
Multifactorial Basis and Therapeutic Strategies in Metabolism-Related Diseases

 ,
,  , , and
, , and
Abstract
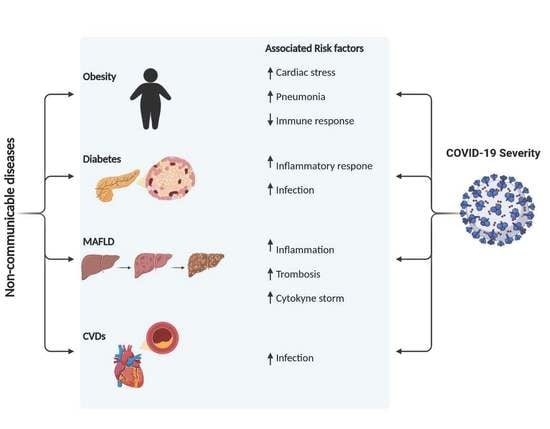
1. Introduction
2. Obesity
2.1. Management of Obesity
2.1.1. Lifestyle Interventions
2.1.2. Pharmacotherapy
2.1.3. Bariatric Surgery
2.1.4. New Drugs and Strategies
2.1.5. COVID-19 and Obesity
3. Diabetes Mellitus
3.1. Type 1 Diabetes Mellitus
3.2. Type 2 Diabetes Mellitus
3.3. Gestational Diabetes Mellitus
3.4. Maturity Onset Diabetes of the Young
3.5. Other Types of Diabetes
3.6. Management of Diabetes Mellitus
3.6.1. Dual Therapies
3.6.2. T1D Therapies
3.6.3. T2D Therapies
3.7. COVID-19 and Diabetes
4. Metabolic Associated Fatty Liver Disease (MAFLD)
4.1. Management of MAFLD
4.2. Anti-Diabetic Drugs
4.3. Antilipidemic Agents
4.4. Antioxidant Agents
4.5. Others
4.6. COVID-19 and MAFLD
5. Cardiovascular Diseases (CVDs)
5.1. Management of CVDs
5.1.1. Primary and Secondary Prevention Strategies
5.1.2. Potential Therapeutic Targets
5.1.3. Emerging Strategies
5.2. COVID-19 and CVDs
6. Conclusions
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Available online: www.who.int/news-room/fact-sheets/ (accessed on 10 October 2020).
- World Health Organization. Global Health Estimates 2016, Deaths by Cause, Age, Sex, by Country and by Region, 2000–2016. Available online: https://www.who.int/healthinfo/global_burden_disease/GHE2016_Deaths_WBInc_2000_2016.xls (accessed on 12 August 2020).
- Sohail, M.U.; Yassine, H.M.; Sohail, A.; Thani, A.A. Impact of Physical Exercise on Gut Microbiome Inflammation the Pathobiology of Metabolic, Disorders. Rev. Diabet Stud. 2019, 15, 35–48. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, M.S. Pathogenesis of Nonalcoholic Steatohepatitis and Hormone-Based Therapeutic Approaches. Front. Endocrinol. 2018, 9, 485. [Google Scholar] [CrossRef] [PubMed]
- NCD Risk Factor Collaboration (NCD-RisC). Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016, a pooled analysis of 2416 population-based measurement studies in 128.9 million children, adolescents, and adults. Lancet 2017, 390, 2627–2642. [Google Scholar] [CrossRef]
- Collaboration NCDRF: Worldwide trends in diabetes since 1980, a pooled analysis of 751 population-based studies with 4.4 million participants. Lancet 2016, 387, 1513–1530. [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Characteristics of COVID-19 Patients Dying in Italy. Available online: https://www.epicentro.iss.it/en/coronavirus/bollettino/ReportCOVID-2019_29_april_2020.pdf (accessed on 28 January 2021).
- III IdSC: Informe COVID-19 nº 28. 04 de Mayo de 2020. Informe Sobre la Situación de COVID-19 en España. 2020. Available online: https://www.isciii.es/QueHacemos/Servicios/VigilanciaSaludPublicaRENAVE/EnfermedadesTransmisibles/Paginas/-COVID-19.-Informes-previos.aspx (accessed on 28 January 2021).
- Epidemiology Working Group for NCIP Epidemic Response, Chinese Center for Disease Control and Prevention. The epidemiological characteristics of an outbreak of 2019 novel coronavirus diseases (COVID-19) in China. Zhonghua Liu Xing Bing Xue Za Zhi 2020, 41, 145–151. [Google Scholar]
- Richardson, S.; Hirsch, J.S.; Narasimhan, M.; Crawford, J.M.; McGinn, T.; Davidson, K.W.; the Northwell COVID-19 Research Consortium; Barnaby, D.P.; Becker, L.B.; Chelico, J.D.; et al. Presenting Characteristics, Comorbidities, and Outcomes Among 5700 Patients Hospitalized With COVID-19 in the New York City Area. JAMA 2020, 323, 2052–2059. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.D.; Meng, K.; Guan, H.Q.; Leng, L.; Zhu, R.R.; Wang, B.Y.; He, M.A.; Cheng, L.X.; Huang, K.; Zeng, Q.T. [Clinical characteristics outcomes of 112 cardiovascular disease patients infected by 2019-nCoV]. Zhonghua Xin Xue Guan Bing Za Zhi 2020, 48, 450–545. [Google Scholar] [PubMed]
- Kluge, H.H.P.; Wickramasinghe, K.; Rippin, H.L.; Mendes, R.; Peters, D.H.; Kontsevaya, A.; Breda, J. Prevention control of non-communicable diseases in the COVID-19, response. Lancet 2020, 395, 1678–1680. [Google Scholar] [CrossRef]
- Kushner, R.F. Weight Loss Strategies for Treatment of Obesity: Lifestyle Management and Pharmacotherapy. Prog. Cardiovasc. Dis. 2018, 61, 246–252. [Google Scholar] [CrossRef]
- Nathan, D.M.; Buse, J.B.; Davidson, M.B.; Ferrannini, E.; Holman, R.R.; Sherwin, R.; Zinman, B.; American Diabetes Association and the European Association for the Study of Diabetes. Medical management of hyperglycemia in type 2 diabetes: A consensus algorithm for the initiation and adjustment of therapy: A consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2009, 32, 193–203. [Google Scholar] [CrossRef]
- Suliburska, J.; Skrypnik, K.; Szulinska, M.; Kupsz, J.; Markuszewski, L.; Bogdanski, P. Diuretics, Ca-Antagonists, and Angiotensin-Converting Enzyme Inhibitors Affect Zinc Status in Hypertensive Patients on Monotherapy: A Randomized Trial. Nutrients 2018, 10, 1284. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Scotti, L.; Monteiro, A.F.M.; de Oliveira Viana, J.; Mendonca Junior, F.J.B.; Ishiki, H.M.; Tchouboun, E.N.; Santos, R.; Scotti, M.T. Multi-Target Drugs Against Metabolic Disorders. Endocr. Metab. Immune Disord. Drug. Targets 2019, 19, 402–418. [Google Scholar] [CrossRef] [PubMed]
- De Lorenzo, A.; Romano, L.; Di Renzo, L.; Di Lorenzo, N.; Cenname, G.; Gualtieri, P. Obesity: A preventable, treatable, but relapsing disease. Nutrition 2020, 71, 110615. [Google Scholar] [CrossRef] [PubMed]
- Chooi, Y.C.; Ding, C.; Magkos, F. The epidemiology of obesity. Metabolism 2019, 92, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.B. Obesity and mortality: Watch your waist, not just your weight. Arch. Intern. Med. 2007, 167, 875–876. [Google Scholar] [CrossRef] [PubMed]
- Di Bonito, P.; Pacifico, L.; Licenziati, M.R.; Maffeis, C.; Morandi, A.; Manco, M.; Del Giudice, E.M.; Di Sessa, A.; Campana, G.; Moio, N.; et al. Elevated blood pressure, cardiometabolic risk and target organ damage in youth with overweight and obesity. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 1840–1847. [Google Scholar] [CrossRef]
- Di Bonito, P.; Pacifico, L.; Chiesa, C.; Valerio, G.; Miraglia Del Giudice, E.; Maffeis, C.; Morandi, A.; Invitti, C.; Licenziati, M.R.; Loche, S.; et al. Impaired fasting glucose and impaired glucose tolerance in children and adolescents with overweight/obesity. J. Endocrinol. Investig. 2017, 40, 409–416. [Google Scholar] [CrossRef]
- Di Bonito, P.; Valerio, G.; Licenziati, M.R.; Campana, G.; Del Giudice, E.M.; Di Sessa, A.; Morandi, A.; Maffeis, C.; Chiesa, C.; Pacifico, L.; et al. Uric acid, impaired fasting glucose and impaired glucose tolerance in youth with overweight and obesity. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 675–680. [Google Scholar] [CrossRef]
- Simmonds, M.; Burch, J.; Llewellyn, A.; Griffiths, C.; Yang, H.; Owen, C.; Duffy, S.; Woolacott, N. The use of measures of obesity in childhood for predicting obesity and the development of obesity-related diseases in adulthood: A systematic review and meta-analysis. Health Technol. Assess 2015, 19, 1–336. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Yaniv, G.; Levine, H.; Leiba, A.; Goldberger, N.; Derazne, E.; Ben-Ami Shor, D.; Tzur, D.; Afek, A.; Shamiss, A.; et al. Body-Mass Index in 2.3 Million Adolescents and Cardiovascular Death in Adulthood. N. Engl. J. Med. 2016, 374, 2430–2440. [Google Scholar] [CrossRef] [PubMed]
- Church, T.S.; Thomas, D.M.; Tudor-Locke, C.; Katzmarzyk, P.T.; Earnest, C.P.; Rodarte, R.Q.; Martin, C.K.; Blair, S.N.; Bouchard, C. Trends over 5 decades in U.S. occupation-related physical activity and their associations with obesity. PLoS ONE 2011, 6, e19657. [Google Scholar] [CrossRef] [PubMed]
- Swinburn, B.A.; Sacks, G.; Hall, K.D.; McPherson, K.; Finegood, D.T.; Moodie, M.L.; Gortmaker, S.L. The global obesity pandemic: Shaped by global drivers and local environments. Lancet 2011, 378, 804–814. [Google Scholar] [CrossRef]
- Janesick, A.S.; Shioda, T.; Blumberg, B. Transgenerational inheritance of prenatal obesogen exposure. Mol. Cell Endocrinol. 2014, 398, 31–35. [Google Scholar] [CrossRef]
- Bergin, J.E.; Neale, M.C.; Eaves, L.J.; Martin, N.G.; Heath, A.C.; Maes, H.H. Genetic and environmental transmission of body mass index fluctuation. Behav. Genet. 2012, 42, 867–874. [Google Scholar] [CrossRef][Green Version]
- Elks, C.E.; den Hoed, M.; Zhao, J.H.; Sharp, S.J.; Wareham, N.J.; Loos, R.J.; Ong, K.K. Variability in the heritability of body mass index: A systematic review and meta-regression. Front. Endocrinol. 2012, 3, 29. [Google Scholar] [CrossRef]
- Locke, A.E.; Kahali, B.; Berndt, S.I.; Justice, A.E.; Pers, T.H.; Day, F.R.; Powell, C.; Vedantam, S.; Buchkovich, M.L.; Yang, J.; et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015, 518, 197–206. [Google Scholar] [CrossRef]
- Shungin, D.; Winkler, T.W.; Croteau-Chonka, D.C.; Ferreira, T.; Locke, A.E.; Magi, R.; Strawbridge, R.J.; Pers, T.H.; Fischer, K.; Justice, A.E.; et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature 2015, 518, 187–196. [Google Scholar] [CrossRef]
- Farooqi, I.S.; Drop, S.; Clements, A.; Keogh, J.M.; Biernacka, J.; Lowenbein, S.; Challis, B.G.; O’Rahilly, S. Heterozygosity for a POMC-null mutation and increased obesity risk in humans. Diabetes 2006, 55, 2549–2553. [Google Scholar] [CrossRef]
- Parton, L.E.; Ye, C.P.; Coppari, R.; Enriori, P.J.; Choi, B.; Zhang, C.Y.; Xu, C.; Vianna, C.R.; Balthasar, N.; Lee, C.E.; et al. Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature 2007, 449, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, M. Peripheral mechanisms in appetite regulation. Gastroenterology 2015, 148, 1219–1233. [Google Scholar] [CrossRef] [PubMed]
- van der Klaauw, A.A.; Farooqi, I.S. The hunger genes: Pathways to obesity. Cell 2015, 161, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.D.; Nogueiras, R.; Andermann, M.L.; Andrews, Z.B.; Anker, S.D.; Argente, J.; Batterham, R.L.; Benoit, S.C.; Bowers, C.Y.; Broglio, F.; et al. Ghrelin. Mol. Metab. 2015, 4, 437–460. [Google Scholar] [CrossRef] [PubMed]
- Makaronidis, J.M.; Batterham, R.L. Obesity body weight regulation the brain: Insights from, fMRI. Br. J. Radiol. 2018, 91, 20170910. [Google Scholar] [CrossRef]
- Manning, S.; Batterham, R.L. The role of gut hormone peptide YY in energy and glucose homeostasis: Twelve years on. Annu. Rev. Physiol. 2014, 76, 585–608. [Google Scholar] [CrossRef] [PubMed]
- Bays, H.E.; Gonzalez-Campoy, J.M.; Henry, R.R.; Bergman, D.A.; Kitabchi, A.E.; Schorr, A.B.; Rodbard, H.W.; Adiposopathy Working, G. Is adiposopathy (sick fat) an endocrine disease? Int. J. Clin. Pract. 2008, 62, 1474–1483. [Google Scholar] [CrossRef]
- Muir, L.A.; Neeley, C.K.; Meyer, K.A.; Baker, N.A.; Brosius, A.M.; Washabaugh, A.R.; Varban, O.A.; Finks, J.F.; Zamarron, B.F.; Flesher, C.G.; et al. Adipose tissue fibrosis, hypertrophy, and hyperplasia: Correlations with diabetes in human obesity. Obesity 2016, 24, 597–605. [Google Scholar] [CrossRef]
- Hammarstedt, A.; Gogg, S.; Hedjazifar, S.; Nerstedt, A.; Smith, U. Impaired Adipogenesis and Dysfunctional Adipose Tissue in Human Hypertrophic Obesity. Physiol. Rev. 2018, 98, 1911–1941. [Google Scholar] [CrossRef]
- Heiss, C.N.; Olofsson, L.E. Gut Microbiota-Dependent Modulation of Energy Metabolism. J. Innate. Immun. 2018, 10, 163–171. [Google Scholar] [CrossRef]
- Sanz, Y.; Moya-Perez, A. Microbiota, inflammation and obesity. Adv. Exp. Med. Biol. 2014, 817, 291–317. [Google Scholar]
- Friedman, J.M.; Halaas, J.L. Leptin and the regulation of body weight in mammals. Nature 1998, 395, 763–770. [Google Scholar] [CrossRef]
- Myers, M.G.; Jr Leibel, R.L.; Seeley, R.J.; Schwartz, M.W. Obesity and leptin resistance: Distinguishing cause from effect. Trends. Endocrinol. Metab. 2010, 21, 643–651. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Strissel, K.J.; Stancheva, Z.; Miyoshi, H.; Perfield, J.W.; DeFuria, J., 2nd; Jick, Z.; Greenberg, A.S.; Obin, M.S. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes 2007, 56, 2910–2918. [Google Scholar] [CrossRef]
- Nishimura, S.; Manabe, I.; Nagasaki, M.; Eto, K.; Yamashita, H.; Ohsugi, M.; Otsu, M.; Hara, K.; Ueki, K.; Sugiura, S.; et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med. 2009, 15, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Sell, H.; Habich, C.; Eckel, J. Adaptive immunity in obesity and insulin resistance. Nat. Rev. Endocrinol. 2012, 8, 709–716. [Google Scholar] [CrossRef]
- Ohmura, K.; Ishimori, N.; Ohmura, Y.; Tokuhara, S.; Nozawa, A.; Horii, S.; Andoh, Y.; Fujii, S.; Iwabuchi, K.; Onoe, K.; et al. Natural killer T cells are involved in adipose tissues inflammation and glucose intolerance in diet-induced obese mice. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Vazquez, I.; Fernandez-Veledo, S.; Kramer, D.K.; Vila-Bedmar, R.; Garcia-Guerra, L.; Lorenzo, M. Insulin resistance associated to obesity: The link TNF-alpha. Arch. Physiol. Biochem. 2008, 114, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Tanti, J.F.; Ceppo, F.; Jager, J.; Berthou, F. Implication of inflammatory signaling pathways in obesity-induced insulin resistance. Front. Endocrinol. 2012, 3, 181. [Google Scholar] [CrossRef]
- Tack, C.J.; Stienstra, R.; Joosten, L.A.; Netea, M.G. Inflammation links excess fat to insulin resistance: The role of the interleukin-1 family. Immunol. Rev. 2012, 249, 239–252. [Google Scholar] [CrossRef]
