
Ketogenic Diets in Pancreatic Cancer and Associated Cachexia: Cellular Mechanisms and Clinical Perspectives


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Pancreatic Cancer: Biology and Current Treatments
3. PDAC-Associated Cachexia
4. Ketogenic Diet in Cancer
5. Ketogenic Diet in Cachexia
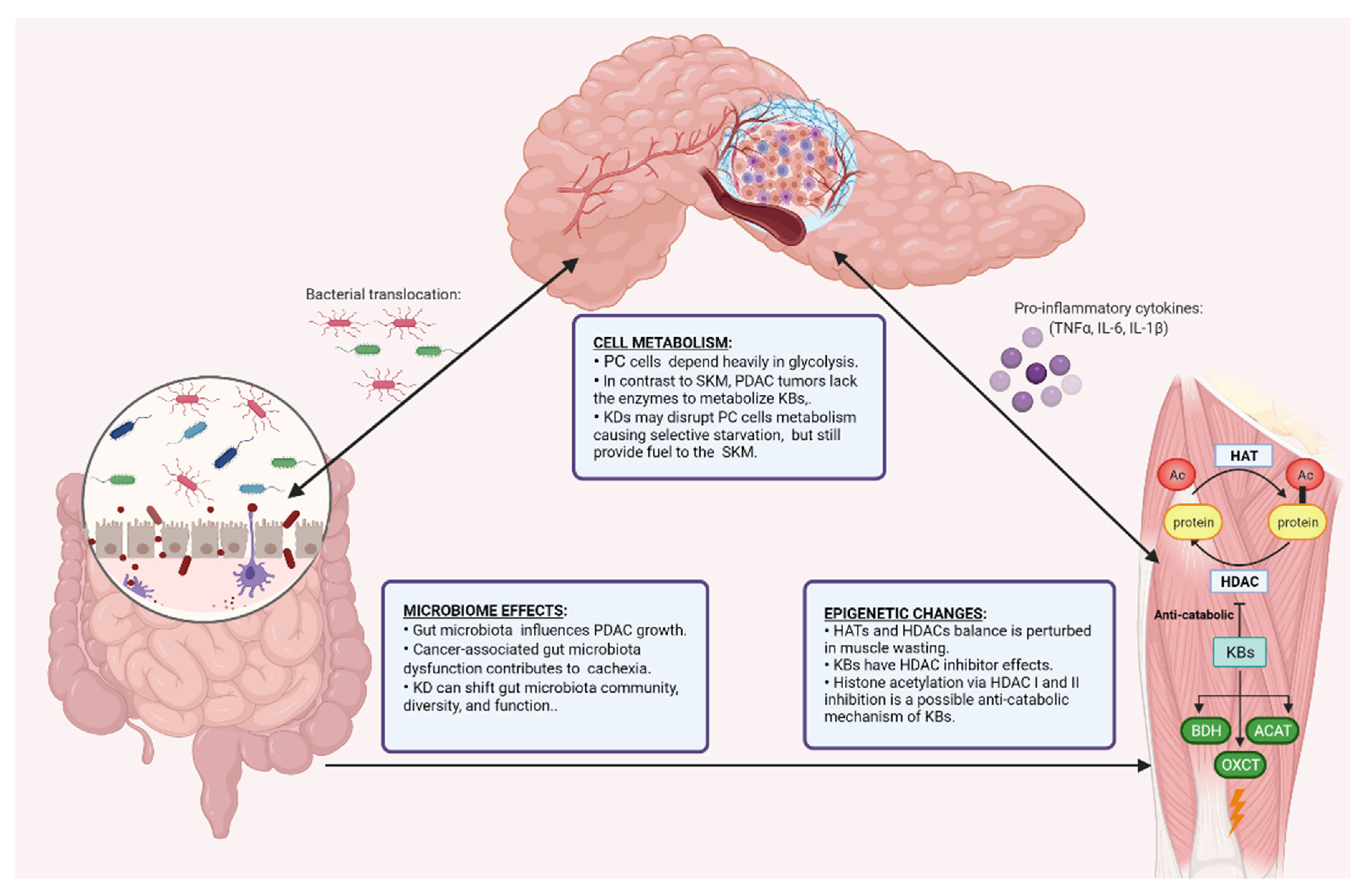
6. Cellular Mechanisms of KDs in PDAC and Cachexia
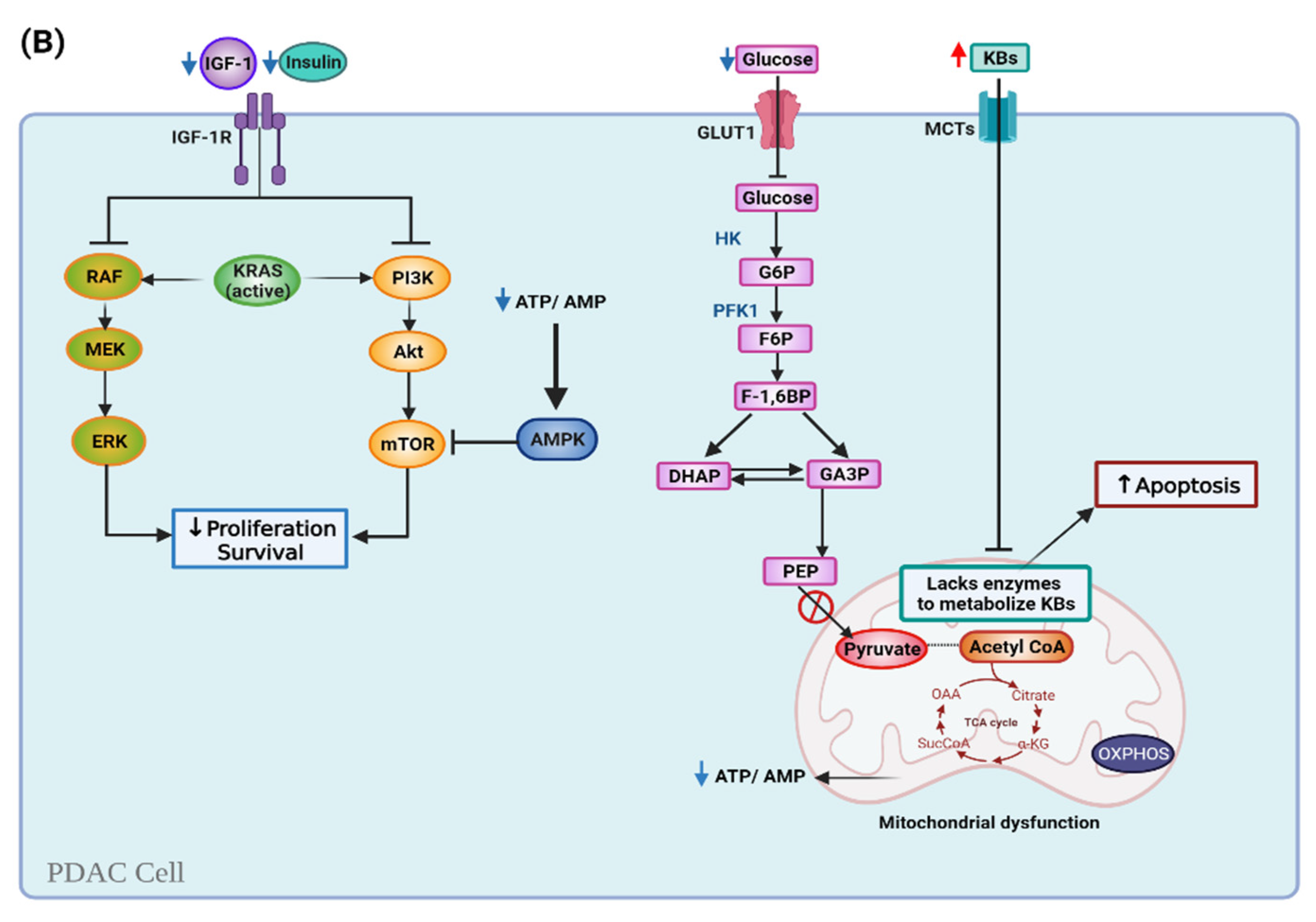
6.1. Ketogenic Diet and Cell Metabolism
6.2. Ketogenic Diet and the Epigenome
6.3. Ketogenic Diet and the Gut Microbiome
7. Clinical Perspectives and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef]
- Smyl, C. Ketogenic Diet and Cancer—A Perspective. In Metabolism in Cancer; Cramer, T., Schmitt, C.A., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 233–240. [Google Scholar]
- Klement, R.J.; Champ, C.E.; Otto, C.; Kämmerer, U. Anti-Tumor Effects of Ketogenic Diets in Mice: A Meta-Analysis. PLoS ONE 2016, 11, e0155050. [Google Scholar] [CrossRef] [PubMed]
- Champ, C.E.; Palmer, J.D.; Volek, J.S.; Werner-Wasik, M.; Andrews, D.W.; Evans, J.J.; Glass, J.; Kim, L.; Shi, W. Targeting metabolism with a ketogenic diet during the treatment of glioblastoma multiforme. J. Neuro-Oncol. 2014, 117, 125–131. [Google Scholar] [CrossRef]
- Erickson, N.; Boscheri, A.; Linke, B.; Huebner, J. Systematic review: Isocaloric ketogenic dietary regimes for cancer patients. Med. Oncol. 2017, 34, 72. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Sakharkar, M.K.; Dhillon, S.K.; Mazumder, M.; Yang, J. Key drug-targeting genes in pancreatic ductal adenocarcinoma. Genes Cancer 2021, 12, 12–24. [Google Scholar] [CrossRef]
- Wirkus, J.; Ead, A.S.; Mackenzie, G.G. Impact of dietary fat composition and quantity in pancreatic carcinogenesis: Recent advances and controversies. Nutr. Res. 2021, 88, 1–18. [Google Scholar] [CrossRef]
- Brosens, L.A.A.; Hackeng, W.M.; Offerhaus, G.J.; Hruban, R.H.; Wood, L.D. Pancreatic adenocarcinoma pathology: Changing “landscape”. J. Gastrointest. Oncol. 2015, 6, 358–374. [Google Scholar] [CrossRef]
- Storz, P.; Crawford, H.C. Carcinogenesis of Pancreatic Ductal Adenocarcinoma. Gastroenterology 2020, 158, 2072–2081. [Google Scholar] [CrossRef]
- Giannis, D.; Moris, D.; Barbas, A.S. Diagnostic, Predictive and Prognostic Molecular Biomarkers in Pancreatic Cancer: An Overview for Clinicians. Cancers 2021, 13, 1071. [Google Scholar] [CrossRef] [PubMed]
- Alzhrani, R.; Alsaab, H.O.; Vanamala, K.; Bhise, K.; Tatiparti, K.; Barari, A.; Sau, S.; Iyer, A.K. Overcoming the Tumor Microenvironmental Barriers of Pancreatic Ductal Adenocarcinomas for Achieving Better Treatment Outcomes. Adv. Ther. 2021, 4, 2000262. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Ahuja, N.; Makary, M.A.; Cameron, J.L.; Eckhauser, F.E.; Choti, M.A.; Hruban, R.H.; Pawlik, T.M.; Wolfgang, C.L. 2564 resected periampullary adenocarcinomas at a single institution: Trends over three decades. HPB 2014, 16, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Kunzmann, V.; Siveke, J.T.; Algül, H.; Goekkurt, E.; Siegler, G.; Martens, U.; Waldschmidt, D.; Pelzer, U.; Fuchs, M.; Kullmann, F.; et al. Nab-paclitaxel plus gemcitabine versus nab-paclitaxel plus gemcitabine followed by FOLFIRINOX induction chemotherapy in locally advanced pancreatic cancer (NEOLAP-AIO-PAK-0113): A multicentre, randomised, phase 2 trial. Lancet Gastroenterol. Hepatol. 2021, 6, 128–138. [Google Scholar] [CrossRef]
- Iyikesici, M.S. Long-Term Survival Outcomes of Metabolically Supported Chemotherapy with Gemcitabine-Based or FOLFIRINOX Regimen Combined with Ketogenic Diet, Hyperthermia, and Hyperbaric Oxygen Therapy in Metastatic Pancreatic Cancer. Complement. Med. Res. 2020, 27, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Parrasia, S.; Zoratti, M.; Szabo, I.; Biasutto, L. Targeting Pancreatic Ductal Adenocarcinoma (PDAC)|Cell Physiol Biochem. Cell. Physiol. Biochem. 2021, 55, 61–90. [Google Scholar]
- Choi, M.; Saif, M.W.; Kim, R. Is there a role for second line therapy in advanced pancreatic cancer? JOP J. Pancreas 2014, 15, 106–109. [Google Scholar] [CrossRef]
- Liao, W.-C.; Chen, P.-R.; Huang, C.-C.; Chang, Y.-T.; Huang, B.-S.; Chang, C.-C.; Wu, M.-S.; Chow, L.-P. Relationship between pancreatic cancer-associated diabetes and cachexia. J. Cachexia Sarcopenia Muscle 2020, 11, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Henderson, S.E.; Makhijani, N.; Mace, T.A. Pancreatic Cancer-Induced Cachexia and Relevant Mouse Models. Pancreas 2018, 47, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Kordes, M.; Larsson, L.; Engstrand, L.; Löhr, J.M. Pancreatic cancer cachexia: Three dimensions of a complex syndrome. Br. J. Cancer 2021, 124, 1623–1636. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, J.; Heiligensetzer, M.; Krakowski-Roosen, H.; Büchler, M.W.; Friess, H.; Martignoni, M.E. Cachexia Worsens Prognosis in Patients with Resectable Pancreatic Cancer. J. Gastrointest. Surg. 2008, 12, 1193. [Google Scholar] [CrossRef]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Primers 2018, 4, 1–18. [Google Scholar] [CrossRef]
