.; Field, C.J. Estimation of choline intake from 24 h dietary intake recalls and contribution of egg and milk consumption to intake among pregnant and lactating women in Alberta. Br. J. Nutr. 2014, 112, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Masih, S.P.; Plumptre, L.; Ly, A.; Berger, H.; Lausman, A.Y.; Croxford, R.; Kim, Y.-I.; O’Connor, D.L. Pregnant Canadian women achieve recommended intakes of one-carbon nutrients through prenatal supplementation but the supplement composition, including choline, requires reconsideration. J. Nutr. 2015, 145, 1824–1834. [Google Scholar] [CrossRef] [Green Version]
- Wallace, T.C.; Fulgoni, V.L. Usual choline intakes are associated with egg and protein food consumption in the United States. Nutrients 2017, 9, 839. [Google Scholar] [CrossRef] [Green Version]
- Bailey, R.L.; Pac, S.G.; Fulgoni, V.L., 3rd; Reidy, K.C.; Catalano, P.M. Estimation of total usual dietary intakes of pregnant women in the United States. JAMA Netw. Open 2019, 2, e195967. [Google Scholar] [CrossRef] [Green Version]
- Ganz, A.B.; Klatt, K.C.; Caudill, M.A. Common genetic variants alter metabolism and influence dietary choline requirements. Nutrients 2017, 9, 837. [Google Scholar] [CrossRef] [Green Version]
- Christensen, K.E.; Deng, L.; Leung, K.Y.; Arning, E.; Bottiglieri, T.; Malysheva, O.V.; Caudill, M.A.; Krupenko, N.I.; Greene, N.D.; Jerome-Majewska, L.; et al. A novel mouse model for genetic variation in 10-formyltetrahydrofolate synthetase exhibits disturbed purine synthesis with impacts on pregnancy and embryonic development. Hum. Mol. Genet. 2013, 22, 3705–3719. [Google Scholar] [CrossRef] [Green Version]
- Christensen, K.E.; Hou, W.; Bahous, R.H.; Deng, L.; Malysheva, O.V.; Arning, E.; Bottiglieri, T.; Caudill, M.A.; Jerome-Majewska, L.A.; Rozen, R. Moderate folic acid supplementation and MTHFD1-synthetase deficiency in mice, a model for the R653Q variant, result in embryonic defects and abnormal placental development. Am. J. Clin. Nutr. 2016, 104, 1459–1469. [Google Scholar] [CrossRef]
- Christensen, K.E.; Bahous, R.H.; Hou, W.; Deng, L.; Malysheva, O.V.; Arning, E.; Bottiglieri, T.; Caudill, M.A.; Jerome-Majewska, L.A.; Rozen, R. Low dietary folate interacts with MTHFD1 synthetase deficiency in mice, a model for the R653Q variant, to increase incidence of developmental delays and defects. J. Nutr. 2018, 148, 501–509. [Google Scholar] [CrossRef]
- Li, D.; Pickell, L.; Liu, Y.; Wu, Q.; Cohn, J.S.; Rozen, R. Maternal methylenetetrahydrofolate reductase deficiency and low dietary folate lead to adverse reproductive outcomes and congenital heart defects in mice. Am. J. Clin. Nutr. 2005, 82, 188–195. [Google Scholar] [CrossRef]
- McFarlane, L.; Truong, V.; Palmer, J.S.; Wilhelm, D. Novel PCR assay for determining the genetic sex of mice. Sex. Dev. 2013, 7, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Molloy, A.M.; Scott, J.M. Microbiological assay for serum, plasma, and red cell folate using cryopreserved, microtiter plate method. Methods Enzymol. 1997, 281, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Bensadoun, A.; Weinstein, D. Assay of proteins in the presence of interfering materials. Anal. Biochem. 1976, 70, 241–250. [Google Scholar] [CrossRef]
- Koc, H.; Mar, M.H.; Ranasinghe, A.; Swenberg, J.A.; Zeisel, S.H. Quantitation of choline and its metabolites in tissues and foods by liquid chromatography/electrospray ionization-isotope dilution mass spectrometry. Anal. Chem. 2002, 74, 4734–4740. [Google Scholar] [CrossRef]
- Holm, P.I.; Ueland, P.M.; Kvalheim, G.; Lien, E.A. Determination of choline, betaine, and dimethylglycine in plasma by a high-throughput method based on normal-phase chromatography-tandem mass spectrometry. Clin. Chem. 2003, 49, 286–294. [Google Scholar] [CrossRef]