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology 2010, 51, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.M.; Danaei, G.; Farzadfar, F.; Stevens, G.A.; Woodward, M.; Wormser, D.; Kaptoge, S.; Whitlock, G.; Qiao, Q.; Lewington, S.; et al. The age-specific quantitative effects of metabolic risk factors on cardiovascular diseases and diabetes: A pooled analysis. PLoS ONE 2013, 8, e65174. [Google Scholar] [CrossRef]
- Van Gaal, L.F.; Mertens, I.L.; De Block, C.E. Mechanisms linking obesity with cardiovascular disease. Nature 2006, 444, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Anandacoomarasamy, A.; Caterson, I.; Sambrook, P.; Fransen, M.; March, L. The impact of obesity on the musculoskeletal system. Int. J. Obes. 2008, 32, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Wearing, S.C.; Hennig, E.M.; Byrne, N.M.; Steele, J.R.; Hills, A.P. Musculoskeletal disorders associated with obesity: A biomechanical perspective. Obes. Rev. 2006, 7, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Doerstling, S.S.; O’Flanagan, C.H.; Hursting, S.D. Obesity and Cancer Metabolism: A Perspective on Interacting Tumor-Intrinsic and Extrinsic Factors. Front. Oncol. 2017, 7, 216. [Google Scholar] [CrossRef]
- Park, J.; Morley, T.S.; Kim, M.; Clegg, D.J.; Scherer, P.E. Obesity and cancer--mechanisms underlying tumour progression and recurrence. Nat. Rev. Endocrinol. 2014, 10, 455–465. [Google Scholar] [CrossRef]
- Wildman, R.P.; Muntner, P.; Reynolds, K.; McGinn, A.P.; Rajpathak, S.; Wylie-Rosett, J.; Sowers, M.R. The obese without cardiometabolic risk factor clustering and the normal weight with cardiometabolic risk factor clustering: Prevalence and correlates of 2 phenotypes among the US population (NHANES 1999–2004). Arch. Intern. Med. 2008, 168, 1617–1624. [Google Scholar] [CrossRef]
- Luchsinger, J.A.; Cheng, D.; Tang, M.X.; Schupf, N.; Mayeux, R. Central obesity in the elderly is related to late-onset Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2012, 26, 101–105. [Google Scholar] [CrossRef]
- Razay, G.; Vreugdenhil, A.; Wilcock, G. Obesity, abdominal obesity and Alzheimer disease. Dement. Geriatr. Cogn. Disord. 2006, 22, 173–176. [Google Scholar] [CrossRef]
- Bray, G.A.; Kim, K.K.; Wilding, J.P.H.; World Obesity, F. Obesity: A chronic relapsing progressive disease process. A position statement of the World Obesity Federation. Obes. Rev. 2017, 18, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, K.R.; Redden, D.T.; Wang, C.; Westfall, A.O.; Allison, D.B. Years of life lost due to obesity. JAMA 2003, 289, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Woolf, A.D.; Pfleger, B. Burden of major musculoskeletal conditions. Bull. World Health Organ. 2003, 81, 646–656. [Google Scholar] [PubMed]
- Jantaratnotai, N.; Mosikanon, K.; Lee, Y.; McIntyre, R.S. The interface of depression and obesity. Obes. Res. Clin. Pract. 2017, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Luppino, F.S.; de Wit, L.M.; Bouvy, P.F.; Stijnen, T.; Cuijpers, P.; Penninx, B.W.; Zitman, F.G. Overweight, obesity, and depression: A systematic review and meta-analysis of longitudinal studies. Arch. Gen. Psychiatry 2010, 67, 220–229. [Google Scholar] [CrossRef]
- Monda, V.; La Marra, M.; Perrella, R.; Caviglia, G.; Iavarone, A.; Chieffi, S.; Messina, G.; Carotenuto, M.; Monda, M.; Messina, A. Obesity and brain illness: From cognitive and psychological evidences to obesity paradox. Diabetes Metab. Syndr. Obes. 2017, 10, 473–479. [Google Scholar] [CrossRef]
- Martins, L.B.; Monteze, N.M.; Calarge, C.; Ferreira, A.V.M.; Teixeira, A.L. Pathways linking obesity to neuropsychiatric disorders. Nutrition 2019, 66, 16–21. [Google Scholar] [CrossRef]
- Tchang, B.G.; Saunders, K.H.; Igel, L.I. Best Practices in the Management of Overweight and Obesity. Med. Clin. N. Am. 2021, 105, 149–174. [Google Scholar] [CrossRef]
- Ryan, D.H.; Kahan, S. Guideline Recommendations for Obesity Management. Med. Clin. N. Am. 2018, 102, 49–63. [Google Scholar] [CrossRef]
- Stewart, T.; Han, H.; Allen, R.H.; Bathalon, G.; Ryan, D.H.; Newton, R.L., Jr.; Williamson, D.A. HEALTH: Efficacy of an internet/population-based behavioral weight management program for the U.S. Army. J. Diabetes Sci. Technol. 2011, 5, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Apovian, C.M.; Aronne, L.J.; Bessesen, D.H.; McDonnell, M.E.; Murad, M.H.; Pagotto, U.; Ryan, D.H.; Still, C.D. Pharmacological management of obesity: An endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 2015, 100, 342–362. [Google Scholar] [CrossRef]
- Bluher, M. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Loffler, A.; Luck, T.; Then, F.S.; Luck-Sikorski, C.; Pabst, A.; Kovacs, P.; Bottcher, Y.; Breitfeld, J.; Tonjes, A.; Horstmann, A.; et al. Effects of psychological eating behaviour domains on the association between socio-economic status and BMI. Public Health Nutr. 2017, 20, 2706–2712. [Google Scholar] [CrossRef]
- Garvey, W.T.; Mechanick, J.I.; Brett, E.M.; Garber, A.J.; Hurley, D.L.; Jastreboff, A.M.; Nadolsky, K.; Pessah-Pollack, R.; Plodkowski, R. Reviewers of the AACEOCPG: American Association of Clinical Endocrinologists and American College of Endocrinology Comprehensive Clinical Practice Guidelines for Medical Care of Patients with Obesity. Endocr Pract. 2016, 3, 1–203. [Google Scholar] [CrossRef]
- Gonzalez-Muniesa, P.; Martinez-Gonzalez, M.A.; Hu, F.B.; Despres, J.P.; Matsuzawa, Y.; Loos, R.J.F.; Moreno, L.A.; Bray, G.A.; Martinez, J.A. Obesity. Nat. Rev. Dis. Primers 2017, 3, 17034. [Google Scholar] [CrossRef] [PubMed]
- American College of Cardiology/American Heart Association Task Force on Practice Guidelines OEP: Executive summary: Guidelines (2013) for the management of overweight and obesity in adults: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Obesity Society published by the Obesity Society and American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Based on a systematic review from the The Obesity Expert Panel, 2013. Obesity 2014, 22, S5–S39.
- Bray, G.A.; Siri-Tarino, P.W. The Role of Macronutrient Content in the Diet for Weight Management. Endocrinol. Metab. Clin. N. Am. 2016, 45, 581–604. [Google Scholar] [CrossRef]
- Golpour-Hamedani, S.; Mohammadifard, N.; Khosravi, A.; Feizi, A.; Safavi, S.M. Dietary approaches to stop hypertension diet and obesity: A cross-sectional study of Iranian children and adolescents. ARYA Atheroscler. 2017, 13, 7–13. [Google Scholar]
- D’Innocenzo, S.; Biagi, C.; Lanari, M. Obesity and the Mediterranean Diet: A Review of Evidence of the Role and Sustainability of the Mediterranean Diet. Nutrients 2019, 11, 1306. [Google Scholar] [CrossRef]
- de la Iglesia, R.; Lopez-Legarrea, P.; Abete, I.; Bondia-Pons, I.; Navas-Carretero, S.; Forga, L.; Martinez, J.A.; Zulet, M.A. A new dietary strategy for long-term treatment of the metabolic syndrome is compared with the American Heart Association (AHA) guidelines: The MEtabolic Syndrome REduction in NAvarra (RESMENA) project. Br. J. Nutr. 2014, 111, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Cantero, I.; Abete, I.; Monreal, J.I.; Martinez, J.A.; Zulet, M.A. Fruit Fiber Consumption Specifically Improves Liver Health Status in Obese Subjects under Energy Restriction. Nutrients 2017, 9, 667. [Google Scholar] [CrossRef] [PubMed]
- de la Iglesia, R.; Lopez-Legarrea, P.; Celada, P.; Sanchez-Muniz, F.J.; Martinez, J.A.; Zulet, M.A. Beneficial effects of the RESMENA dietary pattern on oxidative stress in patients suffering from metabolic syndrome with hyperglycemia are associated to dietary TAC and fruit consumption. Int. J. Mol. Sci. 2013, 14, 6903–6919. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Longo, V.D.; Harvie, M. Impact of intermittent fasting on health and disease processes. Ageing. Res. Rev. 2017, 39, 46–58. [Google Scholar] [CrossRef]
- Stockman, M.C.; Thomas, D.; Burke, J.; Apovian, C.M. Intermittent Fasting: Is the Wait Worth the Weight? Curr. Obes. Rep. 2018, 7, 172–185. [Google Scholar] [CrossRef]
- Varady, K.A.; Bhutani, S.; Church, E.C.; Klempel, M.C. Short-term modified alternate-day fasting: A novel dietary strategy for weight loss and cardioprotection in obese adults. Am. J. Clin. Nutr. 2009, 90, 1138–1143. [Google Scholar] [CrossRef]
- St-Onge, M.P.; Ard, J.; Baskin, M.L.; Chiuve, S.E.; Johnson, H.M.; Kris-Etherton, P.; Varady, K.; on behalf of the American Heart Association Obesity Committee of the Council on Lifestyle and Cardiometabolic Health; Council on Cardiovascular Disease in the Young; Council on Clinical Cardiology; et al. Meal Timing and Frequency: Implications for Cardiovascular Disease Prevention: A Scientific Statement from the American Heart Association. Circulation 2017, 135, e96–e121. [Google Scholar] [CrossRef]
- Jensen, M.D.; Ryan, D.H.; Apovian, C.M.; Ard, J.D.; Comuzzie, A.G.; Donato, K.A.; Hu, F.B.; Hubbard, V.S.; Jakicic, J.M.; Kushner, R.F.; et al. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. J. Am. Coll. Cardiol. 2014, 63, 2985–3023. [Google Scholar] [CrossRef]
- Donnelly, J.E.; Blair, S.N.; Jakicic, J.M.; Manore, M.M.; Rankin, J.W.; Smith, B.K. Appropriate physical activity intervention strategies for weight loss and prevention of weight regain for adults. Med. Sci. Sports Exerc. 2009, 41, 459–471. [Google Scholar] [CrossRef]
- Hill, J.O.; Wyatt, H.R. Role of physical activity in preventing and treating obesity. J. Appl. Physiol. 2005, 99, 765–770. [Google Scholar] [CrossRef]
- Swift, D.L.; McGee, J.E.; Earnest, C.P.; Carlisle, E.; Nygard, M.; Johannsen, N.M. The Effects of Exercise and Physical Activity on Weight Loss and Maintenance. Prog. Cardiovasc. Dis. 2018, 61, 206–213. [Google Scholar] [CrossRef]
- Villareal, D.T.; Aguirre, L.; Gurney, A.B.; Waters, D.L.; Sinacore, D.R.; Colombo, E.; Armamento-Villareal, R.; Qualls, C. Aerobic or Resistance Exercise, or Both, in Dieting Obese Older Adults. N. Engl. J. Med. 2017, 376, 1943–1955. [Google Scholar] [CrossRef] [PubMed]
- Jakicic, J.M.; Rogers, R.J.; Davis, K.K.; Collins, K.A. Role of Physical Activity and Exercise in Treating Patients with Overweight and Obesity. Clin. Chem. 2018, 64, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Toplak, H.; Woodward, E.; Yumuk, V.; Oppert, J.M.; Halford, J.C.; Fruhbeck, G. 2014 EASO Position Statement on the Use of Anti-Obesity Drugs. Obes. Facts 2015, 8, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Igel, L.I.; Kumar, R.B.; Saunders, K.H.; Aronne, L.J. Practical Use of Pharmacotherapy for Obesity. Gastroenterology 2017, 152, 1765–1779. [Google Scholar] [CrossRef]
- Rothman, R.B.; Baumann, M.H.; Dersch, C.M.; Romero, D.V.; Rice, K.C.; Carroll, F.I.; Partilla, J.S. Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse 2001, 39, 32–41. [Google Scholar] [CrossRef]
- Richard, D.; Ferland, J.; Lalonde, J.; Samson, P.; Deshaies, Y. Influence of topiramate in the regulation of energy balance. Nutrition 2000, 16, 961–966. [Google Scholar] [CrossRef]
- Wilding, J.; Van Gaal, L.; Rissanen, A.; Vercruysse, F.; Fitchet, M.; Group, O.-S. A randomized double-blind placebo-controlled study of the long-term efficacy and safety of topiramate in the treatment of obese subjects. Int. J. Obes. Relat. Metab. Disord. 2004, 28, 1399–1410. [Google Scholar] [CrossRef]
- Fleming, J.W.; McClendon, K.S.; Riche, D.M. New obesity agents: Lorcaserin and phentermine/topiramate. Ann. Pharmacother. 2013, 47, 1007–1016. [Google Scholar] [CrossRef]
- Singh, J.; Kumar, R. Phentermine-topiramate: First combination drug for obesity. Int. J. Appl. Basic Med. Res. 2015, 5, 157–158. [Google Scholar] [CrossRef]
- Gadde, K.M.; Allison, D.B.; Ryan, D.H.; Peterson, C.A.; Troupin, B.; Schwiers, M.L.; Day, W.W. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): A randomised, placebo-controlled, phase 3 trial. Lancet 2011, 377, 1341–1352. [Google Scholar] [CrossRef]
- Garvey, W.T.; Ryan, D.H.; Look, M.; Gadde, K.M.; Allison, D.B.; Peterson, C.A.; Schwiers, M.; Day, W.W.; Bowden, C.H. Two-year sustained weight loss and metabolic benefits with controlled-release phentermine/topiramate in obese and overweight adults (SEQUEL): A randomized, placebo-controlled, phase 3 extension study. Am. J. Clin. Nutr. 2012, 95, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Bouza, C.; Angeles, M.; Munoz, A.; Amate, J.M. Efficacy and safety of naltrexone and acamprosate in the treatment of alcohol dependence: A systematic review. Addiction 2004, 99, 811–828. [Google Scholar] [PubMed]
- Greenway, F.L.; Whitehouse, M.J.; Guttadauria, M.; Anderson, J.W.; Atkinson, R.L.; Fujioka, K.; Gadde, K.M.; Gupta, A.K.; O’Neil, P.; Schumacher, D.; et al. Rational design of a combination medication for the treatment of obesity. Obesity 2009, 17, 30–39. [Google Scholar] [CrossRef]
- Foley, K.F.; DeSanty, K.P.; Kast, R.E. Bupropion: Pharmacology and therapeutic applications. Expert Rev. Neurother. 2006, 6, 1249–1265. [Google Scholar] [CrossRef]
- Hasegawa, H.; Meeusen, R.; Sarre, S.; Diltoer, M.; Piacentini, M.F.; Michotte, Y. Acute dopamine/norepinephrine reuptake inhibition increases brain and core temperature in rats. J. Appl. Physiol. 2005, 99, 1397–1401. [Google Scholar] [CrossRef]
- Greenway, F.L.; Fujioka, K.; Plodkowski, R.A.; Mudaliar, S.; Guttadauria, M.; Erickson, J.; Kim, D.D.; Dunayevich, E.; Group C-IS. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-I): A multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2010, 376, 595–605. [Google Scholar] [CrossRef]
- Farr, O.M.; Tsoukas, M.A.; Triantafyllou, G.; Dincer, F.; Filippaios, A.; Ko, B.J.; Mantzoros, C.S. Short-term administration of the GLP-1 analog liraglutide decreases circulating leptin and increases GIP levels and these changes are associated with alterations in CNS responses to food cues: A randomized, placebo-controlled, crossover study. Metabolism 2016, 65, 945–953. [Google Scholar] [CrossRef]