- Kays, J.K.; Shahda, S.; Stanley, M.; Bell, T.M.; O’Neill, B.H.; Kohli, M.D.; Couch, M.E.; Koniaris, L.G.; Zimmers, T.A. Three cachexia phenotypes and the impact of fat-only loss on survival in FOLFIRINOX therapy for pancreatic cancer. J. Cachexia Sarcopenia Muscle 2018, 9, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhou, F.; Cao, Z.; Tang, Y.; Huang, Y.; Li, Y.; Yi, B.; Yang, J.; Du, P.; Zhu, D.; et al. Development and Validation of a Nomogram Based on Nutritional Indicators and Tumor Markers for Prognosis Prediction of Pancreatic Ductal Adenocarcinoma. Front. Oncol. 2021, 11, 682969. [Google Scholar] [CrossRef]
- Ferrucci, L.M.; Bell, D.; Thornton, J.; Black, G.; McCorkle, R.; Heimburger, D.C.; Saif, M.W. Nutritional status of patients with locally advanced pancreatic cancer. Support. Care Cancer 2011, 19, 1729–1734. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Oh, D.-Y.; Kim, T.-Y.; Lee, K.-H.; Han, S.-W.; Im, S.-A.; Kim, T.-Y.; Bang, Y.-J. Skeletal Muscle Depletion Predicts the Prognosis of Patients with Advanced Pancreatic Cancer Undergoing Palliative Chemotherapy, Independent of Body Mass Index. PLoS ONE 2015, 10, e0139749. [Google Scholar] [CrossRef]
- Mueller, T.C.; Burmeister, M.A.; Bachmann, J.; Martignoni, M.E. Cachexia and pancreatic cancer: Are there treatment options? World J. Gastroenterol. 2014, 20, 9361–9373. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, A.; Shahda, S.; Kays, J.K.; Perkins, S.M.; Cheng, L.; Schloss, K.N.H.; Schloss, D.E.I.; Koniaris, L.G.; Zimmers, T.A. Identification of Potential Serum Protein Biomarkers and Pathways for Pancreatic Cancer Cachexia Using an Aptamer-Based Discovery Platform. Cancers 2020, 12, 3787. [Google Scholar] [CrossRef]
- Pin, F.; Couch, M.E.; Bonetto, A. Preservation of muscle mass as a strategy to reduce the toxic effects of cancer chemotherapy on body composition. Curr. Opin. Support. Palliat. Care 2018, 12, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Solheim, T.S.; Laird, B.J.A.; Balstad, T.R.; Stene, G.B.; Bye, A.; Johns, N.; Pettersen, C.H.; Fallon, M.; Fayers, P.; Fearon, K.; et al. A randomized phase II feasibility trial of a multimodal intervention for the management of cachexia in lung and pancreatic cancer. J. Cachexia Sarcopenia Muscle 2017, 8, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Yakovenko, A.; Cameron, M.; Trevino, J.G. Molecular therapeutic strategies targeting pancreatic cancer induced cachexia. World J. Gastrointest. Surg. 2018, 10, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Rupert, J.E.; Narasimhan, A.; Jengelley, D.H.A.; Jiang, Y.; Liu, J.; Au, E.; Silverman, L.M.; Sandusky, G.; Bonetto, A.; Cao, S.; et al. Tumor-derived IL-6 and trans-signaling among tumor, fat, and muscle mediate pancreatic cancer cachexia. J. Exp. Med. 2021, 218, e20190450. [Google Scholar] [CrossRef] [PubMed]
- White, J.P.; Puppa, M.J.; Gao, S.; Sato, S.; Welle, S.L.; Carson, J.A. Muscle mTORC1 suppression by IL-6 during cancer cachexia: A role for AMPK. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E1042–E1052. [Google Scholar] [CrossRef]
- Duval, A.P.; Jeanneret, C.; Santoro, T.; Dormond, O. mTOR and Tumor Cachexia. Int. J. Mol. Sci. 2018, 19, 2225. [Google Scholar] [CrossRef]
- Li, Y.; Jin, H.; Chen, Y.; Huang, T.; Mi, Y.; Zou, Z. Cancer cachexia: Molecular mechanism and pharmacological management. Biochem. J. 2021, 478, 1663–1688. [Google Scholar] [CrossRef] [PubMed]
- Masi, T.; Patel, B.M. Altered glucose metabolism and insulin resistance in cancer-induced cachexia: A sweet poison. Pharm. Rep. 2021, 73, 17–30. [Google Scholar] [CrossRef]
- Siddiqui, J.A.; Pothuraju, R.; Jain, M.; Batra, S.K.; Nasser, M.W. Advances in cancer cachexia: Intersection between affected organs, mediators, and pharmacological interventions. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188359. [Google Scholar] [CrossRef]
- Kuchta, K.; Cameron, S. Phytotherapy for Cachexia: Where Do We Stand? Front. Pharm. 2020, 11. [Google Scholar] [CrossRef]
- Denley, S.M.; Jamieson, N.B.; McCall, P.; Oien, K.A.; Morton, J.P.; Carter, C.R.; Edwards, J.; McKay, C.J. Activation of the IL-6R/Jak/stat pathway is associated with a poor outcome in resected pancreatic ductal adenocarcinoma. J. Gastrointest. Surg. 2013, 17, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Hagg, A.; Kharoud, S.; Goodchild, G.; Goodman, C.A.; Chen, J.L.; Thomson, R.E.; Qian, H.; Gregorevic, P.; Harrison, C.A.; Walton, K.L. TMEPAI/PMEPA1 Is a Positive Regulator of Skeletal Muscle Mass. Front. Physiol. 2020, 11, 560225. [Google Scholar] [CrossRef]