- Kim, J.K.; Harada, K.; Bamba, T.; Fukusaki, E.; Kobayashi, A. Stable isotope dilution-based accurate comparative quantification of nitrogen-containing metabolites in Arabidopsis thaliana T87 cells using in vivo (15)N-isotope enrichment. Biosci. Biotechnol. Biochem. 2005, 69, 1331–1340. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Yan, J.; West, A.A.; Perry, C.A.; Malysheva, O.V.; Devapatla, S.; Pressman, E.; Vermeylen, F.; Caudill, M.A. Maternal choline intake alters the epigenetic state of fetal cortisol-regulating genes in humans. FASEB J. 2012, 26, 3563–3574. [Google Scholar] [CrossRef] [Green Version]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Abreu, S.; Solgadi, A.; Chaminade, P. Optimization of normal phase chromatographic conditions for lipid analysis and comparison of associated detection techniques. J. Chromatogr. A 2017, 1514, 54–71. [Google Scholar] [CrossRef]
- Lian, J.; van der Veen, J.N.; Watts, R.; Jacobs, R.L.; Lehner, R. Carboxylesterase 1d (Ces1d) does not contribute to cholesteryl ester hydrolysis in the liver. J. Lipid Res. 2021, 62, 100093. [Google Scholar] [CrossRef]
- Gardam, M.A.; Mejia, N.R.; MacKenzie, R.E. The NADP-dependent trifunctional methylenetetrahydrofolate dehydrogenase purified from mouse liver is immunologically distinct from the mouse NAD-dependent bifunctional enzyme. Biochem. Cell. Biol. 1988, 66, 66–70. [Google Scholar] [CrossRef]
- Frosst, P.; Blom, H.J.; Milos, R.; Goyette, P.; Sheppard, C.A.; Matthews, R.G.; Boers, G.J.; den Heijer, M.; Kluijtmans, L.A.; van den Heuvel, L.P.; et al. A candidate genetic risk factor for vascular disease: A common mutation in methylenetetrahydrofolate reductase. Nat. Genet. 1995, 10, 111–113. [Google Scholar] [CrossRef]
- Rao, P.V.; Garrow, T.A.; John, F.; Garland, D.; Millian, N.S.; Zigler, J.S., Jr. Betaine-homocysteine methyltransferase is a developmentally regulated enzyme crystallin in rhesus monkey lens. J. Biol. Chem. 1998, 273, 30669–30674. [Google Scholar] [CrossRef] [Green Version]
- R Core Team R. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: http://www.R-project.org/ (accessed on 14 December 2020).
- RStudio Team RStudio. Integrated Development for R; RStudio, Inc.: Boston, MA, USA, 2020; Available online: http://www.rstudio.com/ (accessed on 14 December 2020).
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting linear mixed-effects models using lme4. J. Stat. Soft. 2015, 67, 48. [Google Scholar] [CrossRef]
- Lenth, R.V. Emmeans: Estimated Marginal Means, aka Least-Squares Means; R Package Version 1.5.3. 2020. Available online: https://CRAN.R-project.org/package=emmeans (accessed on 14 December 2020).
- Van der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta Biomembranes 2017, 1859, 1558–1572. [Google Scholar] [CrossRef]
- Moessinger, C.; Klizaite, K.; Steinhagen, A.; Philippou-Massier, J.; Shevchenko, A.; Hoch, M.; Ejsing, C.S.; Thiele, C. Two different pathways of phosphatidylcholine synthesis, the Kennedy Pathway and the Lands Cycle, differentially regulate cellular triacylglycerol storage. BMC Cell Biol. 2014, 15, 43. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.; Strahler, J.R.; Andrews, P.C.; Matthews, R.G. Regulation of human methylenetetrahydrofolate reductase by phosphorylation. Proc. Natl. Acad. Sci. USA 2005, 102, 10454–10459. [Google Scholar] [CrossRef] [Green Version]
- Tran, P.; Leclerc, D.; Chan, M.; Pai, A.; Hiou-Tim, F.; Wu, Q.; Goyette, P.; Artigas, C.; Milos, R.; Rozen, R. Multiple transcription start sites and alternative splicing in the methylenetetrahydrofolate reductase gene result in two enzyme isoforms. Mamm. Genome 2002, 13, 483. [Google Scholar] [CrossRef]
- Zheng, Y.; Ramsamooj, S.; Li, Q.; Johnson, J.L.; Yaron, T.M.; Sharra, K.; Cantley, L.C. Regulation of folate and methionine metabolism by multisite phosphorylation of human methylenetetrahydrofolate reductase. Sci. Rep. 2019, 9, 4190. [Google Scholar] [CrossRef]