- Farr, O.M.; Upadhyay, J.; Rutagengwa, C.; DiPrisco, B.; Ranta, Z.; Adra, A.; Bapatla, N.; Douglas, V.P.; Douglas, K.A.A.; Nolen-Doerr, E.; et al. Longer-term liraglutide administration at the highest dose approved for obesity increases reward-related orbitofrontal cortex activation in response to food cues: Implications for plateauing weight loss in response to anti-obesity therapies. Diabetes Obes. Metab. 2019, 21, 2459–2464. [Google Scholar] [CrossRef]
- Pi-Sunyer, X.; Astrup, A.; Fujioka, K.; Greenway, F.; Halpern, A.; Krempf, M.; Lau, D.C.; le Roux, C.W.; Violante Ortiz, R.; Jensen, C.B.; et al. A Randomized, Controlled Trial of 3.0 mg of Liraglutide in Weight Management. N. Engl. J. Med. 2015, 373, 11–22. [Google Scholar] [CrossRef]
- Van Can, J.; Sloth, B.; Jensen, C.B.; Flint, A.; Blaak, E.E.; Saris, W.H. Effects of the once-daily GLP-1 analog liraglutide on gastric emptying, glycemic parameters, appetite and energy metabolism in obese, non-diabetic adults. Int. J. Obes. 2014, 38, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Le Roux, C.W.; Heneghan, H.M. Bariatric Surgery for Obesity. Med. Clin. N. Am. 2018, 102, 165–182. [Google Scholar] [CrossRef] [PubMed]
- Saunders, K.H.; Igel, L.I.; Saumoy, M.; Sharaiha, R.Z.; Aronne, L.J. Devices and Endoscopic Bariatric Therapies for Obesity. Curr. Obes. Rep. 2018, 7, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Wada, N.; Hirako, S.; Takenoya, F.; Kageyama, H.; Okabe, M.; Shioda, S. Leptin and its receptors. J. Chem. Neuroanat. 2014, 61–62, 191–199. [Google Scholar] [CrossRef]
- Chou, K.; Perry, C.M. Metreleptin: First global approval. Drugs 2013, 73, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Tchang, B.G.; Shukla, A.P.; Aronne, L.J. Metreleptin and generalized lipodystrophy and evolving therapeutic perspectives. Expert Opin. Biol. Ther. 2015, 15, 1061–1075. [Google Scholar] [CrossRef]
- Kissileff, H.R.; Thornton, J.C.; Torres, M.I.; Pavlovich, K.; Mayer, L.S.; Kalari, V.; Leibel, R.L.; Rosenbaum, M. Leptin reverses declines in satiation in weight-reduced obese humans. Am. J. Clin. Nutr. 2012, 95, 309–317. [Google Scholar] [CrossRef]
- Samsom, M.; Szarka, L.A.; Camilleri, M.; Vella, A.; Zinsmeister, A.R.; Rizza, R.A. Pramlintide, an amylin analog, selectively delays gastric emptying: Potential role of vagal inhibition. Am. J. Physiol. Gastrointest Liver Physiol. 2000, 278, G946–G951. [Google Scholar] [CrossRef]
- Mack, C.M.; Soares, C.J.; Wilson, J.K.; Athanacio, J.R.; Turek, V.F.; Trevaskis, J.L.; Roth, J.D.; Smith, P.A.; Gedulin, B.; Jodka, C.M.; et al. Davalintide (AC2307), a novel amylin-mimetic peptide: Enhanced pharmacological properties over native amylin to reduce food intake and body weight. Int. J. Obes. 2010, 34, 385–395. [Google Scholar] [CrossRef]
- Christou, G.A.; Katsiki, N.; Blundell, J.; Fruhbeck, G.; Kiortsis, D.N. Semaglutide as a promising antiobesity drug. Obes. Rev. 2019, 20, 805–815. [Google Scholar] [CrossRef]
- Lau, J.; Bloch, P.; Schaffer, L.; Pettersson, I.; Spetzler, J.; Kofoed, J.; Madsen, K.; Knudsen, L.B.; McGuire, J.; Steensgaard, D.B.; et al. Discovery of the Once-Weekly Glucagon-Like Peptide-1 (GLP-1) Analogue Semaglutide. J. Med. Chem. 2015, 58, 7370–7380. [Google Scholar] [CrossRef] [PubMed]
- Avgerinos, I.; Michailidis, T.; Liakos, A.; Karagiannis, T.; Matthews, D.R.; Tsapas, A.; Bekiari, E. Oral semaglutide for type 2 diabetes: A systematic review and meta-analysis. Diabetes Obes. Metab. 2020, 22, 335–345. [Google Scholar] [CrossRef]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug. Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Norregaard, P.K.; Deryabina, M.A.; Tofteng Shelton, P.; Fog, J.U.; Daugaard, J.R.; Eriksson, P.O.; Larsen, L.F.; Jessen, L. A novel GIP analogue, ZP4165, enhances glucagon-like peptide-1-induced body weight loss and improves glycaemic control in rodents. Diabetes Obes Metab. 2018, 20, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Pocai, A. Action and therapeutic potential of oxyntomodulin. Mol. Metab. 2014, 3, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.J.; Konkar, A.; Hornigold, D.C.; Trevaskis, J.L.; Jackson, R.; Fritsch Fredin, M.; Jansson-Lofmark, R.; Naylor, J.; Rossi, A.; Bednarek, M.A.; et al. Robust anti-obesity and metabolic effects of a dual GLP-1/glucagon receptor peptide agonist in rodents and non-human primates. Diabetes Obes. Metab. 2016, 18, 1176–1190. [Google Scholar] [CrossRef]
- Thomas, M.K.; Nikooienejad, A.; Bray, R.; Cui, X.; Wilson, J.; Duffin, K.; Milicevic, Z.; Haupt, A.; Robins, D.A. Dual GIP and GLP-1 Receptor Agonist Tirzepatide Improves Beta-cell Function and Insulin Sensitivity in Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2021, 106, 388–396. [Google Scholar] [CrossRef]
- Dragano, N.R.V.; Ferno, J.; Dieguez, C.; Lopez, M.; Milbank, E. Recent Updates on Obesity Treatments: Available Drugs and Future Directions. Neuroscience 2020, 437, 215–239. [Google Scholar] [CrossRef]
- Salamone, J.D.; McLaughlin, P.J.; Sink, K.; Makriyannis, A.; Parker, L.A. Cannabinoid CB1 receptor inverse agonists and neutral antagonists: Effects on food intake, food-reinforced behavior and food aversions. Physiol. Behav. 2007, 91, 383–388. [Google Scholar] [CrossRef]
- Cluny, N.L.; Vemuri, V.K.; Chambers, A.P.; Limebeer, C.L.; Bedard, H.; Wood, J.T.; Lutz, B.; Zimmer, A.; Parker, L.A.; Makriyannis, A.; et al. A novel peripherally restricted cannabinoid receptor antagonist, AM6545, reduces food intake and body weight, but does not cause malaise, in rodents. Br. J. Pharmacol. 2010, 161, 629–642. [Google Scholar] [CrossRef]
- Randall, P.A.; Vemuri, V.K.; Segovia, K.N.; Torres, E.F.; Hosmer, S.; Nunes, E.J.; Santerre, J.L.; Makriyannis, A.; Salamone, J.D. The novel cannabinoid CB1 antagonist AM6545 suppresses food intake and food-reinforced behavior. Pharmacol. Biochem. Behav. 2010, 97, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Kato, T.; Ogino, H.; Ashina, S.; Kato, K. Cetilistat (ATL-962), a novel pancreatic lipase inhibitor, ameliorates body weight gain and improves lipid profiles in rats. Horm. Metab. Res. 2008, 40, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Altabas, V.; Zjacic-Rotkvic, V. Anti-ghrelin antibodies in appetite suppression: Recent advances in obesity pharmacotherapy. Immunotargets Ther. 2015, 4, 123–130. [Google Scholar] [PubMed]
- Arimura, A.; Smith, W.D.; Schally, A.V. Blockade of the stress-induced decrease in blood GH by anti-somatostatin serum in rats. Endocrinology 1976, 98, 540–543. [Google Scholar] [CrossRef] [PubMed]
- Haffer, K.N. Effects of novel vaccines on weight loss in diet-induced-obese (DIO) mice. J. Anim. Sci. Biotechnol. 2012, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Na, H.N.; Kim, H.; Nam, J.H. Prophylactic and therapeutic vaccines for obesity. Clin. Exp. Vaccine Res. 2014, 3, 37–41. [Google Scholar] [CrossRef]
- Na, H.N.; Nam, J.H. Proof-of-concept for a virus-induced obesity vaccine; vaccination against the obesity agent adenovirus 36. Int. J. Obes. 2014, 38, 1470–1474. [Google Scholar] [CrossRef]
- Srivastava, S.; Veech, R.L. Brown and Brite: The Fat Soldiers in the Anti-obesity Fight. Front. Physiol. 2019, 10, 38. [Google Scholar] [CrossRef]
- Wang, S.; Pan, M.H.; Hung, W.L.; Tung, Y.C.; Ho, C.T. From white to beige adipocytes: Therapeutic potential of dietary molecules against obesity and their molecular mechanisms. Food Funct. 2019, 10, 1263–1279. [Google Scholar] [CrossRef]
- Rosell, M.; Kaforou, M.; Frontini, A.; Okolo, A.; Chan, Y.W.; Nikolopoulou, E.; Millership, S.; Fenech, M.E.; MacIntyre, D.; Turner, J.O.; et al. Brown and white adipose tissues: Intrinsic differences in gene expression and response to cold exposure in mice. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E945–E964. [Google Scholar] [CrossRef]
- Kawada, T.; Watanabe, T.; Takaishi, T.; Tanaka, T.; Iwai, K. Capsaicin-induced beta-adrenergic action on energy metabolism in rats: Influence of capsaicin on oxygen consumption, the respiratory quotient, and substrate utilization. Proc. Soc. Exp. Biol. Med. 1986, 183, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Baskaran, P.; Krishnan, V.; Ren, J.; Thyagarajan, B. Capsaicin induces browning of white adipose tissue and counters obesity by activating TRPV1 channel-dependent mechanisms. Br. J. Pharmacol. 2016, 173, 2369–2389. [Google Scholar] [CrossRef] [PubMed]
- Di Pierro, F.; Bressan, A.; Ranaldi, D.; Rapacioli, G.; Giacomelli, L.; Bertuccioli, A. Potential role of bioavailable curcumin in weight loss and omental adipose tissue decrease: Preliminary data of a randomized, controlled trial in overweight people with metabolic syndrome. Preliminary study. Eur. Rev. Med Pharmacol. Sci. 2015, 19, 4195–4202. [Google Scholar]
- Laiglesia, L.M.; Lorente-Cebrian, S.; Prieto-Hontoria, P.L.; Fernandez-Galilea, M.; Ribeiro, S.M.; Sainz, N.; Martinez, J.A.; Moreno-Aliaga, M.J. Eicosapentaenoic acid promotes mitochondrial biogenesis and beige-like features in subcutaneous adipocytes from overweight subjects. J. Nutr. Biochem. 2016, 37, 76–82. [Google Scholar] [CrossRef]
- Yan, J.; Zhao, Y.; Zhao, B. Green tea catechins prevent obesity through modulation of peroxisome proliferator-activated receptors. Sci. China. Life. Sci. 2013, 56, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liang, X.; Yang, Q.; Fu, X.; Rogers, C.J.; Zhu, M.; Rodgers, B.D.; Jiang, Q.; Dodson, M.V.; Du, M. Resveratrol induces brown-like adipocyte formation in white fat through activation of AMP-activated protein kinase (AMPK) alpha1. Int. J. Obes. 2015, 39, 967–976. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, H.; Li, B.; Meng, X.; Wang, J.; Zhang, Y.; Yao, S.; Ma, Q.; Jin, L.; Yang, J.; et al. Berberine activates thermogenesis in white and brown adipose tissue. Nat. Commun. 2014, 5, 5493. [Google Scholar] [CrossRef]
- Cypess, A.M.; Weiner, L.S.; Roberts-Toler, C.; Franquet Elia, E.; Kessler, S.H.; Kahn, P.A.; English, J.; Chatman, K.; Trauger, S.A.; Doria, A.; et al. Activation of human brown adipose tissue by a beta3-adrenergic receptor agonist. Cell Metab. 2015, 21, 33–38. [Google Scholar] [CrossRef]
- Merlin, J.; Sato, M.; Nowell, C.; Pakzad, M.; Fahey, R.; Gao, J.; Dehvari, N.; Summers, R.J.; Bengtsson, T.; Evans, B.A.; et al. The PPARgamma agonist rosiglitazone promotes the induction of brite adipocytes, increasing beta-adrenoceptor-mediated mitochondrial function and glucose uptake. Cell Signal 2018, 42, 54–66. [Google Scholar] [CrossRef]
- Loh, R.K.C.; Formosa, M.F.; Eikelis, N.; Bertovic, D.A.; Anderson, M.J.; Barwood, S.A.; Nanayakkara, S.; Cohen, N.D.; La Gerche, A.; Reutens, A.T.; et al. Pioglitazone reduces cold-induced brown fat glucose uptake despite induction of browning in cultured human adipocytes: A randomised, controlled trial in humans. Diabetologia 2018, 61, 220–230. [Google Scholar] [CrossRef]
- Rachid, T.L.; Silva-Veiga, F.M.; Graus-Nunes, F.; Bringhenti, I.; Mandarim-de-Lacerda, C.A.; Souza-Mello, V. Differential actions of PPAR-alpha and PPAR-beta/delta on beige adipocyte formation: A study in the subcutaneous white adipose tissue of obese male mice. PLoS ONE 2018, 13, e0191365. [Google Scholar] [CrossRef] [PubMed]
- Pettersson-Klein, A.T.; Izadi, M.; Ferreira, D.M.S.; Cervenka, I.; Correia, J.C.; Martinez-Redondo, V.; Southern, M.; Cameron, M.; Kamenecka, T.; Agudelo, L.Z.; et al. Small molecule PGC-1alpha1 protein stabilizers induce adipocyte Ucp1 expression and uncoupled mitochondrial respiration. Mol. Metab. 2018, 9, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Lee, S.H.; Lee, S.Y.; Kim, J.K.; Jhun, J.Y.; Na, H.S.; Kim, S.Y.; Choi, J.Y.; Yang, C.W.; Park, S.H.; et al. Metformin ameliorates experimental-obesity-associated autoimmune arthritis by inducing FGF21 expression and brown adipocyte differentiation. Exp. Mol. Med. 2018, 50, e432. [Google Scholar] [CrossRef]
- Tokubuchi, I.; Tajiri, Y.; Iwata, S.; Hara, K.; Wada, N.; Hashinaga, T.; Nakayama, H.; Mifune, H.; Yamada, K. Beneficial effects of metformin on energy metabolism and visceral fat volume through a possible mechanism of fatty acid oxidation in human subjects and rats. PLoS ONE 2017, 12, e0171293. [Google Scholar] [CrossRef] [PubMed]
- Popkin, B.M.; Du, S.; Green, W.D.; Beck, M.A.; Algaith, T.; Herbst, C.H.; Alsukait, R.F.; Alluhidan, M.; Alazemi, N.; Shekar, M. Individuals with obesity and COVID-19, A global perspective on the epidemiology and biological relationships. Obes. Rev. 2020, 21, e13128. [Google Scholar] [CrossRef]
- Stefan, N.; Birkenfeld, A.L.; Schulze, M.B.; Ludwig, D.S. Obesity and impaired metabolic health in patients with COVID-19. Nat. Rev. Endocrinol. 2020, 16, 341–342. [Google Scholar] [CrossRef]
- Sheetz, M.J.; King, G.L. Molecular understanding of hyperglycemia’s adverse effects for diabetic complications. JAMA 2002, 288, 2579–2588. [Google Scholar] [CrossRef]
- Saucillo, D.C.; Gerriets, V.A.; Sheng, J.; Rathmell, J.C.; Maciver, N.J. Leptin metabolically licenses T cells for activation to link nutrition and immunity. J. Immunol. 2014, 192, 136–144. [Google Scholar] [CrossRef]
- Tsai, S.; Clemente-Casares, X.; Zhou, A.C.; Lei, H.; Ahn, J.J.; Chan, Y.T.; Choi, O.; Luck, H.; Woo, M.; Dunn, S.E.; et al. Insulin Receptor-Mediated Stimulation Boosts T Cell Immunity during Inflammation and Infection. Cell Metab. 2018, 28, 922–934.e924. [Google Scholar] [CrossRef]
- American Diabetes A: 2. Classification and Diagnosis of Diabetes. Diabetes Care 2017, 40, S11–S24. [CrossRef]
- IDF Diabetes Atlas. Available online: https://www.diabetesatlas.org/en/ (accessed on 10 October 2020).