- Gerber, M.H.; Underwood, P.W.; Judge, S.M.; Delitto, D.; Delitto, A.E.; Nosacka, R.L.; DiVita, B.B.; Thomas, R.M.; Permuth, J.B.; Hughes, S.J.; et al. Local and Systemic Cytokine Profiling for Pancreatic Ductal Adenocarcinoma to Study Cancer Cachexia in an Era of Precision Medicine. Int. J. Mol. Sci. 2018, 19, 3836. [Google Scholar] [CrossRef]
- Gorjao, R.; Dos Santos, C.M.M.; Serdan, T.D.A.; Diniz, V.L.S.; Alba-Loureiro, T.C.; Cury-Boaventura, M.F.; Hatanaka, E.; Levada-Pires, A.C.; Sato, F.T.; Pithon-Curi, T.C.; et al. New insights on the regulation of cancer cachexia by N-3 polyunsaturated fatty acids. Pharmacol. Ther. 2019, 196, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Silva, K.A.; Dong, J.; Dong, Y.; Dong, Y.; Schor, N.; Tweardy, D.J.; Zhang, L.; Mitch, W.E. Inhibition of Stat3 activation suppresses caspase-3 and the ubiquitin-proteasome system, leading to preservation of muscle mass in cancer cachexia. J. Biol. Chem. 2015, 290, 11177–11187. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Huo, R.; Zhi, Q.; Zhan, M.; Chen, X.; Hua, Z.C. Increased expression of zinc transporter ZIP4, ZIP11, ZnT1, and ZnT6 predicts poor prognosis in pancreatic cancer. J. Trace Elem. Med. Biol. 2021, 65, 126734. [Google Scholar] [CrossRef]
- Poulia, K.A.; Sarantis, P.; Antoniadou, D.; Koustas, E.; Papadimitropoulou, A.; Papavassiliou, A.G.; Karamouzis, M.V. Pancreatic Cancer and Cachexia-Metabolic Mechanisms and Novel Insights. Nutrients 2020, 12, 1543. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, Z.; Zhang, Y.; Ni, X.; Zhang, G.; Cui, X.; Liu, M.; Xu, C.; Zhang, Q.; Zhu, H.; et al. ZIP4 Promotes Muscle Wasting and Cachexia in Mice With Orthotopic Pancreatic Tumors by Stimulating RAB27B-Regulated Release of Extracellular Vesicles From Cancer Cells. Gastroenterology 2019, 156, 722–734.e726. [Google Scholar] [CrossRef] [PubMed]
- Cousins, R.J. Gastrointestinal factors influencing zinc absorption and homeostasis. Int. J. Vitam. Nutr. Res. 2010, 80, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Biswas, A.K.; Ma, W.; Kandpal, M.; Coker, C.; Grandgenett, P.M.; Hollingsworth, M.A.; Jain, R.; Tanji, K.; Lόpez-Pintado, S.; et al. Metastatic cancers promote cachexia through ZIP14 upregulation in skeletal muscle. Nat. Med. 2018, 24, 770–781. [Google Scholar] [CrossRef]
- Shakri, A.R.; Zhong, T.J.; Ma, W.; Coker, C.; Kim, S.; Calluori, S.; Scholze, H.; Szabolcs, M.; Caffrey, T.; Grandgenett, P.M.; et al. Upregulation of ZIP14 and Altered Zinc Homeostasis in Muscles in Pancreatic Cancer Cachexia. Cancers 2019, 12, 3. [Google Scholar] [CrossRef]
- Cohen, C.W.; Fontaine, K.R.; Arend, R.C.; Alvarez, R.D.; Leath Iii, C.A.; Huh, W.K.; Bevis, K.S.; Kim, K.H.; Straughn, J.M.; Gower, B.A. A Ketogenic Diet Reduces Central Obesity and Serum Insulin in Women with Ovarian or Endometrial Cancer. J. Nutr. 2018, 148, 1253–1260. [Google Scholar] [CrossRef] [PubMed]
- Gershuni, V.M.; Yan, S.L.; Medici, V. Nutritional Ketosis for Weight Management and Reversal of Metabolic Syndrome. Curr Nutr. Rep. 2018, 7, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Bandera-Merchan, B.; Boughanem, H.; Crujeiras, A.B.; Macias-Gonzalez, M.; Tinahones, F.J. Ketotherapy as an epigenetic modifier in cancer. Rev. Endocr. Metab. Disord. 2020, 21, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.; Bonucci, A.; Maggi, E.; Corsi, M.; Businaro, R. Anti-Oxidant and Anti-Inflammatory Activity of Ketogenic Diet: New Perspectives for Neuroprotection in Alzheimer’s Disease. Antioxid 2018, 7, 63. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.G.; Veznedaroglu, E. Ketogenic Diet for Malignant Gliomas: A Review. Curr. Nutr. Rep. 2020, 9, 258–263. [Google Scholar] [CrossRef]
- Schwartz, K.A.; Noel, M.; Nikolai, M.; Chang, H.T. Investigating the Ketogenic Diet As Treatment for Primary Aggressive Brain Cancer: Challenges and Lessons Learned. Front. Nutr. 2018, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Morscher, R.J.; Aminzadeh-Gohari, S.; Feichtinger, R.G.; Mayr, J.A.; Lang, R.; Neureiter, D.; Sperl, W.; Kofler, B. Inhibition of Neuroblastoma Tumor Growth by Ketogenic Diet and/or Calorie Restriction in a CD1-Nu Mouse Model. PLoS ONE 2015, 10, e0129802. [Google Scholar] [CrossRef]
- Talib, W.H.; Mahmod, A.I.; Kamal, A.; Rashid, H.M.; Alashqar, A.M.D.; Khater, S.; Jamal, D.; Waly, M. Ketogenic Diet in Cancer Prevention and Therapy: Molecular Targets and Therapeutic Opportunities. Curr. Issues Mol. Biol. 2021, 43, 558–589. [Google Scholar] [CrossRef] [PubMed]
- Khodadadi, S.; Sobhani, N.; Mirshekar, S.; Ghiasvand, R.; Pourmasoumi, M.; Miraghajani, M.; Dehsoukhteh, S.S. Tumor Cells Growth and Survival Time with the Ketogenic Diet in Animal Models: A Systematic Review. Int. J. Prev. Med. 2017, 8, 35. [Google Scholar] [CrossRef]