- Jadavji, N.M.; Deng, L.; Malysheva, O.; Caudill, M.A.; Rozen, R. MTHFR deficiency or reduced intake of folate or choline in pregnant mice results in impaired short-term memory and increased apoptosis in the hippocampus of wild-type offspring. Neuroscience 2015, 300, 1–9. [Google Scholar] [CrossRef]
- Di Pietro, E.; Wang, X.L.; MacKenzie, R.E. The expression of mitochondrial methylenetetrahydrofolate dehydrogenase-cyclohydrolase supports a role in rapid cell growth. Biochim. Biophys. Acta 2004, 1674, 78–84. [Google Scholar] [CrossRef]
- Pickell, L.; Wu, Q.; Wang, X.L.; Leclerc, D.; Friedman, H.; Peterson, A.C.; Rozen, R. Targeted insertion of two Mthfr promoters in mice reveals temporal- and tissue-specific regulation. Mamm. Genome 2011, 22, 635–647. [Google Scholar] [CrossRef]
- Wang, L.; Magdaleno, S.; Tabas, I.; Jackowski, S. Early embryonic lethality in mice with targeted deletion of the CTP: Phosphocholine cytidylyltransferase alpha gene (Pcyt1a). Mol. Cell Biol. 2005, 25, 3357–3363. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Aoyama, C.; Young, S.G.; Vance, D.E. Early embryonic lethality caused by disruption of the gene for choline kinase alpha, the first enzyme in phosphatidylcholine biosynthesis. J. Biol. Chem. 2008, 283, 1456–1462. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Agellon, L.B.; Vance, D.E. Choline redistribution during adaptation to choline deprivation. J. Biol. Chem. 2007, 282, 10283–10289. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, R.P.; Kelly, K.B.; Al Rajabi, A.; Jacobs, R.L. Novel insights on interactions between folate and lipid metabolism. Biofactors 2014, 40, 277–283. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Jiang, X.; West, A.A.; Perry, C.A.; Malysheva, O.V.; Brenna, J.T.; Stabler, S.P.; Allen, R.H.; Gregory, J.F., 3rd; Caudill, M.A. Pregnancy alters choline dynamics: Results of a randomized trial using stable isotope methodology in pregnant and nonpregnant women. Am. J. Clin. Nutr. 2013, 98, 1459–1467. [Google Scholar] [CrossRef]
- DeLong, C.J.; Shen, Y.J.; Thomas, M.J.; Cui, Z. Molecular distinction of phosphatidylcholine synthesis between the CDP-choline pathway and phosphatidylethanolamine methylation pathway. J. Biol. Chem. 1999, 274, 29683–29688. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; He, F.; Wu, C.; Li, P.; Li, N.; Deng, J.; Zhu, G.; Ren, W.; Peng, Y. Betaine in inflammation: Mechanistic aspects and applications. Front. Immunol. 2018, 9, 1070. [Google Scholar] [CrossRef] [Green Version]
- Tscherner, A.K.; Macaulay, A.D.; Ortman, C.S.; Baltz, J.M. Initiation of cell volume regulation and unique cell volume regulatory mechanisms in mammalian oocytes and embryos. J. Cell Physiol. 2021, 236, 7117–7133. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Christensen, K.E.; Malysheva, O.V.; Carlin, S.; Matias, F.; MacFarlane, A.J.; Jacobs, R.L.; Caudill, M.A.; Rozen, R. Mild Choline Deficiency and MTHFD1 Synthetase Deficiency Interact to Increase Incidence of Developmental Delays and Defects in Mice. Nutrients 2022, 14, 127. https://doi.org/10.3390/nu14010127
Christensen KE, Malysheva OV, Carlin S, Matias F, MacFarlane AJ, Jacobs RL, Caudill MA, Rozen R. Mild Choline Deficiency and MTHFD1 Synthetase Deficiency Interact to Increase Incidence of Developmental Delays and Defects in Mice. Nutrients. 2022; 14(1):127. https://doi.org/10.3390/nu14010127
Chicago/Turabian StyleChristensen, Karen E., Olga V. Malysheva, Stephanie Carlin, Fernando Matias, Amanda J. MacFarlane, René L. Jacobs, Marie A. Caudill, and Rima Rozen. 2022. "Mild Choline Deficiency and MTHFD1 Synthetase Deficiency Interact to Increase Incidence of Developmental Delays and Defects in Mice" Nutrients 14, no. 1: 127. https://doi.org/10.3390/nu14010127
APA StyleChristensen, K. E., Malysheva, O. V., Carlin, S., Matias, F., MacFarlane, A. J., Jacobs, R. L., Caudill, M. A., & Rozen, R. (2022). Mild Choline Deficiency and MTHFD1 Synthetase Deficiency Interact to Increase Incidence of Developmental Delays and Defects in Mice. Nutrients, 14(1), 127. https://doi.org/10.3390/nu14010127