- White, M.F. Insulin signaling in health and disease. Science 2003, 302, 1710–1711. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, H.K.; Zierath, J.R. Insulin signaling and glucose transport in insulin resistant human skeletal muscle. Cell Biochem. Biophys. 2007, 48, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Michael, M.D.; Kulkarni, R.N.; Postic, C.; Previs, S.F.; Shulman, G.I.; Magnuson, M.A.; Kahn, C.R. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol. Cell 2000, 6, 87–97. [Google Scholar] [CrossRef]
- Laviola, L.; Perrini, S.; Cignarelli, A.; Natalicchio, A.; Leonardini, A.; De Stefano, F.; Cuscito, M.; De Fazio, M.; Memeo, V.; Neri, V.; et al. Insulin signaling in human visceral and subcutaneous adipose tissue in vivo. Diabetes 2006, 55, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef]
- Insel, R.A.; Dunne, J.L.; Atkinson, M.A.; Chiang, J.L.; Dabelea, D.; Gottlieb, P.A.; Greenbaum, C.J.; Herold, K.C.; Krischer, J.P.; Lernmark, A.; et al. Staging presymptomatic type 1 diabetes: A scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care 2015, 38, 1964–1974. [Google Scholar] [CrossRef]
- Barrett, J.C.; Clayton, D.G.; Concannon, P.; Akolkar, B.; Cooper, J.D.; Erlich, H.A.; Julier, C.; Morahan, G.; Nerup, J.; Nierras, C.; et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 2009, 41, 703–707. [Google Scholar] [CrossRef]
- Noble, J.A.; Valdes, A.M.; Varney, M.D.; Carlson, J.A.; Moonsamy, P.; Fear, A.L.; Lane, J.A.; Lavant, E.; Rappner, R.; Louey, A.; et al. HLA class I and genetic susceptibility to type 1 diabetes: Results from the Type 1 Diabetes Genetics Consortium. Diabetes 2010, 59, 2972–2979. [Google Scholar] [CrossRef]
- Saberzadeh-Ardestani, B.; Karamzadeh, R.; Basiri, M.; Hajizadeh-Saffar, E.; Farhadi, A.; Shapiro, A.M.J.; Tahamtani, Y.; Baharvand, H. Type 1 Diabetes Mellitus: Cellular and Molecular Pathophysiology at A Glance. Cell J. 2018, 20, 294–301. [Google Scholar]
- Katsarou, A.; Gudbjornsdottir, S.; Rawshani, A.; Dabelea, D.; Bonifacio, E.; Anderson, B.J.; Jacobsen, L.M.; Schatz, D.A.; Lernmark, A. Type 1 diabetes mellitus. Nat. Rev. Dis. Primers 2017, 3, 17016. [Google Scholar] [CrossRef]
- Ozougwu, J.C.; Obimba, K.C.; Belonwu, C.D.; Unakalamba, C. The pathogenesis and pathophysiology of type 1 and type 2 diabetes mellitus. J. Physiol. Pathophysiol. 2013, 4, 46–57. [Google Scholar] [CrossRef]
- Schellenberg, E.S.; Dryden, D.M.; Vandermeer, B.; Ha, C.; Korownyk, C. Lifestyle interventions for patients with and at risk for type 2 diabetes: A systematic review and meta-analysis. Ann. Intern. Med. 2013, 159, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bao, W.; Liu, J.; Ouyang, Y.Y.; Wang, D.; Rong, S.; Xiao, X.; Shan, Z.L.; Zhang, Y.; Yao, P.; et al. Inflammatory markers and risk of type 2 diabetes: A systematic review and meta-analysis. Diabetes Care 2013, 36, 166–175. [Google Scholar] [CrossRef]
- Esteve, E.; Ricart, W.; Fernandez-Real, J.M. Gut microbiota interactions with obesity, insulin resistance and type 2 diabetes: Did gut microbiote co-evolve with insulin resistance? Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Ding, E.L.; Song, Y.; Manson, J.E.; Hunter, D.J.; Lee, C.C.; Rifai, N.; Buring, J.E.; Gaziano, J.M.; Liu, S. Sex hormone-binding globulin and risk of type 2 diabetes in women and men. N. Engl. J. Med. 2009, 361, 1152–1163. [Google Scholar] [CrossRef]
- Kulkarni, R.N.; Bruning, J.C.; Winnay, J.N.; Postic, C.; Magnuson, M.A.; Kahn, C.R. Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell 1999, 96, 329–339. [Google Scholar] [CrossRef]
- Honka, H.; Makinen, J.; Hannukainen, J.C.; Tarkia, M.; Oikonen, V.; Teras, M.; Fagerholm, V.; Ishizu, T.; Saraste, A.; Stark, C.; et al. Validation of [18F] fluorodeoxyglucose and positron emission tomography (PET) for the measurement of intestinal metabolism in pigs, and evidence of intestinal insulin resistance in patients with morbid obesity. Diabetologia 2013, 56, 893–900. [Google Scholar] [CrossRef]
- Artunc, F.; Schleicher, E.; Weigert, C.; Fritsche, A.; Stefan, N.; Haring, H.U. The impact of insulin resistance on the kidney and vasculature. Nat. Rev. Nephrol. 2016, 12, 721–737. [Google Scholar] [CrossRef]
- Blazquez, E.; Velazquez, E.; Hurtado-Carneiro, V.; Ruiz-Albusac, J.M. Insulin in the brain: Its pathophysiological implications for States related with central insulin resistance, type 2 diabetes and Alzheimer’s disease. Front. Endocrinol. 2014, 5, 161. [Google Scholar] [CrossRef]
- Meijer, R.I.; De Boer, M.P.; Groen, M.R.; Eringa, E.C.; Rattigan, S.; Barrett, E.J.; Smulders, Y.M.; Serne, E.H. Insulin-induced microvascular recruitment in skin and muscle are related and both are associated with whole-body glucose uptake. Microcirculation 2012, 19, 494–500. [Google Scholar] [CrossRef]
- Loria, P.; Lonardo, A.; Anania, F. Liver and diabetes. A vicious circle. Hepatol. Res. 2013, 43, 51–64. [Google Scholar] [CrossRef]
- DeFronzo, R.A. Insulin resistance, lipotoxicity, type 2 diabetes and atherosclerosis: The missing links. The Claude Bernard Lecture 2009. Diabetologia 2010, 53, 1270–1287. [Google Scholar] [CrossRef]
- Gastaldelli, A.; Gaggini, M.; DeFronzo, R.A. Role of Adipose Tissue Insulin Resistance in the Natural History of Type 2 Diabetes: Results from the San Antonio Metabolism Study. Diabetes 2017, 66, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Romeo, G.R.; Lee, J.; Shoelson, S.E. Metabolic syndrome, insulin resistance, and roles of inflammation--mechanisms and therapeutic targets. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1771–1776. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Camporez, J.G.; Kursawe, R.; Titchenell, P.M.; Zhang, D.; Perry, C.J.; Jurczak, M.J.; Abudukadier, A.; Han, M.S.; Zhang, X.M.; et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 2015, 160, 745–758. [Google Scholar] [CrossRef]
- Harding, J.L.; Pavkov, M.E.; Magliano, D.J.; Shaw, J.E.; Gregg, E.W. Global trends in diabetes complications: A review of current evidence. Diabetologia 2019, 62, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Duckworth, W.; Abraira, C.; Moritz, T.; Reda, D.; Emanuele, N.; Reaven, P.D.; Zieve, F.J.; Marks, J.; Davis, S.N.; Hayward, R.; et al. Glucose control and vascular complications in veterans with type 2 diabetes. N. Engl. J. Med. 2009, 360, 129–139. [Google Scholar] [CrossRef]
- van Dieren, S.; Beulens, J.W.; van der Schouw, Y.T.; Grobbee, D.E.; Neal, B. The global burden of diabetes and its complications: An emerging pandemic. Eur. J. Cardiovasc. Prev. Rehabil. 2010, 17, S3–S8. [Google Scholar]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef]
- Di Cianni, G.; Miccoli, R.; Volpe, L.; Lencioni, C.; Del Prato, S. Intermediate metabolism in normal pregnancy and in gestational diabetes. Diabetes Metab. Res. Rev. 2003, 19, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Plows, J.F.; Stanley, J.L.; Baker, P.N.; Reynolds, C.M.; Vickers, M.H. The Pathophysiology of Gestational Diabetes Mellitus. Int. J. Mol. Sci. 2018, 19, 3342. [Google Scholar] [CrossRef]
- Hattersley, A.T.; Patel, K.A. Precision diabetes: Learning from monogenic diabetes. Diabetologia 2017, 60, 769–777. [Google Scholar] [CrossRef]
- Urakami, T. Maturity-onset diabetes of the young (MODY): Current perspectives on diagnosis and treatment. Diabetes Metab. Syndr. Obes. 2019, 12, 1047–1056. [Google Scholar] [CrossRef]
- Vaxillaire, M.; Froguel, P.; Bonnefond, A. How Recent Advances in Genomics Improve Precision Diagnosis and Personalized Care of Maturity-Onset Diabetes of the Young. Curr. Diab. Rep. 2019, 19, 79. [Google Scholar] [CrossRef]
- Christ-Crain, M.; Bichet, D.G.; Fenske, W.K.; Goldman, M.B.; Rittig, S.; Verbalis, J.G.; Verkman, A.S. Diabetes insipidus. Nat. Rev. Dis. Primers 2019, 5, 54. [Google Scholar] [CrossRef]
- Chaudhary, N.; Tyagi, N. Diabetes mellitus: An Overview. Int. J. Res. Dev. Pharm. Life Sci. 2018, 7, 3030–3033. [Google Scholar] [CrossRef]
- Gullo, L.; Pezzilli, R.; Morselli-Labate, A.M.; Italian Pancreatic Cancer Study Group. Diabetes and the risk of pancreatic cancer. N. Engl. J. Med. 1994, 331, 81–84. [Google Scholar] [CrossRef]
- Yi, Y.; Norris, A.W.; Wang, K.; Sun, X.; Uc, A.; Moran, A.; Engelhardt, J.F.; Ode, K.L. Abnormal Glucose Tolerance in Infants and Young Children with Cystic Fibrosis. Am. J. Respir. Crit. Care. Med. 2016, 194, 974–980. [Google Scholar] [CrossRef]
- Kirti, K.; Tarr, J.M.; Ahmad, S.I.; Eva, M.K.; Rakesh, C. Diabetes, An Old Disease, a New Insigh, 1st ed.; Springer: New York, NY, USA, 2013. [Google Scholar]
- Pandit, M.K.; Burke, J.; Gustafson, A.B.; Minocha, A.; Peiris, A.N. Drug-induced disorders of glucose tolerance. Ann. Intern. Med. 1993, 118, 529–539. [Google Scholar] [CrossRef]
- Campbell, M.D.; Sathish, T.; Zimmet, P.Z.; Thankappan, K.R.; Oldenburg, B.; Owens, D.R.; Shaw, J.E.; Tapp, R.J. Benefit of lifestyle-based T2DM prevention is influenced by prediabetes phenotype. Nat. Rev. Endocrinol. 2020, 16, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Veni, D.K.; Gupta, N.V. Diabetes mellitus treatment: A rapid review on innovative therapies. Asian J. Pharm. Clin. Res. 2019, 12, 46–53. [Google Scholar] [CrossRef][Green Version]
- Janez, A.; Guja, C.; Mitrakou, A.; Lalic, N.; Tankova, T.; Czupryniak, L.; Tabak, A.G.; Prazny, M.; Martinka, E.; Smircic-Duvnjak, L. Insulin Therapy in Adults with Type 1 Diabetes Mellitus: A Narrative Review. Diabetes Ther. 2020, 11, 387–409. [Google Scholar] [CrossRef]
- Gough, S.C.; Bode, B.; Woo, V.; Rodbard, H.W.; Linjawi, S.; Poulsen, P.; Damgaard, L.H.; Buse, J.B.; Investigators NNt. Efficacy and safety of a fixed-ratio combination of insulin degludec and liraglutide (IDegLira) compared with its components given alone: Results of a phase 3, open-label, randomised, 26-week, treat-to-target trial in insulin-naive patients with type 2 diabetes. Lancet Diabetes Endocrinol. 2014, 2, 885–893. [Google Scholar] [PubMed]
- Foretz, M.; Guigas, B.; Viollet, B. Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 569–589. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Aroda, V.R.; Henry, R.R.; Han, J.; Huang, W.; DeYoung, M.B.; Darsow, T.; Hoogwerf, B.J. Efficacy of GLP-1 receptor agonists and DPP-4 inhibitors: Meta-analysis and systematic review. Clin. Ther. 2012, 34, 1247–1258.e1222. [Google Scholar] [CrossRef]
- Wang, W.; Liu, H.; Xiao, S.; Liu, S.; Li, X.; Yu, P. Effects of Insulin Plus Glucagon-Like Peptide-1 Receptor Agonists (GLP-1RAs) in Treating Type 1 Diabetes Mellitus: A Systematic Review and Meta-Analysis. Diabetes Ther. 2017, 8, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Feutren, G.; Papoz, L.; Assan, R.; Vialettes, B.; Karsenty, G.; Vexiau, P.; Du Rostu, H.; Rodier, M.; Sirmai, J.; Lallemand, A.; et al. Cyclosporin increases the rate and length of remissions in insulin-dependent diabetes of recent onset. Results of a multicentre double-blind trial. Lancet 1986, 2, 119–124. [Google Scholar] [CrossRef]
- Warshauer, J.T.; Bluestone, J.A.; Anderson, M.S. New Frontiers in the Treatment of Type 1 Diabetes. Cell Metab. 2020, 31, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Edelman, S.; Maier, H.; Wilhelm, K. Pramlintide in the treatment of diabetes mellitus. BioDrugs 2008, 22, 375–386. [Google Scholar] [CrossRef]
- Aref, A.; Zayan, T.; Pararajasingam, R.; Sharma, A.; Halawa, A. Pancreatic transplantation: Brief review of the current evidence. World J. Transplant. 2019, 9, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Proks, P.; Reimann, F.; Green, N.; Gribble, F.; Ashcroft, F. Sulfonylurea stimulation of insulin secretion. Diabetes 2002, 51, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Kalra, S.; Aamir, A.H.; Raza, A.; Das, A.K.; Azad Khan, A.K.; Shrestha, D.; Qureshi, M.F.; Md, F.; Pathan, M.F.; Jawad, F.; et al. Place of sulfonylureas in the management of type 2 diabetes mellitus in South Asia: A consensus statement. Indian J. Endocrinol. Metab. 2015, 19, 577–596. [Google Scholar] [CrossRef]
- Hauner, H. The mode of action of thiazolidinediones. Diabetes Metab. Res. Rev. 2002, 2, S10–S15. [Google Scholar] [CrossRef] [PubMed]
- Eldor, R.; DeFronzo, R.A.; Abdul-Ghani, M. In vivo actions of peroxisome proliferator-activated receptors: Glycemic control, insulin sensitivity, and insulin secretion. Diabetes Care 2013, 36, 162–174. [Google Scholar] [CrossRef]
- Van de Laar, F.A.; Lucassen, P.L.; Akkermans, R.P.; Van de Lisdonk, E.H.; Rutten, G.E.; Van Weel, C. Alpha-glucosidase inhibitors for type 2 diabetes mellitus. Cochrane Database Syst. Rev. 2005, CD003639, 1–176. [Google Scholar] [CrossRef]
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons from the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72314 Cases from the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef]
- Pal, R.; Bhansali, A. COVID-19, diabetes mellitus and ACE2: The conundrum. Diabetes Res. Clin. Pract. 2020, 162, 108132. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Li, M.; Dong, Y.; Zhou, H.; Zhang, Z.; Tian, C.; Qin, R.; Wang, H.; Shen, Y.; Du, K.; et al. Diabetes is a risk factor for the progression and prognosis of COVID-19. Diabetes Metab. Res. Rev. 2020, e3319, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cuschieri, S.; Grech, S. COVID-19 and diabetes: The why, the what and the how. J. Diabetes Complicat. 2020, 34, 107637. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.K.; Lin, S.S.; Ji, X.J.; Guo, L.M. Binding of SARS coronavirus to its receptor damages islets and causes acute diabetes. Acta Diabetol. 2010, 47, 193–199. [Google Scholar] [CrossRef]