- Otto, C.; Kaemmerer, U.; Illert, B.; Muehling, B.; Pfetzer, N.; Wittig, R.; Voelker, H.U.; Thiede, A.; Coy, J.F. Growth of human gastric cancer cells in nude mice is delayed by a ketogenic diet supplemented with omega-3 fatty acids and medium-chain triglycerides. BMC Cancer 2008, 8, 122. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Liu, C.; Jin, L.; Zhang, R.; Wang, T.; Wang, Q.; Chen, J.; Yang, F.; Siebert, H.-C.; Zheng, X. Ketogenic Diet Elicits Antitumor Properties through Inducing Oxidative Stress, Inhibiting MMP-9 Expression, and Rebalancing M1/M2 Tumor-Associated Macrophage Phenotype in a Mouse Model of Colon Cancer. J. Agric. Food Chem. 2020, 68, 11182–11196. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-H.; Liu, C.-L.; Chiu, W.-C.; Twu, Y.-C.; Liao, Y.-J. HMGCS2 Mediates Ketone Production and Regulates the Proliferation and Metastasis of Hepatocellular Carcinoma. Cancers 2019, 11, 1876. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.K.; Gebregiworgis, T.; Purohit, V.; Chaika, N.V.; Gunda, V.; Radhakrishnan, P.; Mehla, K.; Pipinos, I.I.; Powers, R.; Yu, F.; et al. Metabolic reprogramming induced by ketone bodies diminishes pancreatic cancer cachexia. Cancer Metab. 2014, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Fineberg, S.; Pearlman, A.; Feinman, R.D.; Fine, E.J. The effect of a ketogenic diet and synergy with rapamycin in a mouse model of breast cancer. PLoS ONE 2020, 15, e0233662. [Google Scholar] [CrossRef] [PubMed]
- Abdelwahab, M.G.; Fenton, K.E.; Preul, M.C.; Rho, J.M.; Lynch, A.; Stafford, P.; Scheck, A.C. The Ketogenic Diet Is an Effective Adjuvant to Radiation Therapy for the Treatment of Malignant Glioma. PLoS ONE 2012, 7, e36197. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Tonouchi, H.; Sasayama, A.; Ashida, K. A Ketogenic Formula Prevents Tumor Progression and Cancer Cachexia by Attenuating Systemic Inflammation in Colon 26 Tumor-Bearing Mice. Nutrients 2018, 10, 206. [Google Scholar] [CrossRef]
- Rieger, J.; Bähr, O.; Maurer, G.D.; Hattingen, E.; Franz, K.; Brucker, D.; Walenta, S.; Kämmerer, U.; Coy, J.F.; Weller, M.; et al. ERGO: A pilot study of ketogenic diet in recurrent glioblastoma. Int. J. Oncol. 2014, 44, 1843–1852. [Google Scholar] [CrossRef]
- Mukherjee, P.; Augur, Z.M.; Li, M.; Hill, C.; Greenwood, B.; Domin, M.A.; Kondakci, G.; Narain, N.R.; Kiebish, M.A.; Bronson, R.T.; et al. Therapeutic benefit of combining calorie-restricted ketogenic diet and glutamine targeting in late-stage experimental glioblastoma. Commun. Biol. 2019, 2, 200. [Google Scholar] [CrossRef]
- Aminzadeh-Gohari, S.; Feichtinger, R.G.; Vidali, S.; Locker, F.; Rutherford, T.; O’Donnel, M.; Stöger-Kleiber, A.; Mayr, J.A.; Sperl, W.; Kofler, B. A ketogenic diet supplemented with medium-chain triglycerides enhances the anti-tumor and anti-angiogenic efficacy of chemotherapy on neuroblastoma xenografts in a CD1-nu mouse model. Oncotarget 2017, 8, 64728–64744. [Google Scholar] [CrossRef]
- Talib, W.H. A ketogenic diet combined with melatonin overcomes cisplatin and vincristine drug resistance in breast carcinoma syngraft. Nutrition 2020, 72, 110659. [Google Scholar] [CrossRef]
- Aggarwal, A.; Yuan, Z.; Barletta, J.A.; Lorch, J.H.; Nehs, M.A. Ketogenic diet combined with antioxidant N-acetylcysteine inhibits tumor growth in a mouse model of anaplastic thyroid cancer. Surgery 2020, 167, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, B.D.; Pauli, C.; Du, X.; Wang, D.G.; Li, X.; Wu, D.; Amadiume, S.C.; Goncalves, M.D.; Hodakoski, C.; Lundquist, M.R.; et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 2018, 560, 499–503. [Google Scholar] [CrossRef]
- Allen, B.G.; Bhatia, S.K.; Buatti, J.M.; Brandt, K.E.; Lindholm, K.E.; Button, A.M.; Szweda, L.I.; Smith, B.J.; Spitz, D.R.; Fath, M.A. Ketogenic Diets Enhance Oxidative Stress and Radio-Chemo-Therapy Responses in Lung Cancer Xenografts. Clin. Cancer Res. 2013, 19, 3905–3913. [Google Scholar] [CrossRef] [PubMed]
- Zahra, A.; Fath, M.A.; Opat, E.; Mapuskar, K.A.; Bhatia, S.K.; Ma, D.C.; Rodman, S.N.; Snyders, T.P.; Chenard, C.A.; Eichenberger-Gilmore, J.M.; et al. Consuming a Ketogenic Diet while Receiving Radiation and Chemotherapy for Locally Advanced Lung and Pancreatic Cancer: The University of Iowa Experience of Two Phase I Clinical Trials. Radiat. Res. 2017, 187, 743–754. [Google Scholar] [CrossRef]
- Ferrere, G.; Tidjani Alou, M.; Liu, P.; Goubet, A.G.; Fidelle, M.; Kepp, O.; Durand, S.; Iebba, V.; Fluckiger, A.; Daillere, R.; et al. Ketogenic diet and ketone bodies enhance the anticancer effects of PD-1 blockade. JCI Insight 2021, 6, e145207. [Google Scholar] [CrossRef]