- Brunt, E.M.; Wong, V.W.; Nobili, V.; Day, C.P.; Sookoian, S.; Maher, J.J.; Bugianesi, E.; Sirlin, C.B.; Neuschwander-Tetri, B.A.; Rinella, M.E. Nonalcoholic fatty liver disease. Nat. Rev. Dis. Primers. 2015, 1, 15080. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, J.V.; Colombo, M.; Cortez-Pinto, H.; Huang, T.T.; Miller, V.; Ninburg, M.; Schattenberg, J.M.; Seim, L.; Wong, V.W.S.; Zelber-Sagi, S. NAFLD—Sounding the alarm on a silent epidemic. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 377–379. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Ress, C.; Kaser, S. Mechanisms of intrahepatic triglyceride accumulation. World J. Gastroenterol. 2016, 22, 1664–1673. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef]
- Trauner, M.; Arrese, M.; Wagner, M. Fatty liver and lipotoxicity. Biochim. Biophys. Acta 2010, 1801, 299–310. [Google Scholar] [CrossRef]
- Anderson, N.; Borlak, J. Molecular mechanisms and therapeutic targets in steatosis and steatohepatitis. Pharmacol. Rev. 2008, 60, 311–357. [Google Scholar] [CrossRef] [PubMed]
- Morigny, P.; Houssier, M.; Mouisel, E.; Langin, D. Adipocyte lipolysis and insulin resistance. Biochimie 2016, 125, 259–266. [Google Scholar] [CrossRef]
- Berk, P.D. Regulatable fatty acid transport mechanisms are central to the pathophysiology of obesity, fatty liver, and metabolic syndrome. Hepatology 2008, 48, 1362–1376. [Google Scholar] [CrossRef] [PubMed]
- Diraison, F.; Moulin, P.; Beylot, M. Contribution of hepatic de novo lipogenesis and reesterification of plasma non esterified fatty acids to plasma triglyceride synthesis during non-alcoholic fatty liver disease. Diabetes Metab. 2003, 29, 478–485. [Google Scholar] [CrossRef]
- Wang, Y.; Viscarra, J.; Kim, S.J.; Sul, H.S. Transcriptional regulation of hepatic lipogenesis. Nat. Rev. Mol. Cell Biol. 2015, 16, 678–689. [Google Scholar] [CrossRef]
- Hodson, L.; Gunn, P.J. Publisher Correction: The regulation of hepatic fatty acid synthesis and partitioning: The effect of nutritional state. Nat. Rev. Endocrinol. 2020, 16, 340. [Google Scholar] [CrossRef]
- Cooper, A.D. Hepatic uptake of chylomicron remnants. J. Lipid Res. 1997, 38, 2173–2192. [Google Scholar] [CrossRef]
- Tessari, P.; Coracina, A.; Cosma, A.; Tiengo, A. Hepatic lipid metabolism and non-alcoholic fatty liver disease. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 291–302. [Google Scholar] [CrossRef]
- Fabbrini, E.; Mohammed, B.S.; Magkos, F.; Korenblat, K.M.; Patterson, B.W.; Klein, S. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology 2008, 134, 424–431. [Google Scholar] [CrossRef]
- Hannou, S.A.; Haslam, D.E.; McKeown, N.M.; Herman, M.A. Fructose metabolism and metabolic disease. J. Clin. Investig. 2018, 128, 545–555. [Google Scholar] [CrossRef]
- Softic, S.; Cohen, D.E.; Kahn, C.R. Role of Dietary Fructose and Hepatic De Novo Lipogenesis in Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Svegliati-Baroni, G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J. Hepatol. 2018, 68, 280–295. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Vatner, D.F.; Shulman, G.I. Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol. 2017, 13, 572–587. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.W. Malonyl-CoA: The regulator of fatty acid synthesis and oxidation. J. Clin. Investig. 2012, 122, 1958–1959. [Google Scholar] [CrossRef]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell. Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef]
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering resolution of inflammation in, NASH. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 349–364. [Google Scholar] [CrossRef]
- Kesar, V.; Odin, J.A. Toll-like receptors and liver disease. Liver Int. 2014, 34, 184–196. [Google Scholar] [CrossRef]
- Arrese, M.; Cabrera, D.; Kalergis, A.M.; Feldstein, A.E. Innate Immunity Inflammation in, NAFLD/NASH. Dig Dis. Sci. 2016, 61, 1294–1303. [Google Scholar] [CrossRef]
- Petrasek, J.; Dolganiuc, A.; Csak, T.; Kurt-Jones, E.A.; Szabo, G. Type I interferons protect from Toll-like receptor 9-associated liver injury and regulate IL-1 receptor antagonist in mice. Gastroenterology 2011, 140, 697–708.e694. [Google Scholar] [CrossRef]
- Chatterjee, S.; Rana, R.; Corbett, J.; Kadiiska, M.B.; Goldstein, J.; Mason, R.P. P2X7 receptor-NADPH oxidase axis mediates protein radical formation and Kupffer cell activation in carbon tetrachloride-mediated steatohepatitis in obese mice. Free Radic. Biol. Med. 2012, 52, 1666–1679. [Google Scholar] [CrossRef]
- Morinaga, H.; Mayoral, R.; Heinrichsdorff, J.; Osborn, O.; Franck, N.; Hah, N.; Walenta, E.; Bandyopadhyay, G.; Pessentheiner, A.R.; Chi, T.J.; et al. Characterization of distinct subpopulations of hepatic macrophages in HFD/obese mice. Diabetes 2015, 64, 1120–1130. [Google Scholar] [CrossRef]
- Kiechl, S.; Wittmann, J.; Giaccari, A.; Knoflach, M.; Willeit, P.; Bozec, A.; Moschen, A.R.; Muscogiuri, G.; Sorice, G.P.; Kireva, T.; et al. Blockade of receptor activator of nuclear factor-kappaB (RANKL) signaling improves hepatic insulin resistance and prevents development of diabetes mellitus. Nat. Med. 2013, 19, 358–363. [Google Scholar] [CrossRef]
- Han, M.S.; Park, S.Y.; Shinzawa, K.; Kim, S.; Chung, K.W.; Lee, J.H.; Kwon, C.H.; Lee, K.W.; Lee, J.H.; Park, C.K.; et al. Lysophosphatidylcholine as a death effector in the lipoapoptosis of hepatocytes. J. Lipid Res. 2008, 49, 84–97. [Google Scholar] [CrossRef]
- Rocha, M.; Apostolova, N.; Diaz-Rua, R.; Muntane, J.; Victor, V.M. Mitochondria and T2D: Role of Autophagy, ER Stress, and Inflammasome. Trends Endocrinol. Metab. 2020, 31, 725–741. [Google Scholar] [CrossRef]
- Begriche, K.; Massart, J.; Robin, M.A.; Bonnet, F.; Fromenty, B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 2013, 58, 1497–1507. [Google Scholar] [CrossRef]
- Morris, E.M.; Rector, R.S.; Thyfault, J.P.; Ibdah, J.A. Mitochondria and redox signaling in steatohepatitis. Antioxid. Redox Signal. 2011, 15, 485–504. [Google Scholar] [CrossRef] [PubMed]
- Schmid, A.I.; Szendroedi, J.; Chmelik, M.; Krssak, M.; Moser, E.; Roden, M. Liver ATP synthesis is lower and relates to insulin sensitivity in patients with type 2 diabetes. Diabetes Care 2011, 34, 448–453. [Google Scholar] [CrossRef]
- Scorrano, L.; Oakes, S.A.; Opferman, J.T.; Cheng, E.H.; Sorcinelli, M.D.; Pozzan, T.; Korsmeyer, S.J. BAX and BAK regulation of endoplasmic reticulum Ca2+: A control point for apoptosis. Science 2003, 300, 135–139. [Google Scholar] [CrossRef]
- Nazal, L.; Riquelme, A.; Solis, N.; Pizarro, M.; Escalona, A.; Burotto, M.; Mendez, J.I.; Saint-Jean, C.; Concha, M.J.; Giovanni, S.; et al. Hypoadiponectinemia and its association with liver fibrosis in morbidly obese patients. Obes. Surg. 2010, 20, 1400–1407. [Google Scholar] [CrossRef] [PubMed]
- Arab, J.P.; Arrese, M.; Trauner, M. Recent Insights into the Pathogenesis of Nonalcoholic Fatty Liver Disease. Annu. Rev. Pathol. 2018, 13, 321–350. [Google Scholar] [CrossRef] [PubMed]
- Ikejima, K.; Okumura, K.; Lang, T.; Honda, H.; Abe, W.; Yamashina, S.; Enomoto, N.; Takei, Y.; Sato, N. The role of leptin in progression of non-alcoholic fatty liver disease. Hepatol. Res. 2005, 33, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed]
- Mancina, R.M.; Matikainen, N.; Maglio, C.; Soderlund, S.; Lundbom, N.; Hakkarainen, A.; Rametta, R.; Mozzi, E.; Fargion, S.; Valenti, L.; et al. Paradoxical dissociation between hepatic fat content and de novo lipogenesis due to PNPLA3 sequence variant. J. Clin. Endocrinol. Metab. 2015, 100, E821–E825. [Google Scholar] [CrossRef]
- Pirazzi, C.; Adiels, M.; Burza, M.A.; Mancina, R.M.; Levin, M.; Stahlman, M.; Taskinen, M.R.; Orho-Melander, M.; Perman, J.; Pujia, A.; et al. Patatin-like phospholipase domain-containing 3 (PNPLA3) I148M (rs738409) affects hepatic VLDL secretion in humans and in vitro. J. Hepatol. 2012, 57, 1276–1282. [Google Scholar] [CrossRef] [PubMed]
- Kalia, H.S.; Gaglio, P.J. The Prevalence and Pathobiology of Nonalcoholic Fatty Liver Disease in Patients of Different Races or Ethnicities. Clin. Liver. Dis. 2016, 20, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Patman, G.L.; Leathart, J.B.; Piguet, A.C.; Burt, A.D.; Dufour, J.F.; Day, C.P.; Daly, A.K.; Reeves, H.L.; Anstee, Q.M. Carriage of the PNPLA3 rs738409 C >G polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J. Hepatol. 2014, 61, 75–81. [Google Scholar] [CrossRef]
- Bruschi, F.V.; Claudel, T.; Tardelli, M.; Caligiuri, A.; Stulnig, T.M.; Marra, F.; Trauner, M. The PNPLA3 I148M variant modulates the fibrogenic phenotype of human hepatic stellate cells. Hepatology 2017, 65, 1875–1890. [Google Scholar] [CrossRef]
- Sookoian, S.; Pirola, C.J. Genetic predisposition in nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 2017, 23, 1–12. [Google Scholar] [CrossRef]
- Marzuillo, P.; Grandone, A.; Perrone, L.; Miraglia Del Giudice, E. Understanding the pathophysiological mechanisms in the pediatric non-alcoholic fatty liver disease: The role of genetics. World J. Hepatol. 2015, 7, 1439–1443. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Charatcharoenwitthaya, P.; Treeprasertsuk, S.; Benson, J.T.; Enders, F.B.; Angulo, P. The natural history of non-alcoholic fatty liver disease in children: A follow-up study for up to 20 years. Gut 2009, 58, 1538–1544. [Google Scholar] [CrossRef]
- Di Sessa, A.; Umano, G.R.; Miraglia Del Giudice, E. The Association between Non-Alcoholic Fatty Liver Disease and Cardiovascular Risk in Children. Children 2017, 4, 57. [Google Scholar] [CrossRef] [PubMed]
- Di Sessa, A.; Umano, G.R.; Miraglia Del Giudice, E.; Santoro, N. From the liver to the heart: Cardiac dysfunction in obese children with non-alcoholic fatty liver disease. World J. Hepatol. 2017, 9, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Koot, B.G.; de Groot, E.; van der Baan-Slootweg, O.H.; Bohte, A.E.; Nederveen, A.J.; Jansen, P.L.; Stoker, J.; Benninga, M.A. Nonalcoholic fatty liver disease and cardiovascular risk in children with obesity. Obesity 2015, 23, 1239–1243. [Google Scholar] [CrossRef] [PubMed]
- Di Sessa, A.; Guarino, S.; Umano, G.R.; Arenella, M.; Alfiero, S.; Quaranta, G.; Miraglia Del Giudice, E.; Marzuillo, P. MAFLD in Obese Children: A Challenging Definition. Children 2021, 8, 247. [Google Scholar] [CrossRef]
- Barrera, F.; George, J. The role of diet nutritional intervention for the management of patients with, NAFLD. Clin. Liver Dis. 2014, 18, 91–112. [Google Scholar] [CrossRef]
- El-Agroudy, N.N.; Kurzbach, A.; Rodionov, R.N.; O’Sullivan, J.; Roden, M.; Birkenfeld, A.L.; Pesta, D.H. Are Lifestyle Therapies Effective for NAFLD Treatment? Trends. Endocrinol. Metab. 2019, 30, 701–709. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010, 52, 774–788. [Google Scholar] [CrossRef]
- Li, Y.; Liu, L.; Wang, B.; Wang, J.; Chen, D. Metformin in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Biomed. Rep. 2013, 1, 57–64. [Google Scholar] [CrossRef]
- Yu, J.G.; Javorschi, S.; Hevener, A.L.; Kruszynska, Y.T.; Norman, R.A.; Sinha, M.; Olefsky, J.M. The effect of thiazolidinediones on plasma adiponectin levels in normal, obese, and type 2 diabetic subjects. Diabetes 2002, 51, 2968–2974. [Google Scholar] [CrossRef]
- Cuthbertson, D.J.; Irwin, A.; Gardner, C.J.; Daousi, C.; Purewal, T.; Furlong, N.; Goenka, N.; Thomas, E.L.; Adams, V.L.; Pushpakom, S.P.; et al. Improved glycaemia correlates with liver fat reduction in obese, type 2 diabetes, patients given glucagon-like peptide-1 (GLP-1) receptor agonists. PLoS ONE 2012, 7, e50117. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.; Gaunt, P.; Aithal, G.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; Abouda, G.; Aldersley, M.A.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef]
- Miyazaki, M.; Kato, M.; Tanaka, K.; Tanaka, M.; Kohjima, M.; Nakamura, K.; Enjoji, M.; Nakamuta, M.; Kotoh, K.; Takayanagi, R. Increased hepatic expression of dipeptidyl peptidase-4 in non-alcoholic fatty liver disease and its association with insulin resistance and glucose metabolism. Mol. Med. Rep. 2012, 5, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Scheen, A.J. Beneficial effects of SGLT2 inhibitors on fatty liver in type 2 diabetes: A common comorbidity associated with severe complications. Diabetes Metab. 2019, 45, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Mudaliar, S.; Henry, R.R.; Sanyal, A.J.; Morrow, L.; Marschall, H.U.; Kipnes, M.; Adorini, L.; Sciacca, C.I.; Clopton, P.; Castelloe, E.; et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013, 145, 574–582.e571. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Saha, P.K.; Chan, L.; Moore, D.D. Farnesoid X receptor is essential for normal glucose homeostasis. J. Clin. Investig. 2006, 116, 1102–1109. [Google Scholar] [CrossRef]
- Watanabe, M.; Houten, S.M.; Wang, L.; Moschetta, A.; Mangelsdorf, D.J.; Heyman, R.A.; Moore, D.D.; Auwerx, J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Investig. 2004, 113, 1408–1418. [Google Scholar] [CrossRef]
- Wagner, M.; Zollner, G.; Trauner, M. Nuclear bile acid receptor farnesoid X receptor meets nuclear factor-kappaB: New insights into hepatic inflammation. Hepatology 2008, 48, 1383–1386. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, J.; Liu, Q.; Harnish, D.C. Farnesoid X receptor agonist WAY-362450 attenuates liver inflammation and fibrosis in murine model of non-alcoholic steatohepatitis. J. Hepatol. 2009, 51, 380–388. [Google Scholar] [CrossRef]
- Gomez-Dominguez, E.; Gisbert, J.P.; Moreno-Monteagudo, J.A.; Garcia-Buey, L.; Moreno-Otero, R. A pilot study of atorvastatin treatment in dyslipemid, non-alcoholic fatty liver patients. Aliment Pharmacol. Ther. 2006, 23, 1643–1647. [Google Scholar] [CrossRef]
- Okada, Y.; Yamaguchi, K.; Nakajima, T.; Nishikawa, T.; Jo, M.; Mitsumoto, Y.; Kimura, H.; Nishimura, T.; Tochiki, N.; Yasui, K.; et al. Rosuvastatin ameliorates high-fat and high-cholesterol diet-induced nonalcoholic steatohepatitis in rats. Liver Int. 2013, 33, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.S.; Liu, S.; Chen, X.M.; Huang, Z.G.; Zhang, D.W. Effects of n-3 polyunsaturated fatty acids from seal oils on nonalcoholic fatty liver disease associated with hyperlipidemia. World J. Gastroenterol. 2008, 14, 6395–6400. [Google Scholar] [CrossRef] [PubMed]
- Hossain, N.; Kanwar, P.; Mohanty, S.R. AComprehensive Updated Review of Pharmaceutical Nonpharmaceutical Treatment for NAFLD. Gastroenterol. Res. Pract. 2016, 2016, 7109270. [Google Scholar] [CrossRef] [PubMed]
- Nakade, Y.; Murotani, K.; Inoue, T.; Kobayashi, Y.; Yamamoto, T.; Ishii, N.; Ohashi, T.; Ito, K.; Fukuzawa, Y.; Yoneda, M. Ezetimibe for the treatment of non-alcoholic fatty liver disease: A meta-analysis. Hepatol. Res. 2017, 47, 1417–1428. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, M.; Fujita, K.; Nozaki, Y.; Endo, H.; Takahashi, H.; Hosono, K.; Suzuki, K.; Mawatari, H.; Kirikoshi, H.; Inamori, M.; et al. Efficacy of ezetimibe for the treatment of non-alcoholic steatohepatitis: An open-label, pilot study. Hepatol. Res. 2010, 40, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Gilat, T.; Leikin-Frenkel, A.; Goldiner, L.; Laufer, H.; Halpern, Z.; Konikoff, F.M. Arachidyl amido cholanoic acid (Aramchol) is a cholesterol solubilizer and prevents the formation of cholesterol gallstones in inbred mice. Lipids 2001, 36, 1135–1140. [Google Scholar] [CrossRef]
- Safadi, R.; Konikoff, F.M.; Mahamid, M.; Zelber-Sagi, S.; Halpern, M.; Gilat, T.; Oren, R.; Group, F. The fatty acid-bile acid conjugate Aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2014, 12, 2085–2091.e2081. [Google Scholar] [CrossRef]
- Imai, N.; Cohen, D.E. Trimming the Fat: Acetyl-CoACarboxylase Inhibition for the Management of NAFLD. Hepatology 2018, 68, 2062–2065. [Google Scholar] [CrossRef]
- Goedeke, L.; Bates, J.; Vatner, D.F.; Perry, R.J.; Wang, T.; Ramirez, R.; Li, L.; Ellis, M.W.; Zhang, D.; Wong, K.E.; et al. Acetyl-CoA Carboxylase Inhibition Reverses NAFLD and Hepatic Insulin Resistance but Promotes Hypertriglyceridemia in Rodents. Hepatology 2018, 68, 2197–2211. [Google Scholar] [CrossRef]
- Sumida, Y.; Yoneda, M. Current future pharmacological therapies for, NAFLD/NASH. J. Gastroenterol. 2018, 53, 362–376. [Google Scholar] [CrossRef]
- Harrison, S.A.; Torgerson, S.; Hayashi, P.; Ward, J.; Schenker, S. Vitamin E and vitamin C treatment improves fibrosis in patients with nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2003, 98, 2485–2490. [Google Scholar] [CrossRef]
- Dufour, J.F.; Oneta, C.M.; Gonvers, J.J.; Bihl, F.; Cerny, A.; Cereda, J.M.; Zala, J.F.; Helbling, B.; Steuerwald, M.; Zimmermann, A.; et al. Randomized placebo-controlled trial of ursodeoxycholic acid with vitamin e in nonalcoholic steatohepatitis. Clin. Gastroenterol. Hepatol. 2006, 4, 1537–1543. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T.; Yoneda, M.; Nakamura, K.; Makino, I.; Terano, A. Plasma transforming growth factor-beta1 level and efficacy of alpha-tocopherol in patients with non-alcoholic steatohepatitis: A pilot study. Aliment Pharmacol. Ther. 2001, 15, 1667–1672. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.R., 3rd; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.A.; Appel, L.J.; Guallar, E. Meta-analysis: High-dosage vitamin E supplementation may increase all-cause, mortality. Ann. Intern. Med. 2005, 142, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.A.; Thompson, I.M.; Jr Tangen, C.M.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the risk of prostate cancer: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2011, 306, 1549–1556. [Google Scholar] [CrossRef]
- Khoshbaten, M.; Aliasgarzadeh, A.; Masnadi, K.; Tarzamani, M.K.; Farhang, S.; Babaei, H.; Kiani, J.; Zaare, M.; Najafipoor, F. N-acetylcysteine improves liver function in patients with non-alcoholic Fatty liver disease. Hepat. Mon. 2010, 10, 12–16. [Google Scholar]
- Kathirvel, E.; Morgan, K.; Nandgiri, G.; Sandoval, B.C.; Caudill, M.A.; Bottiglieri, T.; French, S.W.; Morgan, T.R. Betaine improves nonalcoholic fatty liver and associated hepatic insulin resistance: A potential mechanism for hepatoprotection by betaine. Am. J. Physiol. Gastrointest. Liver. Physiol. 2010, 299, G1068–G1077. [Google Scholar] [CrossRef]
- Cariou, B.; Hanf, R.; Lambert-Porcheron, S.; Zair, Y.; Sauvinet, V.; Noel, B.; Flet, L.; Vidal, H.; Staels, B.; Laville, M. Dual peroxisome proliferator-activated receptor alpha/delta agonist GFT505 improves hepatic and peripheral insulin sensitivity in abdominally obese subjects. Diabetes Care 2013, 36, 2923–2930. [Google Scholar] [CrossRef]
- Staels, B.; Rubenstrunk, A.; Noel, B.; Rigou, G.; Delataille, P.; Millatt, L.J.; Baron, M.; Lucas, A.; Tailleux, A.; Hum, D.W.; et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2013, 58, 1941–1952. [Google Scholar] [CrossRef]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-alpha and -delta, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159.e1145. [Google Scholar] [CrossRef]
- Zein, C.O.; Yerian, L.M.; Gogate, P.; Lopez, R.; Kirwan, J.P.; Feldstein, A.E.; McCullough, A.J. Pentoxifylline improves nonalcoholic steatohepatitis: A randomized placebo-controlled trial. Hepatology 2011, 54, 1610–1619. [Google Scholar] [CrossRef] [PubMed]
- Iacono, A.; Raso, G.M.; Canani, R.B.; Calignano, A.; Meli, R. Probiotics as an emerging therapeutic strategy to treat NAFLD: Focus on molecular and biochemical mechanisms. J. Nutr. Biochem. 2011, 22, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Perumpail, B.J.; Li, A.A.; John, N.; Sallam, S.; Shah, N.D.; Kwong, W.; Cholankeril, G.; Kim, D.; Ahmed, A. The Therapeutic Implications of the Gut Microbiome Probiotics in Patients with, NAFLD. Diseases 2019, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, M.; Shabat, Y.; Ben Ya’acov, A.; Lalazar, G.; Adar, T.; Wong, V.; Muller, B.; Rawlin, G.; Ilan, Y. Alleviation of insulin resistance liver damage by oral administration of Imm124-E is mediated by increased Tregs associated with increased serum, G.L.P.-1.; adiponectin: Results of a phase I/II clinical trial in, NASH. J. Inflamm. Res. 2012, 5, 141–150. [Google Scholar] [PubMed]
- Bramante, C.; Tignanelli, C.J.; Dutta, N.; Jones, E.; Tamariz, L.; Clark, J.M.; Usher, M.; Metlon-Meaux, G.; Ikramuddin, S. Non-alcoholic fatty liver disease (NAFLD) and risk of hospitalization for Covid-19. MedRxiv 2020, 1–7. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus Panel. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1991. [Google Scholar] [CrossRef]
- Zhou, Y.J.; Zheng, K.I.; Wang, X.B.; Sun, Q.F.; Pan, K.H.; Wang, T.Y.; Ma, H.L.; Chen, Y.P.; George, J.; Zheng, M.H. Metabolic-associated fatty liver disease is associated with severity of COVID-19. Liver. Int. 2020, 40, 2160–2163. [Google Scholar] [CrossRef]
- Sharma, P.; Kumar, A. Metabolic dysfunction associated fatty liver disease increases risk of severe Covid-19. Diabetes Metab. Syndr. 2020, 14, 825–827. [Google Scholar] [CrossRef]
- Targher, G.; Mantovani, A.; Byrne, C.D.; Wang, X.B.; Yan, H.D.; Sun, Q.F.; Pan, K.H.; Zheng, K.I.; Chen, Y.P.; Eslam, M.; et al. Risk of severe illness from COVID-19 in patients with metabolic dysfunction-associated fatty liver disease and increased fibrosis scores. Gut 2020, 69, 1545–1547. [Google Scholar] [CrossRef]
- Ji, D.; Qin, E.; Xu, J.; Zhang, D.; Cheng, G.; Wang, Y.; Lau, G. Non-alcoholic fatty liver diseases in patients with COVID-19, A retrospective study. J. Hepatol. 2020, 73, 451–453. [Google Scholar] [CrossRef]
- Hegyi, P.J.; Váncsa, S.; Ocskay, K.; Dembrovszky, F.; Kiss, S.; Farkas, N.; Erőss, B.; Szakács, Z.; Hegyi, P.; Pár, G. Metabolic Associated Fatty Liver Disease Is Associated with an Increased Risk of Severe COVID-19, A Systematic Review with Meta-Analysis. Front. Med. 2021, 8, 277. [Google Scholar] [CrossRef] [PubMed]
- Jose, R.J. Manuel A: COVID-19 cytokine storm: The interplay between inflammation and coagulation. Lancet Respir. Med. 2020, 8, E46–E47. [Google Scholar] [CrossRef]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Flora, G.D.; Nayak, M.K. A Brief Review of Cardiovascular Diseases, Associated Risk Factors and Current Treatment Regimes. Curr. Pharm. Des. 2019, 25, 4063–4084. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, K.; VanderZanden, A.; Shepard, D.; Leach-Kemon, K. Cardiovascular Disease Worldwide, 1990–2013. JAMA 2015, 314, 1905. [Google Scholar] [CrossRef]
- Friedrich, M.J. Global Obesity Epidemic Worsening. JAMA 2017, 318, 603. [Google Scholar] [CrossRef]
- Chen, L.; Magliano, D.J.; Zimmet, P.Z. The worldwide epidemiology of type 2 diabetes mellitus--present and future perspectives. Nat. Rev. Endocrinol. 2011, 8, 228–236. [Google Scholar] [CrossRef]
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef]
- Yaniv, Y.; Juhaszova, M.; Nuss, H.B.; Wang, S.; Zorov, D.B.; Lakatta, E.G.; Sollott, S.J. Matching ATP supply and demand in mammalian heart: In vivo, in vitro, and in silico perspectives. Ann. N. Y. Acad. Sci. 2010, 1188, 133–142. [Google Scholar] [CrossRef]
- O’Neill, S.; O’Driscoll, L. Metabolic syndrome: A closer look at the growing epidemic and its associated pathologies. Obes. Rev. 2015, 16, 1–12. [Google Scholar] [CrossRef]
- Yamazoe, M.; Hisamatsu, T.; Miura, K.; Kadowaki, S.; Zaid, M.; Kadota, A.; Torii, S.; Miyazawa, I.; Fujiyoshi, A.; Arima, H.; et al. Relationship of Insulin Resistance to Prevalence and Progression of Coronary Artery Calcification Beyond Metabolic Syndrome Components: Shiga Epidemiological Study of Subclinical Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1703–1708. [Google Scholar] [CrossRef]
- Perrone-Filardi, P.; Paolillo, S.; Costanzo, P.; Savarese, G.; Trimarco, B.; Bonow, R.O. The role of metabolic syndrome in heart failure. Eur. Heart J. 2015, 36, 2630–2634. [Google Scholar] [CrossRef]
- Flegal, K.M.; Graubard, B.I.; Williamson, D.F.; Gail, M.H. Cause-specific excess deaths associated with underweight, overweight, and obesity. JAMA 2007, 298, 2028–2037. [Google Scholar] [CrossRef] [PubMed]
- Alpert, M.A. Obesity cardiomyopathy: Pathophysiology and evolution of the clinical syndrome. Am. J. Med. Sci. 2001, 321, 225–236. [Google Scholar] [CrossRef]
- Grundy, S.M. Obesity, metabolic syndrome, and cardiovascular disease. J. Clin. Endocrinol. Metab. 2004, 89, 2595–2600. [Google Scholar] [CrossRef] [PubMed]
- Turkbey, E.B.; McClelland, R.L.; Kronmal, R.A.; Burke, G.L.; Bild, D.E.; Tracy, R.P.; Arai, A.E.; Lima, J.A.; Bluemke, D.A. The impact of obesity on the left ventricle: The Multi-Ethnic Study of Atherosclerosis (MESA). JACC Cardiovasc. Imaging 2010, 3, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Sassoon, D.J.; Goodwill, A.G.; Noblet, J.N.; Conteh, A.M.; Herring, B.P.; McClintick, J.N.; Tune, J.D.; Mather, K.J. Obesity alters molecular and functional cardiac responses to ischemia/reperfusion and glucagon-like peptide-1 receptor agonism. Basic Res. Cardiol. 2016, 111, 43. [Google Scholar] [CrossRef]
- Pascual, F.; Coleman, R.A. Fuel availability and fate in cardiac metabolism: A tale of two substrates. Biochim. Biophys. Acta 2016, 1861, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Coort, S.L.; Luiken, J.J.; van der Vusse, G.J.; Bonen, A.; Glatz, J.F. Increased FAT (fatty acid translocase)/CD36-mediated long-chain fatty acid uptake in cardiac myocytes from obese Zucker rats. Biochem. Soc. Trans. 2004, 32, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Ormazabal, V.; Nair, S.; Elfeky, O.; Aguayo, C.; Salomon, C.; Zuniga, F.A. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc. Diabetol. 2018, 17, 122. [Google Scholar] [CrossRef]
- Eddy, D.; Schlessinger, L.; Kahn, R.; Peskin, B.; Schiebinger, R. Relationship of insulin resistance and related metabolic variables to coronary artery disease: A mathematical analysis. Diabetes Care 2009, 32, 361–366. [Google Scholar] [CrossRef]
- Zhou, Y.T.; Grayburn, P.; Karim, A.; Shimabukuro, M.; Higa, M.; Baetens, D.; Orci, L.; Unger, R.H. Lipotoxic heart disease in obese rats: Implications for human obesity. Proc. Natl. Acad. Sci. USA 2000, 97, 1784–1789. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, E.; Picatoste, B.; Gonzalez-Bris, A.; Oteo, M.; Cruz, F.; Caro-Vadillo, A.; Egido, J.; Tunon, J.; Morcillo, M.A.; Lorenzo, O. Sitagliptin improved glucose assimilation in detriment of fatty-acid utilization in experimental type-II diabetes: Role of GLP-1 isoforms in Glut4 receptor trafficking. Cardiovasc. Diabetol. 2018, 17, 12. [Google Scholar] [CrossRef] [PubMed]
- Bonora, E.; Kiechl, S.; Willeit, J.; Oberhollenzer, F.; Egger, G.; Targher, G.; Alberiche, M.; Bonadonna, R.C.; Muggeo, M. Prevalence of insulin resistance in metabolic disorders: The Bruneck Study. Diabetes 1998, 47, 1643–1649. [Google Scholar] [CrossRef] [PubMed]
- Tenenbaum, A.; Adler, Y.; Boyko, V.; Tenenbaum, H.; Fisman, E.Z.; Tanne, D.; Lapidot, M.; Schwammenthal, E.; Feinberg, M.S.; Matas, Z.; et al. Insulin resistance is associated with increased risk of major cardiovascular events in patients with preexisting coronary artery disease. Am. Heart J. 2007, 153, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.; Hama, S.; Liu, Y.; Siahmansur, T.; Schofield, J.; Syed, A.A.; France, M.; Pemberton, P.; Adam, S.; Ho, J.H.; et al. Effect of Roux-en-Y Bariatric Surgery on Lipoproteins, Insulin Resistance, and Systemic and Vascular Inflammation in Obesity and Diabetes. Front. Immunol. 2017, 8, 1512. [Google Scholar] [CrossRef] [PubMed]
- De Luca, C.; Olefsky, J.M. Inflammation and insulin resistance. FEBS Lett. 2008, 582, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Austin, M.A.; Hokanson, J.E.; Edwards, K.L. Hypertriglyceridemia as a cardiovascular risk factor. Am. J. Cardiol. 1998, 81, 7B–12B. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The role of mitochondrial dysfunction in cardiovascular disease: A brief review. Ann. Med. 2018, 50, 121–127. [Google Scholar] [CrossRef]
- Hoppel, C.L.; Tandler, B.; Fujioka, H.; Riva, A. Dynamic organization of mitochondria in human heart and in myocardial disease. Int. J. Biochem. Cell Biol. 2009, 41, 1949–1956. [Google Scholar] [CrossRef]