- Hagihara, K.; Kajimoto, K.; Osaga, S.; Nagai, N.; Shimosegawa, E.; Nakata, H.; Saito, H.; Nakano, M.; Takeuchi, M.; Kanki, H.; et al. Promising Effect of a New Ketogenic Diet Regimen in Patients with Advanced Cancer. Nutrients 2020, 12, 1473. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.M.; Yun, B.; Kim, M.; Song, M.; Kim, Y.-h.; Lee, S.H.; Lee, H.; Lee, S.M.; Lee, S.-M. Postoperative serum metabolites of patients on a low carbohydrate ketogenic diet after pancreatectomy for pancreatobiliary cancer: A nontargeted metabolomics pilot study. Sci. Rep. 2019, 9, 16820. [Google Scholar] [CrossRef]
- Poff, A.M.; Ari, C.; Seyfried, T.N.; D’Agostino, D.P. The Ketogenic Diet and Hyperbaric Oxygen Therapy Prolong Survival in Mice with Systemic Metastatic Cancer. PLoS ONE 2013, 8, e65522. [Google Scholar] [CrossRef]
- Oliveira, C.L.P.; Mattingly, S.; Schirrmacher, R.; Sawyer, M.B.; Fine, E.J.; Prado, C.M. A Nutritional Perspective of Ketogenic Diet in Cancer: A Narrative Review. J. Acad. Nutr. Diet. 2018, 118, 668–688. [Google Scholar] [CrossRef]
- Klein, P.; Tyrlikova, I.; Zuccoli, G.; Tyrlik, A.; Maroon, J.C. Treatment of glioblastoma multiforme with “classic” 4:1 ketogenic diet total meal replacement. Cancer Metab. 2020, 8, 24. [Google Scholar] [CrossRef]
- Mitchell, T.; Clarke, L.; Goldberg, A.; Bishop, K.S. Pancreatic Cancer Cachexia: The Role of Nutritional Interventions. Healthcare 2019, 7, 89. [Google Scholar] [CrossRef]
- Paoli, A.; Cancellara, P.; Pompei, P.; Moro, T. Ketogenic Diet and Skeletal Muscle Hypertrophy: A Frenemy Relationship? J. Hum. Kinet. 2019, 68, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.-Y.; Linden, M.A.; Fuller, S.E.; Goldsmith, F.R.; Simon, J.; Batdorf, H.M.; Scott, M.C.; Essajee, N.M.; Brown, J.M.; Noland, R.C. Combined effects of a ketogenic diet and exercise training alter mitochondrial and peroxisomal substrate oxidative capacity in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E1053–E1067. [Google Scholar] [CrossRef]
- Wallace, M.A.; Aguirre, N.W.; Marcotte, G.R.; Marshall, A.G.; Baehr, L.M.; Hughes, D.C.; Hamilton, K.L.; Roberts, M.N.; Lopez-Dominguez, J.A.; Miller, B.F.; et al. The ketogenic diet preserves skeletal muscle with aging in mice. Aging Cell 2021, 20, e13322. [Google Scholar] [CrossRef] [PubMed]
- Nakao, R.; Abe, T.; Yamamoto, S.; Oishi, K. Ketogenic diet induces skeletal muscle atrophy via reducing muscle protein synthesis and possibly activating proteolysis in mice. Sci. Rep. 2019, 9, 19652. [Google Scholar] [CrossRef] [PubMed]
- Branco, A.F.; Ferreira, A.; Simões, R.F.; Magalhães-Novais, S.; Zehowski, C.; Cope, E.; Silva, A.M.; Pereira, D.; Sardão, V.A.; Cunha-Oliveira, T. Ketogenic diets: From cancer to mitochondrial diseases and beyond. Eur. J. Clin. Investig. 2016, 46, 285–298. [Google Scholar] [CrossRef]
- Mann, K.M.; Ying, H.; Juan, J.; Jenkins, N.A.; Copeland, N.G. KRAS-related proteins in pancreatic cancer. Pharm. Ther. 2016, 168, 29–42. [Google Scholar] [CrossRef]
- Hu, M.; Chen, X.; Ma, L.; Ma, Y.; Li, Y.; Song, H.; Xu, J.; Zhou, L.; Li, X.; Jiang, Y.; et al. AMPK Inhibition Suppresses the Malignant Phenotype of Pancreatic Cancer Cells in Part by Attenuating Aerobic Glycolysis. J. Cancer 2019, 10, 1870–1878. [Google Scholar] [CrossRef]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef]
- Chan, A.K.C.; Bruce, J.I.E.; Siriwardena, A.K. Glucose metabolic phenotype of pancreatic cancer. World J. Gastroenterol. 2016, 22, 3471–3485. [Google Scholar] [CrossRef] [PubMed]
- Madden, S.K.; de Araujo, A.D.; Gerhardt, M.; Fairlie, D.P.; Mason, J.M. Taking the Myc out of cancer: Toward therapeutic strategies to directly inhibit c-Myc. Mol. Cancer 2021, 20, 3. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.; Bollig, A.; Wu, J.; Liao, D.J. Gene expression profiles in primary pancreatic tumors and metastatic lesions of Ela-c-myc transgenic mice. Mol. Cancer 2008, 7, 11. [Google Scholar] [CrossRef]
- Azoitei, N.; Becher, A.; Steinestel, K.; Rouhi, A.; Diepold, K.; Genze, F.; Simmet, T.; Seufferlein, T. PKM2 promotes tumor angiogenesis by regulating HIF-1α through NF-κB activation. Mol. Cancer 2016, 15, 3. [Google Scholar] [CrossRef] [PubMed]
- Barrea, L.; Caprio, M.; Tuccinardi, D.; Moriconi, E.; Di Renzo, L.; Muscogiuri, G.; Colao, A.; Savastano, S.; Obesity Programs of nutrition, Education, Research and Assessment (OPERA) Group. Could ketogenic diet “starve” cancer? Emerging evidence. Crit. Rev. Food Sci. Nutr. 2020, 1–22. [Google Scholar] [CrossRef]
- Kumar, S.; Behl, T.; Sachdeva, M.; Sehgal, A.; Kumari, S.; Kumar, A.; Kaur, G.; Yadav, H.N.; Bungau, S. Implicating the effect of ketogenic diet as a preventive measure to obesity and diabetes mellitus. Life Sci. 2021, 264, 118661. [Google Scholar] [CrossRef]
- O’Flanagan, C.H.; Smith, L.A.; McDonell, S.B.; Hursting, S.D. When less may be more: Calorie restriction and response to cancer therapy. BMC Med. 2017, 15, 106. [Google Scholar] [CrossRef]