- Stadhouders, A.M.; Jap, P.H.; Winkler, H.P.; Eppenberger, H.M.; Wallimann, T. Mitochondrial creatine kinase: A major constituent of pathological inclusions seen in mitochondrial myopathies. Proc. Natl. Acad. Sci. USA 1994, 91, 5089–5093. [Google Scholar] [CrossRef]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure: Implications beyond ATP production. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, F.D. Cardiovascular disease: Different strategies for primary and secondary prevention? Heart 2004, 90, 1217–1223. [Google Scholar] [CrossRef] [PubMed]
- Leong, D.P.; Joseph, P.G.; McKee, M.; Anand, S.S.; Teo, K.K.; Schwalm, J.D.; Yusuf, S. Reducing the Global Burden of Cardiovascular Disease, Part 2: Prevention and Treatment of Cardiovascular Disease. Circ. Res. 2017, 121, 695–710. [Google Scholar] [CrossRef]
- Pietrzak, E.; Cotea, C.; Pullman, S. Primary and secondary prevention of cardiovascular disease: Is there a place for Internet-based interventions? J. Cardiopulm. Rehabil. Prev. 2014, 34, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Gaede, P.; Vedel, P.; Larsen, N.; Jensen, G.V.; Parving, H.H.; Pedersen, O. Multifactorial intervention and cardiovascular disease in patients with type 2 diabetes. N. Engl. J. Med. 2003, 348, 383–393. [Google Scholar] [CrossRef]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jodar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef] [PubMed]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Czernichow, S.; Ninomiya, T.; Huxley, R.; Kengne, A.P.; Batty, G.D.; Grobbee, D.E.; Woodward, M.; Neal, B.; Chalmers, J. Impact of blood pressure lowering on cardiovascular outcomes in normal weight, overweight, and obese individuals: The Perindopril Protection Against Recurrent Stroke Study trial. Hypertension 2010, 55, 1193–1198. [Google Scholar] [CrossRef]
- Law, M.R.; Morris, J.K.; Wald, N.J. Use of blood pressure lowering drugs in the prevention of cardiovascular disease: Meta-analysis of 147 randomised trials in the context of expectations from prospective epidemiological studies. BMJ 2009, 338, b1665. [Google Scholar] [CrossRef]
- Cannon, C.P.; Blazing, M.A.; Giugliano, R.P.; McCagg, A.; White, J.A.; Theroux, P.; Darius, H.; Lewis, B.S.; Ophuis, T.O.; Jukema, J.W.; et al. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N. Engl. J. Med. 2015, 372, 2387–2397. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Leiter, L.A.; Wiviott, S.D.; Giugliano, R.P.; Deedwania, P.; De Ferrari, G.M.; Murphy, S.A.; Kuder, J.F.; Gouni-Berthold, I.; Lewis, B.S.; et al. Cardiovascular safety and efficacy of the PCSK9 inhibitor evolocumab in patients with and without diabetes and the effect of evolocumab on glycaemia and risk of new-onset diabetes: A prespecified analysis of the FOURIER randomised controlled trial. Lancet Diabetes Endocrinol. 2017, 5, 941–950. [Google Scholar] [CrossRef]
- Schupke, S.; Neumann, F.J.; Menichelli, M.; Mayer, K.; Bernlochner, I.; Wohrle, J.; Richardt, G.; Liebetrau, C.; Witzenbichler, B.; Antoniucci, D.; et al. Ticagrelor or Prasugrel in Patients with Acute Coronary Syndromes. N. Engl. J. Med. 2019, 381, 1524–1534. [Google Scholar] [CrossRef]
- Yusuf, S.; Zhao, F.; Mehta, S.R.; Chrolavicius, S.; Tognoni, G.; Fox, K.K. Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial I: Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N. Engl. J. Med. 2001, 345, 494–502. [Google Scholar]
- Thom, S.; Poulter, N.; Field, J.; Patel, A.; Prabhakaran, D.; Stanton, A.; Grobbee, D.E.; Bots, M.L.; Reddy, K.S.; Cidambi, R.; et al. Effects of a fixed-dose combination strategy on adherence and risk factors in patients with or at high risk of CVD: The UMPIRE randomized clinical trial. JAMA 2013, 310, 918–929. [Google Scholar] [CrossRef] [PubMed]
- Koliaki, C.; Doupis, J. Incretin-based therapy: A powerful and promising weapon in the treatment of type 2 diabetes mellitus. Diabetes Ther. 2011, 2, 101–121. [Google Scholar] [CrossRef] [PubMed]
- Husain, M.; Birkenfeld, A.L.; Donsmark, M.; Dungan, K.; Eliaschewitz, F.G.; Franco, D.R.; Jeppesen, O.K.; Lingvay, I.; Mosenzon, O.; Pedersen, S.D.; et al. Oral Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2019, 381, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Ou, H.T.; Chang, K.C.; Li, C.Y.; Wu, J.S. Risks of cardiovascular diseases associated with dipeptidyl peptidase-4 inhibitors and other antidiabetic drugs in patients with type 2 diabetes: A nation-wide longitudinal study. Cardiovasc. Diabetol. 2016, 15, 41. [Google Scholar] [CrossRef]
- Ou, H.T.; Chang, K.C.; Li, C.Y.; Wu, J.S. Comparative cardiovascular risks of dipeptidyl peptidase 4 inhibitors with other second- and third-line antidiabetic drugs in patients with type 2 diabetes. Br. J. Clin. Pharmacol. 2017, 83, 1556–1570. [Google Scholar] [CrossRef]
- Heerspink, H.J.; Perkins, B.A.; Fitchett, D.H.; Husain, M.; Cherney, D.Z. Sodium Glucose Cotransporter 2 Inhibitors in the Treatment of Diabetes Mellitus: Cardiovascular and Kidney Effects, Potential Mechanisms, and Clinical Applications. Circulation 2016, 134, 752–772. [Google Scholar] [CrossRef]
- Kemp, B.E.; Stapleton, D.; Campbell, D.J.; Chen, Z.P.; Murthy, S.; Walter, M.; Gupta, A.; Adams, J.J.; Katsis, F.; van Denderen, B.; et al. AMP-activated protein kinase, super metabolic regulator. Biochem. Soc. Trans. 2003, 31, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.K.; Steinberg, G.R. AMP-activated protein kinase, fatty acid metabolism, and insulin sensitivity. Curr. Opin. Clin. Nutr. Metab. Care 2017, 20, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Paiva, M.A.; Goncalves, L.M.; Providencia, L.A.; Davidson, S.M.; Yellon, D.M.; Mocanu, M.M. Transitory activation of AMPK at reperfusion protects the ischaemic-reperfused rat myocardium against infarction. Cardiovasc. Drugs Ther. 2010, 24, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Shibata, R.; Ouchi, N.; Ito, M.; Kihara, S.; Shiojima, I.; Pimentel, D.R.; Kumada, M.; Sato, K.; Schiekofer, S.; Ohashi, K.; et al. Adiponectin-mediated modulation of hypertrophic signals in the heart. Nat. Med. 2004, 10, 1384–1389. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.J.; Xie, Z.; Viollet, B.; Zou, M.H. Activation of the AMP-activated kinase by antidiabetes drug metformin stimulates nitric oxide synthesis in vivo by promoting the association of heat shock protein 90 and endothelial nitric oxide synthase. Diabetes 2006, 55, 496–505. [Google Scholar] [CrossRef]
- Fujita, Y.; Hosokawa, M.; Fujimoto, S.; Mukai, E.; Abudukadier, A.; Obara, A.; Ogura, M.; Nakamura, Y.; Toyoda, K.; Nagashima, K.; et al. Metformin suppresses hepatic gluconeogenesis and lowers fasting blood glucose levels through reactive nitrogen species in mice. Diabetologia 2010, 53, 1472–1481. [Google Scholar] [CrossRef]
- Wang, S.; Xu, J.; Song, P.; Viollet, B.; Zou, M.H. In vivo activation of AMP-activated protein kinase attenuates diabetes-enhanced degradation of GTP cyclohydrolase, I. Diabetes 2009, 58, 1893–1901. [Google Scholar] [CrossRef]
- Morrison, A.; Yan, X.; Tong, C.; Li, J. Acute rosiglitazone treatment is cardioprotective against ischemia-reperfusion injury by modulating AMPK, Akt, and JNK signaling in nondiabetic mice. Am. J. Physiol. Heart. Circ. Physiol. 2011, 301, H895–H902. [Google Scholar] [CrossRef]
- Tsimikas, S.; Hall, J.L. Lipoprotein(a) as a potential causal genetic risk factor of cardiovascular disease: A rationale for increased efforts to understand its pathophysiology and develop targeted therapies. J. Am. Coll. Cardiol. 2012, 60, 716–721. [Google Scholar] [CrossRef]
- Saeed, A.; Virani, S.S. Lipoprotein(a) and cardiovascular disease: Current state and future directions for an enigmatic lipoprotein. Front. Biosci. 2018, 23, 1099–1112. [Google Scholar]
- Chapman, M.J.; Redfern, J.S.; McGovern, M.E.; Giral, P. Niacin and fibrates in atherogenic dyslipidemia: Pharmacotherapy to reduce cardiovascular risk. Pharmacol. Ther. 2010, 126, 314–345. [Google Scholar] [CrossRef] [PubMed]
- Yeang, C.; Clopton, P.C.; Tsimikas, S. Lipoprotein(a)-cholesterol levels estimated by vertical auto profile correlate poorly with Lp(a) mass in hyperlipidemic subjects: Implications for clinical practice interpretation of Lp(a)-mediated risk. J. Clin. Lipidol. 2016, 10, 1389–1396. [Google Scholar] [CrossRef] [PubMed]
- Yeang, C.; Hung, M.Y.; Byun, Y.S.; Clopton, P.; Yang, X.; Witztum, J.L.; Tsimikas, S. Effect of therapeutic interventions on oxidized phospholipids on apolipoprotein B100 and lipoprotein(a). J. Clin. Lipidol. 2016, 10, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Akaike, M.; Azuma, H.; Kagawa, A.; Matsumoto, K.; Hayashi, I.; Tamura, K.; Nishiuchi, T.; Iuchi, T.; Takamori, N.; Aihara, K.; et al. Effect of aspirin treatment on serum concentrations of lipoprotein(a) in patients with atherosclerotic diseases. Clin. Chem. 2002, 48, 1454–1459. [Google Scholar] [CrossRef]
- Raal, F.J.; Giugliano, R.P.; Sabatine, M.S.; Koren, M.J.; Langslet, G.; Bays, H.; Blom, D.; Eriksson, M.; Dent, R.; Wasserman, S.M.; et al. Reduction in lipoprotein(a) with PCSK9 monoclonal antibody evolocumab (AMG 145): A pooled analysis of more than 1,300 patients in 4 phase II trials. J. Am. Coll. Cardiol. 2014, 63, 1278–1288. [Google Scholar] [CrossRef]
- Graham, M.J.; Viney, N.; Crooke, R.M.; Tsimikas, S. Antisense inhibition of apolipoprotein (a) to lower plasma lipoprotein (a) levels in humans. J. Lipid Res. 2016, 57, 340–351. [Google Scholar] [CrossRef]
- Merki, E.; Graham, M.; Taleb, A.; Leibundgut, G.; Yang, X.; Miller, E.R.; Fu, W.; Mullick, A.E.; Lee, R.; Willeit, P.; et al. Antisense oligonucleotide lowers plasma levels of apolipoprotein (a) and lipoprotein (a) in transgenic mice. J. Am. Coll. Cardiol 2011, 57, 1611–1621. [Google Scholar] [CrossRef]
- Viney, N.J.; van Capelleveen, J.C.; Geary, R.S.; Xia, S.; Tami, J.A.; Yu, R.Z.; Marcovina, S.M.; Hughes, S.G.; Graham, M.J.; Crooke, R.M.; et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): Two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 2016, 388, 2239–2253. [Google Scholar] [CrossRef]
- Zhang, R. The ANGPTL3-4-8 model, a molecular mechanism for triglyceride trafficking. Open Biol. 2016, 6, 150272. [Google Scholar] [CrossRef]
- Dijk, W.; Kersten, S. Regulation of lipid metabolism by angiopoietin-like proteins. Curr. Opin. Lipidol. 2016, 27, 249–256. [Google Scholar] [CrossRef]
- Shan, L.; Yu, X.C.; Liu, Z.; Hu, Y.; Sturgis, L.T.; Miranda, M.L.; Liu, Q. The angiopoietin-like proteins ANGPTL3 and ANGPTL4 inhibit lipoprotein lipase activity through distinct mechanisms. J. Biol. Chem. 2009, 284, 1419–1424. [Google Scholar] [CrossRef] [PubMed]
- Aryal, B.; Singh, A.K.; Zhang, X.; Varela, L.; Rotllan, N.; Goedeke, L.; Chaube, B.; Camporez, J.P.; Vatner, D.F.; Horvath, T.L.; et al. Absence of ANGPTL4 in adipose tissue improves glucose tolerance and attenuates atherogenesis. JCI Insight 2018, 3, e97918. [Google Scholar] [CrossRef] [PubMed]
- Gusarova, V.; O’Dushlaine, C.; Teslovich, T.M.; Benotti, P.N.; Mirshahi, T.; Gottesman, O.; Van Hout, C.V.; Murray, M.F.; Mahajan, A.; Nielsen, J.B.; et al. Genetic inactivation of ANGPTL4 improves glucose homeostasis and is associated with reduced risk of diabetes. Nat. Commun. 2018, 9, 2252. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; McNutt, M.C.; Banfi, S.; Levin, M.G.; Holland, W.L.; Gusarova, V.; Gromada, J.; Cohen, J.C.; Hobbs, H.H. Hepatic ANGPTL3 regulates adipose tissue energy homeostasis. Proc. Natl. Acad. Sci. USA 2015, 112, 11630–11635. [Google Scholar] [CrossRef] [PubMed]
- Dewey, F.E.; Gusarova, V.; Dunbar, R.L.; O’Dushlaine, C.; Schurmann, C.; Gottesman, O.; McCarthy, S.; Van Hout, C.V.; Bruse, S.; Dansky, H.M.; et al. Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 211–221. [Google Scholar] [CrossRef]
- Lin, L.; Burke, J.; Venkatesh, S.; Sadana, P. AMPK-SIRT1-independent inhibition of ANGPTL3 gene expression is a potential lipid-lowering mechanism of metformin. J. Pharm. Pharmacol. 2019, 71, 1421–1428. [Google Scholar] [CrossRef]
- Go, G.W. Low-Density Lipoprotein Receptor-Related Protein 6 (LRP6) Is a Novel Nutritional Therapeutic Target for Hyperlipidemia, Non-Alcoholic Fatty Liver Disease, and Atherosclerosis. Nutrients 2015, 7, 4453–4464. [Google Scholar] [CrossRef]
- Bastakoty, D.; Saraswati, S.; Joshi, P.; Atkinson, J.; Feoktistov, I.; Liu, J.; Harris, J.L.; Young, P.P. Temporary, Systemic Inhibition of the WNT/beta-Catenin Pathway promotes Regenerative Cardiac Repair following Myocardial Infarct. Cell Stem. Cells Regen. Med. 2016, 2, 2. [Google Scholar] [CrossRef]
- Cheng, P.W.; Chen, Y.Y.; Cheng, W.H.; Lu, P.J.; Chen, H.H.; Chen, B.R.; Yeh, T.C.; Sun, G.C.; Hsiao, M.; Tseng, C.J. Wnt Signaling Regulates Blood Pressure by Downregulating a GSK-3beta-Mediated Pathway to Enhance Insulin Signaling in the Central Nervous System. Diabetes 2015, 64, 3413–3424. [Google Scholar] [CrossRef]
- Bonora, M.; Wieckowski, M.R.; Sinclair, D.A.; Kroemer, G.; Pinton, P.; Galluzzi, L. Targeting mitochondria for cardiovascular disorders: Therapeutic potential and obstacles. Nat. Rev. Cardiol. 2019, 16, 33–55. [Google Scholar] [CrossRef]
- McLachlan, J.; Beattie, E.; Murphy, M.P.; Koh-Tan, C.H.; Olson, E.; Beattie, W.; Dominiczak, A.F.; Nicklin, S.A.; Graham, D. Combined therapeutic benefit of mitochondria-targeted antioxidant, MitoQ10, and angiotensin receptor blocker, losartan, on cardiovascular function. J. Hypertens. 2014, 32, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, S.; Haruyama, A.; Inami, S.; Arikawa, T.; Saito, F.; Watanabe, R.; Sakuma, M.; Abe, S.; Nakajima, T.; Tanaka, A.; et al. Effects of carvedilol vs bisoprolol on inflammation and oxidative stress in patients with chronic heart failure. J. Cardiol. 2020, 75, 140–147. [Google Scholar] [CrossRef]