- Karnevi, E.; Said, K.; Andersson, R.; Rosendahl, A.H. Metformin-mediated growth inhibition involves suppression of the IGF-I receptor signalling pathway in human pancreatic cancer cells. BMC Cancer 2013, 13, 235. [Google Scholar] [CrossRef]
- Luo, Z.; Zang, M.; Guo, W. AMPK as a metabolic tumor suppressor: Control of metabolism and cell growth. Future Oncol. 2010, 6, 457–470. [Google Scholar] [CrossRef]
- Weber, D.D.; Aminzadeh-Gohari, S.; Tulipan, J.; Catalano, L.; Feichtinger, R.G.; Kofler, B. Ketogenic diet in the treatment of cancer—Where do we stand? Mol. Metab. 2020, 33, 102–121. [Google Scholar] [CrossRef]
- Zhou, W.; Mukherjee, P.; Kiebish, M.A.; Markis, W.T.; Mantis, J.G.; Seyfried, T.N. The calorically restricted ketogenic diet, an effective alternative therapy for malignant brain cancer. Nutr. Metab. 2007, 4, 5. [Google Scholar] [CrossRef]
- Römer, M.; Dörfler, J.; Huebner, J. The use of ketogenic diets in cancer patients: A systematic review. Clin. Exp. Med. 2021, 13, 1–36. [Google Scholar] [CrossRef]
- Gray, A.; Dang, B.N.; Moore, T.B.; Clemens, R.; Pressman, P. A review of nutrition and dietary interventions in oncology. Sage Open Med. 2020, 8, 2050312120926877. [Google Scholar] [CrossRef] [PubMed]
- Hao, G.-W.; Chen, Y.-S.; He, D.-M.; Wang, H.-Y.; Wu, G.-H.; Zhang, B. Growth of human colon cancer cells in nude mice is delayed by ketogenic diet with or without omega-3 fatty acids and medium-chain triglycerides. Asian Pac. J. Cancer Prev. 2015, 16, 2061–2068. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.T.; Olson, L.K.; Schwartz, K.A. Ketolytic and glycolytic enzymatic expression profiles in malignant gliomas: Implication for ketogenic diet therapy. Nutr. Metab. 2013, 10, 47. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.-H.; Wang, W.-Q.; Han, X.; Gao, H.-L.; Li, T.-J.; Xu, S.-S.; Li, S.; Xu, H.-X.; Li, H.; Ye, L.-Y.; et al. Advances on diagnostic biomarkers of pancreatic ductal adenocarcinoma: A systems biology perspective. Comput. Struct. Biotechnol. J. 2020, 18, 3606–3614. [Google Scholar] [CrossRef]
- Klement, R.J. The emerging role of ketogenic diets in cancer treatment. Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 129–134. [Google Scholar] [CrossRef]
- Evans, M.; Cogan, K.E.; Egan, B. Metabolism of ketone bodies during exercise and training: Physiological basis for exogenous supplementation. J. Physiol. 2017, 595, 2857–2871. [Google Scholar] [CrossRef]
- Allen, B.G.; Bhatia, S.K.; Anderson, C.M.; Eichenberger-Gilmore, J.M.; Sibenaller, Z.A.; Mapuskar, K.A.; Schoenfeld, J.D.; Buatti, J.M.; Spitz, D.R.; Fath, M.A. Ketogenic diets as an adjuvant cancer therapy: History and potential mechanism. Redox Biol. 2014, 2, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Miller, V.J.; LaFountain, R.A.; Barnhart, E.; Sapper, T.S.; Short, J.; Arnold, W.D.; Hyde, P.N.; Crabtree, C.D.; Kackley, M.L.; Kraemer, W.J.; et al. A ketogenic diet combined with exercise alters mitochondrial function in human skeletal muscle while improving metabolic health. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E995–E1007. [Google Scholar] [CrossRef]
- Liu, K.A.; Lashinger, L.M.; Rasmussen, A.J.; Hursting, S.D. Leucine supplementation differentially enhances pancreatic cancer growth in lean and overweight mice. Cancer Metab. 2014, 2, 6. [Google Scholar] [CrossRef]
- Roberts, M.N.; Wallace, M.A.; Tomilov, A.A.; Zhou, Z.; Marcotte, G.R.; Tran, D.; Perez, G.; Gutierrez-Casado, E.; Koike, S.; Knotts, T.A.; et al. A Ketogenic Diet Extends Longevity and Healthspan in Adult Mice. Cell Metab. 2017, 26, 539–546.e535. [Google Scholar] [CrossRef]
- Douris, N.; Melman, T.; Pecherer, J.M.; Pissios, P.; Flier, J.S.; Cantley, L.C.; Locasale, J.W.; Maratos-Flier, E. Adaptive changes in amino acid metabolism permit normal longevity in mice consuming a low-carbohydrate ketogenic diet. Biochim. Biophys. Acta 2015, 1852, 2056–2065. [Google Scholar] [CrossRef] [PubMed]
- Koutnik, A.P.; D’Agostino, D.P.; Egan, B. Anticatabolic Effects of Ketone Bodies in Skeletal Muscle. Trends Endocrinol. Metab. 2019, 30, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, H.H.; Rittig, N.; Johannsen, M.; Møller, A.B.; Jørgensen, J.O.; Jessen, N.; Møller, N. Effects of 3-hydroxybutyrate and free fatty acids on muscle protein kinetics and signaling during LPS-induced inflammation in humans: Anticatabolic impact of ketone bodies. Am. J. Clin. Nutr. 2018, 108, 857–867. [Google Scholar] [CrossRef]
- Ogura, Y.; Kakehashi, C.; Yoshihara, T.; Kurosaka, M.; Kakigi, R.; Higashida, K.; Fujiwara, S.-E.; Akema, T.; Funabashi, T. Ketogenic diet feeding improves aerobic metabolism property in extensor digitorum longus muscle of sedentary male rats. PLoS ONE 2020, 15, e0241382. [Google Scholar] [CrossRef] [PubMed]
- Kinnaird, A.; Zhao, S.; Wellen, K.E.; Michelakis, E.D. Metabolic control of epigenetics in cancer. Nat. Rev. Cancer 2016, 16, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Paradise, B.D.; Barham, W.; Fernandez-Zapico, M.E. Targeting Epigenetic Aberrations in Pancreatic Cancer, a New Path to Improve Patient Outcomes? Cancers 2018, 10, 128. [Google Scholar] [CrossRef]
- Noberini, R.; Restellini, C.; Savoia, E.O.; Raimondi, F.; Ghiani, L.; Jodice, M.G.; Bertalot, G.; Bonizzi, G.; Capra, M.; Maffini, F.A.; et al. Profiling of Epigenetic Features in Clinical Samples Reveals Novel Widespread Changes in Cancer. Cancers 2019, 11, 723. [Google Scholar] [CrossRef]
- Carr, R.M.; Enriquez-Hesles, E.; Olson, R.L.; Jatoi, A.; Doles, J.; Fernandez-Zapico, M.E. Epigenetics of cancer-associated muscle catabolism. Epigenomics 2017, 9, 1259–1265. [Google Scholar] [CrossRef]
- Bishop, K.S.; Ferguson, L.R. The interaction between epigenetics, nutrition and the development of cancer. Nutrients 2015, 7, 922–947. [Google Scholar] [CrossRef]
- Alamdari, N.; Aversa, Z.; Castillero, E.; Hasselgren, P.O. Acetylation and deacetylation--novel factors in muscle wasting. Metabolism 2013, 62, 1–11. [Google Scholar] [CrossRef]
- Juiz, N.A.; Iovanna, J.; Dusetti, N. Pancreatic Cancer Heterogeneity Can Be Explained Beyond the Genome. Front. Oncol. 2019, 9, 246. [Google Scholar] [CrossRef]
- Benjamin, J.S.; Pilarowski, G.O.; Carosso, G.A.; Zhang, L.; Huso, D.L.; Goff, L.A.; Vernon, H.J.; Hansen, K.D.; Bjornsson, H.T. A ketogenic diet rescues hippocampal memory defects in a mouse model of Kabuki syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, 125–130. [Google Scholar] [CrossRef]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef]
- Shirahata, M.; Tang, W.Y.; Kostuk, E.W. A Short-Term Fasting in Neonates Induces Breathing Instability and Epigenetic Modification in the Carotid Body. Adv. Exp. Med. Biol. 2015, 860, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Zhang, D.; Chung, D.; Tang, Z.; Huang, H.; Dai, L.; Qi, S.; Li, J.; Colak, G.; Chen, Y.; et al. Metabolic Regulation of Gene Expression by Histone Lysine beta-Hydroxybutyrylation. Mol. Cell 2016, 62, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; He, X.; Luan, G.; Li, T. Role of DNA Methylation and Adenosine in Ketogenic Diet for Pharmacoresistant Epilepsy: Focus on Epileptogenesis and Associated Comorbidities. Front. Neurol. 2019, 10, 119. [Google Scholar] [CrossRef] [PubMed]
- Preston, J.; Stylianou, J.; Zeng, Q.; Glover, S.; Scheck, A.C.; Woolf, E.; O’Neill, K.; Syed, N. The ketogenic diet induces epigenetic changes that play key roles in tumour development. Neuro-Oncology 2017, 19, i28. [Google Scholar] [CrossRef][Green Version]
- Kerk, S.A.; Papagiannakopoulos, T.; Shah, Y.M.; Lyssiotis, C.A. Metabolic networks in mutant KRAS-driven tumours: Tissue specificities and the microenvironment. Nat. Rev. Cancer 2021, 21, 510–525. [Google Scholar] [CrossRef]
- Chandra, V.; McAllister, F. Therapeutic potential of microbial modulation in pancreatic cancer. Gut 2021, 70. [Google Scholar] [CrossRef]
- Wei, M.-Y.; Shi, S.; Liang, C.; Meng, Q.-C.; Hua, J.; Zhang, Y.-Y.; Liu, J.; Zhang, B.; Xu, J.; Yu, X.-J. The microbiota and microbiome in pancreatic cancer: More influential than expected. Mol. Cancer 2019, 18, 97. [Google Scholar] [CrossRef]
- Ren, Z.; Jiang, J.; Xie, H.; Li, A.; Lu, H.; Xu, S.; Zhou, L.; Zhang, H.; Cui, G.; Chen, X.; et al. Gut microbial profile analysis by MiSeq sequencing of pancreatic carcinoma patients in China. Oncotarget 2017, 8, 95176–95191. [Google Scholar] [CrossRef]
- Thomas, R.M.; Gharaibeh, R.Z.; Gauthier, J.; Beveridge, M.; Pope, J.L.; Guijarro, M.V.; Yu, Q.; He, Z.; Ohland, C.; Newsome, R.; et al. Intestinal microbiota enhances pancreatic carcinogenesis in preclinical models. Carcinogenesis 2018, 39, 1068–1078. [Google Scholar] [CrossRef]
- Riquelme, E.; Zhang, Y.; Zhang, L.; Montiel, M.; Zoltan, M.; Dong, W.; Quesada, P.; Sahin, I.; Chandra, V.; San Lucas, A.; et al. Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer Outcomes. Cell 2019, 178, 795–806 e712. [Google Scholar] [CrossRef] [PubMed]
- Wyart, E.; Reano, S.; Hsu, M.Y.; Longo, D.L.; Li, M.; Hirsch, E.; Filigheddu, N.; Ghigo, A.; Riganti, C.; Porporato, P.E. Metabolic Alterations in a Slow-Paced Model of Pancreatic Cancer-Induced Wasting. Oxid. Med. Cell. Longev. 2018, 2018, 6419805. [Google Scholar] [CrossRef]
- Olson, C.A.; Vuong, H.E.; Yano, J.M.; Liang, Q.Y.; Nusbaum, D.J.; Hsiao, E.Y. The Gut Microbiota Mediates the Anti-Seizure Effects of the Ketogenic Diet. Cell 2018, 173, 1728–1741.e1713. [Google Scholar] [CrossRef] [PubMed]
- Klement, R.J. Restricting carbohydrates to fight head and neck cancer—Is this realistic? Cancer Biol. Med. 2014, 11, 145–161. [Google Scholar] [CrossRef]
- Ang, Q.Y.; Alexander, M.; Newman, J.C.; Tian, Y.; Cai, J.; Upadhyay, V.; Turnbaugh, J.A.; Verdin, E.; Hall, K.D.; Leibel, R.L.; et al. Ketogenic Diets Alter the Gut Microbiome Resulting in Decreased Intestinal Th17 Cells. Cell 2020, 181, 1263–1275.e1216. [Google Scholar] [CrossRef] [PubMed]
- Ni, F.F.; Li, C.R.; Liao, J.X.; Wang, G.B.; Lin, S.F.; Xia, Y.; Wen, J.L. The effects of ketogenic diet on the Th17/Treg cells imbalance in patients with intractable childhood epilepsy. Seizure 2016, 38, 17–22. [Google Scholar] [CrossRef]
- Yu, Q.; Jobin, C.; Thomas, R.M. Implications of the microbiome in the development and treatment of pancreatic cancer: Thinking outside of the box by looking inside the gut. Neoplasia 2021, 23, 246–256. [Google Scholar] [CrossRef]
- Bindels, L.B.; Neyrinck, A.M.; Loumaye, A.; Catry, E.; Walgrave, H.; Cherbuy, C.; Leclercq, S.; Hul, M.V.; Plovier, H.; Pachikian, B.; et al. Increased gut permeability in cancer cachexia: Mechanisms and clinical relevance. Oncotarget 2018, 9, 18224–18238. [Google Scholar] [CrossRef]
- Klement, R.J. Beneficial effects of ketogenic diets for cancer patients: A realist review with focus on evidence and confirmation. Med. Oncol. 2017, 34, 132. [Google Scholar] [CrossRef]
- Murray, A.J.; Knight, N.S.; Cole, M.A.; Cochlin, L.E.; Carter, E.; Tchabanenko, K.; Pichulik, T.; Gulston, M.K.; Atherton, H.J.; Schroeder, M.A.; et al. Novel ketone diet enhances physical and cognitive performance. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2016, 30, 4021–4032. [Google Scholar] [CrossRef] [PubMed]
- Clarke, K.; Tchabanenko, K.; Pawlosky, R.; Carter, E.; Knight, N.S.; Murray, A.J.; Cochlin, L.E.; King, M.T.; Wong, A.W.; Roberts, A.; et al. Oral 28-day and developmental toxicity studies of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate. Regul. Toxicol. Pharm. 2012, 63, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Clarke, K.; Tchabanenko, K.; Pawlosky, R.; Carter, E.; Todd King, M.; Musa-Veloso, K.; Ho, M.; Roberts, A.; Robertson, J.; Vanitallie, T.B.; et al. Kinetics, safety and tolerability of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate in healthy adult subjects. Regul. Toxicol. Pharm. 2012, 63, 401–408. [Google Scholar] [CrossRef]
- Pezzilli, R.; Caccialanza, R.; Capurso, G.; Brunetti, O.; Milella, M.; Falconi, M. Pancreatic Enzyme Replacement Therapy in Pancreatic Cancer. Cancers 2020, 12, 275. [Google Scholar] [CrossRef] [PubMed]
- Hendifar, A.E.; Petzel, M.Q.B.; Zimmers, T.A.; Denlinger, C.S.; Matrisian, L.M.; Picozzi, V.J.; Rahib, L. Pancreas Cancer-Associated Weight Loss. Oncologist 2019, 24, 691–701. [Google Scholar] [CrossRef]
- Chung, H.-Y.; Park, Y.K. Rationale, Feasibility and Acceptability of Ketogenic Diet for Cancer Treatment. J. Cancer Prev. 2017, 22, 127–134. [Google Scholar] [CrossRef]
- Kiriukova, M.; de la Iglesia Garcia, D.; Panic, N.; Bozhychko, M.; Avci, B.; Maisonneuve, P.; de-Madaria, E.; Capurso, G.; Sandru, V. Pancreatic Cancer Malnutrition and Pancreatic Exocrine Insufficiency in the Course of Chemotherapy in Unresectable Pancreatic Cancer. Front. Med. 2020, 7, 495. [Google Scholar] [CrossRef]
- Brennan, G.T.; Saif, M.W. Pancreatic Enzyme Replacement Therapy: A Concise Review. JOP J. Pancreas 2019, 20, 121–125. [Google Scholar]
- Dominguez-Muñoz, J.E. Management of pancreatic exocrine insufficiency. Curr. Opin. Gastroenterol. 2019, 35, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Vujasinovic, M.; Valente, R.; Del Chiaro, M.; Permert, J.; Löhr, J.M. Pancreatic Exocrine Insufficiency in Pancreatic Cancer. Nutrients 2017, 9, 183. [Google Scholar] [CrossRef]
- Aronica, L.; Volek, J.; Poff, A.; D’Agostino, D.P. Genetic variants for personalised management of very low carbohydrate ketogenic diets. BMJ Nutr. Prev. Health 2020, 3, 363–373. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cortez, N.E.; Mackenzie, G.G. Ketogenic Diets in Pancreatic Cancer and Associated Cachexia: Cellular Mechanisms and Clinical Perspectives. Nutrients 2021, 13, 3202. https://doi.org/10.3390/nu13093202
Cortez NE, Mackenzie GG. Ketogenic Diets in Pancreatic Cancer and Associated Cachexia: Cellular Mechanisms and Clinical Perspectives. Nutrients. 2021; 13(9):3202. https://doi.org/10.3390/nu13093202
Chicago/Turabian StyleCortez, Natalia E., and Gerardo G. Mackenzie. 2021. "Ketogenic Diets in Pancreatic Cancer and Associated Cachexia: Cellular Mechanisms and Clinical Perspectives" Nutrients 13, no. 9: 3202. https://doi.org/10.3390/nu13093202
APA StyleCortez, N. E., & Mackenzie, G. G. (2021). Ketogenic Diets in Pancreatic Cancer and Associated Cachexia: Cellular Mechanisms and Clinical Perspectives. Nutrients, 13(9), 3202. https://doi.org/10.3390/nu13093202