- Daiber, A.; Xia, N.; Steven, S.; Oelze, M.; Hanf, A.; Kroller-Schon, S.; Munzel, T.; Li, H. New Therapeutic Implications of Endothelial Nitric Oxide Synthase (eNOS) Function/Dysfunction in Cardiovascular Disease. Int. J. Mol. Sci. 2019, 20, 187. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, P.; Schulz, E.; Oelze, M.; Muller, J.; Schuhmacher, S.; Alhamdani, M.S.; Debrezion, J.; Hortmann, M.; Reifenberg, K.; Fleming, I.; et al. AT1-receptor blockade by telmisartan upregulates GTP-cyclohydrolase I and protects eNOS in diabetic rats. Free Radic. Biol. Med. 2008, 45, 619–626. [Google Scholar] [CrossRef]
- Broeders, M.A.; Doevendans, P.A.; Bekkers, B.C.; Bronsaer, R.; van Gorsel, E.; Heemskerk, J.W.; Egbrink, M.G.; van Breda, E.; Reneman, R.S.; van Der Zee, R. Nebivolol: A third-generation beta-blocker that augments vascular nitric oxide release: Endothelial beta(2)-adrenergic receptor-mediated nitric oxide production. Circulation 2000, 102, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Oelze, M.; Daiber, A.; Brandes, R.P.; Hortmann, M.; Wenzel, P.; Hink, U.; Schulz, E.; Mollnau, H.; von Sandersleben, A.; Kleschyov, A.L.; et al. Nebivolol inhibits superoxide formation by NADPH oxidase and endothelial dysfunction in angiotensin II-treated rats. Hypertension 2006, 48, 677–684. [Google Scholar] [CrossRef]
- Lu, D.; Thum, T. RNA-based diagnostic and therapeutic strategies for cardiovascular disease. Nat. Rev. Cardiol. 2019, 16, 661–674. [Google Scholar] [CrossRef]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef]
- Small, E.M.; Olson, E.N. Pervasive roles of microRNAs in cardiovascular biology. Nature 2011, 469, 336–342. [Google Scholar] [CrossRef]
- Latronico, M.V.; Condorelli, G. MicroRNAs and cardiac pathology. Nat. Rev. Cardiol. 2009, 6, 419–429. [Google Scholar] [CrossRef]
- Schober, A.; Weber, C. Mechanisms of MicroRNAs in Atherosclerosis. Annu. Rev. Pathol. 2016, 11, 583–616. [Google Scholar] [CrossRef] [PubMed]
- Thum, T. Noncoding RNAs and myocardial fibrosis. Nat. Rev. Cardiol. 2014, 11, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Lucas, T.; Bonauer, A.; Dimmeler, S. RNA Therapeutics in Cardiovascular Disease. Circ. Res. 2018, 123, 205–220. [Google Scholar] [CrossRef]
- Mellis, D.; Caporali, A. MicroRNA-based therapeutics in cardiovascular disease: Screening and delivery to the target. Biochem. Soc. Trans. 2018, 46, 11–21. [Google Scholar] [CrossRef] [PubMed]
- van Rooij, E.; Olson, E.N. MicroRNA therapeutics for cardiovascular disease: Opportunities and obstacles. Nat. Rev. Drug. Discov. 2012, 11, 860–872. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ha, T.; Liu, L.; Zou, J.; Zhang, X.; Kalbfleisch, J.; Gao, X.; Williams, D.; Li, C. Increased expression of microRNA-146a decreases myocardial ischaemia/reperfusion injury. Cardiovasc. Res. 2013, 97, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Xie, J.; Li, R.; Shi, J.; Sun, J.; Gu, R.; Ding, L.; Wang, L.; Xu, B. Overexpression of microRNA-99a attenuates heart remodelling and improves cardiac performance after myocardial infarction. J. Cell Mol. Med. 2014, 18, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Bei, Y.; Chen, P.; Lei, Z.; Fu, S.; Zhang, H.; Xu, J.; Che, L.; Chen, X.; Sluijter, J.P.; et al. Crucial Role of miR-433 in Regulating Cardiac Fibrosis. Theranostics 2016, 6, 2068–2083. [Google Scholar] [CrossRef] [PubMed]
- Beermann, J.; Piccoli, M.T.; Viereck, J.; Thum, T. Non-coding RNAs in Development and Disease: Background, Mechanisms, and Therapeutic Approaches. Physiol. Rev. 2016, 96, 1297–1325. [Google Scholar] [CrossRef]
- Pankratz, F.; Hohnloser, C.; Bemtgen, X.; Jaenich, C.; Kreuzaler, S.; Hoefer, I.; Pasterkamp, G.; Mastroianni, J.; Zeiser, R.; Smolka, C.; et al. MicroRNA-100 Suppresses Chronic Vascular Inflammation by Stimulation of Endothelial Autophagy. Circ/ Res. 2018, 122, 417–432. [Google Scholar] [CrossRef]
- Lesizza, P.; Prosdocimo, G.; Martinelli, V.; Sinagra, G.; Zacchigna, S.; Giacca, M. Single-Dose Intracardiac Injection of Pro-Regenerative MicroRNAs Improves Cardiac Function After Myocardial Infarction. Circ. Res. 2017, 120, 1298–1304. [Google Scholar] [CrossRef] [PubMed]
- Cunha, P.; Romao, A.M.; Mascarenhas-Melo, F.; Teixeira, H.M.; Reis, F. Endocannabinoid system in cardiovascular disorders—New pharmacotherapeutic opportunities. J. Pharm. Bioallied. Sci. 2011, 3, 350–360. [Google Scholar] [PubMed]
- Godlewski, G.; Alapafuja, S.O.; Batkai, S.; Nikas, S.P.; Cinar, R.; Offertaler, L.; Osei-Hyiaman, D.; Liu, J.; Mukhopadhyay, B.; Harvey-White, J.; et al. Inhibitor of fatty acid amide hydrolase normalizes cardiovascular function in hypertension without adverse metabolic effects. Chem. Biol. 2010, 17, 1256–1266. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.S.; Gardiner, S.M. Acute hypertension reveals depressor and vasodilator effects of cannabinoids in conscious rats. Br. J. Pharmacol. 2009, 156, 94–104. [Google Scholar] [CrossRef]
- Jarai, Z.; Wagner, J.A.; Goparaju, S.K.; Wang, L.; Razdan, R.K.; Sugiura, T.; Zimmer, A.M.; Bonner, T.I.; Zimmer, A.; Kunos, G. Cardiovascular effects of 2-arachidonoyl glycerol in anesthetized mice. Hypertension 2000, 35, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Polak, A.; Harasim-Symbor, E.; Malinowska, B.; Kasacka, I.; Pedzinska-Betiuk, A.; Weresa, J.; Chabowski, A. The effects of chronic FAAH inhibition on myocardial lipid metabolism in normotensive and DOCA-salt hypertensive rats. Life Sci. 2017, 183, 1–10. [Google Scholar] [CrossRef]
- Wang, Y.; Kaminski, N.E.; Wang, D.H. Endocannabinoid regulates blood pressure via activation of the transient receptor potential vanilloid type 1 in Wistar rats fed a high-salt diet. J. Pharmacol. Exp. Ther. 2007, 321, 763–769. [Google Scholar] [CrossRef]
- Wheal, A.J.; Bennett, T.; Randall, M.D.; Gardiner, S.M. Cardiovascular effects of cannabinoids in conscious spontaneously hypertensive rats. Br. J. Pharmacol. 2007, 152, 717–724. [Google Scholar] [CrossRef]
- Farah, C.; Michel, L.Y.M.; Balligand, J.L. Nitric oxide signalling in cardiovascular health and disease. Nat. Rev. Cardiol. 2018, 15, 292–316. [Google Scholar] [CrossRef]
- Camargo, A.B.; Manucha, W. [Potential protective role of nitric oxide and Hsp70 linked to functional foods in the atherosclerosis]. Clin. Investig. Arterioscler. 2017, 29, 36–45. [Google Scholar] [CrossRef]
- Mazzei, L.; Docherty, N.G.; Manucha, W. Mediators and mechanisms of heat shock protein 70 based cytoprotection in obstructive nephropathy. Cell Stress Chaperones 2015, 20, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Liu, Y.X.; Yuan, F.; Ma, H.J.; Maslov, L.; Zhang, Y. Enhanced vasorelaxation effect of endogenous anandamide on thoracic aorta in renal vascular hypertension rats. Clin. Exp. Pharmacol. Physiol. 2015, 42, 950–955. [Google Scholar] [CrossRef]
- Stanley, C.P.; Hind, W.H.; Tufarelli, C.; O’Sullivan, S.E. The endocannabinoid anandamide causes endothelium-dependent vasorelaxation in human mesenteric arteries. Pharmacol. Res. 2016, 113, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Baranowska-Kuczko, M.; Kozlowska, H.; Kozlowski, M.; Schlicker, E.; Kloza, M.; Surazynski, A.; Grzęda, E.; Malinowska, B. Mechanisms of endothelium-dependent relaxation evoked by anandamide in isolated human pulmonary arteries. Naunyn Schmiedeberg’s Arch. Pharmacol. 2014, 387, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Quercioli, A.; Pataky, Z.; Vincenti, G.; Makoundou, V.; Di Marzo, V.; Montecucco, F.; Carballo, S.; Thomas, A.; Staub, C.; Steffens, S.; et al. Elevated endocannabinoid plasma levels are associated with coronary circulatory dysfunction in obesity. Eur. Heart J. 2011, 32, 1369–1378. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, F.; Matias, I.; Lenglet, S.; Petrosino, S.; Burger, F.; Pelli, G.; Braunersreuther, V.; Mach, F.; Steffens, S.; Di Marzo, V. Regulation and possible role of endocannabinoids and related mediators in hypercholesterolemic mice with atherosclerosis. Atherosclerosis 2009, 205, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Steffens, S.; Veillard, N.R.; Arnaud, C.; Pelli, G.; Burger, F.; Staub, C.; Karsak, M.; Zimmer, A.; Frossard, J.L.; Mach, F. Low dose oral cannabinoid therapy reduces progression of atherosclerosis in mice. Nature 2005, 434, 782–786. [Google Scholar] [CrossRef]
- Rajesh, M.; Mukhopadhyay, P.; Hasko, G.; Liaudet, L.; Mackie, K.; Pacher, P. Cannabinoid-1 receptor activation induces reactive oxygen species-dependent and -independent mitogen-activated protein kinase activation and cell death in human coronary artery endothelial cells. Br. J. Pharmacol. 2010, 160, 688–700. [Google Scholar] [CrossRef]
- Abdel-Salam, O. Gastric acid inhibitory and gastric protective effects of Cannabis and cannabinoids. Asian Pac. J. Trop. Med. 2016, 9, 413–419. [Google Scholar] [CrossRef]
- Palomba, L.; Silvestri, C.; Imperatore, R.; Morello, G.; Piscitelli, F.; Martella, A.; Cristino, L.; Di Marzo, V. Negative Regulation of Leptin-induced Reactive Oxygen Species (ROS) Formation by Cannabinoid CB1 Receptor Activation in Hypothalamic Neurons. J. Biol. Chem. 2015, 290, 13669–13677. [Google Scholar] [CrossRef]
- Nishiga, M.; Wang, D.W.; Han, Y.; Lewis, D.B.; Wu, J.C. COVID-19 and cardiovascular disease: From basic mechanisms to clinical perspectives. Nat. Rev. Cardiol. 2020, 17, 543–558. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Bosso, M.; Thanaraj, T.A.; Abu-Farha, M.; Alanbaei, M.; Abubaker, J.; Al-Mulla, F. The Two Faces of ACE2: The Role of ACE2 Receptor and Its Polymorphisms in Hypertension and COVID-19. Mol. Ther. Methods. Clin. Dev. 2020, 18, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, X.; Chen, J.; Zhang, H.; Deng, A. Association of Renin-Angiotensin System Inhibitors with Severity or Risk of Death in Patients with Hypertension Hospitalized for Coronavirus Disease 2019 (COVID-19) Infection in Wuhan, China. JAMA Cardiol. 2020, 5, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, H.R.; Adhikari, S.; Pulgarin, C.; Troxel, A.B.; Iturrate, E.; Johnson, S.B.; Hausvater, A.; Newman, J.D.; Berger, J.S.; Bangalore, S.; et al. Renin-Angiotensin-Aldosterone System Inhibitors and Risk of Covid-19. N. Engl. J. Med. 2020, 382, 2441–2448. [Google Scholar] [CrossRef] [PubMed]
- Vaduganathan, M.; Vardeny, O.; Michel, T.; McMurray, J.J.V.; Pfeffer, M.A.; Solomon, S.D. Renin-Angiotensin-Aldosterone System Inhibitors in Patients with Covid-19. N. Engl. J. Med. 2020, 382, 1653–1659. [Google Scholar] [CrossRef] [PubMed]


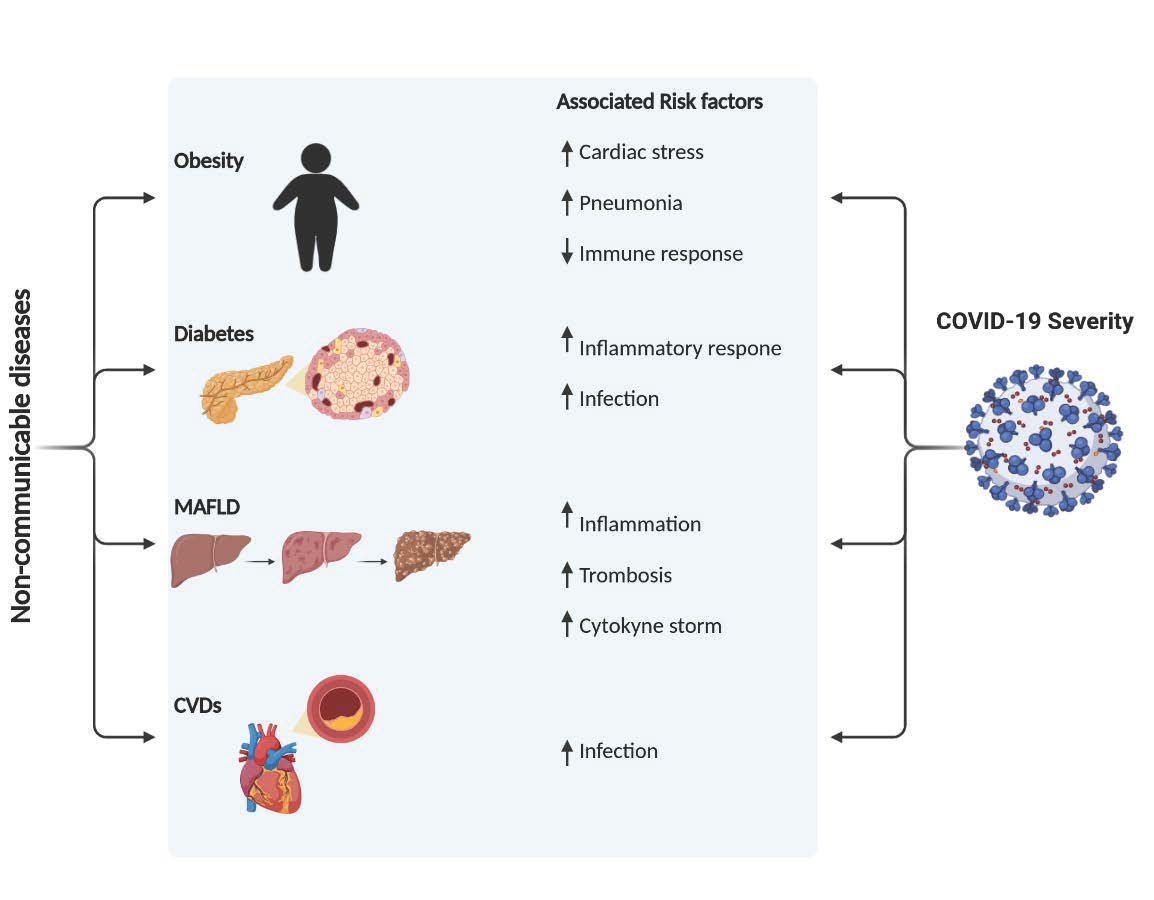{kind=link}
{kind=link}
| Class | Mecanism of Action | Drugs | Current State | Side Effects | Reference |
|---|---|---|---|---|---|
| Lipase inhibitors | Inhibit long-chain FAs absorption | Orlistat | Commercial | Abdominal pain Fecal urgency Flatulence Oil stool | [73,99] |
| Induce weight loss Improve glycemic control Improve lipid profile | Cetilistat | Under studies | Diarrhea Flatulence Oily spotting | [136] | |
| Adrenergic agonists | Increase norepinephrine release in CNS | Phentermine | Commercial | Increase heart rate and blood pressure | [99,100,101,102] |
| GABA receptor modulators | Neuroestabilizer Enhance thermogenesis | Topiramate | Commercial | Alteration of taste GI upset Nausea | [103,104] |
| POMC neurons activators | Induce α-MSH release | Naltrexone/Bupropion | Commercial | Nausea | [107,108,109,110,111,112,113] |
| Delay gastric emptying Enhance insulin secretin | Liraglutide * | Commercial | Transient nausea Vomiting | [114,115] | |
| GLP-1 receptor agonists | Reduce blood glucose levels Induce weight loss | Semaglutide TTP-054 ZYOGI | Commercial | Nausea Vomiting Diarrhea | [124,125,126,127] |
| Induce insulin release Decrease HbA1c levels | ZP4165 | Under studies | Unknown | [128] | |
| Leptin analogues | Lower hepatic steatosis Improve insulin sensitivity | Metreleptin | Commercial | Nausea | [119,120,121] |
| Amylin analogues | Delay gastric emptying | Pramlintide * Davalintide * | Commercial | Nausea | [122,123] |
| Glucagon and GLP-1 receptors agonists | Supresses appetite Increase energy expenditure | Oxyntomodulin | Under studies | Unknown | [129] |
| GLP-1, glucagon and GIP receptors agonists | Induce weight loss | Triagonist 1706 | Under studies | Unknown | [132] |
| CB1 antagonists | Stimulate anorexigenic signaling | AM-6545 | Under studies | Unknown | [134,135] |
| Vaccines | Restrain appetite-stimulating hormones Decrease nutrient absorption | Anti-ghrelin Anti-somastatin Anti-ad36 | Under studies | Unknown | [137,138,139,140,141] |
| Induction of beige-cells | Increase thermogenic gene expression Epigenetic modulators Activation of AMPK pathway | Capsaicin Curcumin PUFAs | Under studies | Unknown | [152,153,154,155,156,157,158] |
| Class | Mecanism of Action | Drugs | Type | Current State | Side Effects | Reference |
|---|---|---|---|---|---|---|
| Hormone | Reduce blood glucose levels | Insulin | 1 and 2 | Commercial | Hypoglicemia | [208,209] |
| Biguanides | Reduce hepatic glucose production | Metformin * Phenformin * Buformin * | 1 and 2 | Commercial | Abdominal discomfort | [169,211] |
| SGLT2 inhibitors | Prevent glucose reabsorption | Canagliflozin * Dapagliflozin * Empagliflozin * | 1 and 2 | Commercial | Urinary tract and genital infections Decrease in blood pressure Weight gain | [193,212,213] |
| DPP4 inhibitors | Improve glycemic control | Sitagliptin * Vildagliptin * Saxagliptin * Linagliptin * Alogliptin * | 1 and 2 | Commercial | Hypoglicemia Loss of consciousness Gastrointestinal side effects | [214] |
| GLP-1 receptor agonist | Promote insulin secretion | Exenatide * Liraglutide * Lixisenatide * Dulaglutide * | 1 and 2 | Commercial | Transient nausea Vomiting | [215] |
| Calcineurin inhibitor | Inhibit T cell activation | Cyclosporin | 1 | Commercial | Nephrotoxicity Increase risk of cancer | [216,217] |
| Amylin analogues | Reduce blood glucose levels Induce weight loss | Pramlintide * | 1 | Commercial | Nausea | [217,219] |
| Sulfonylureas and glinides | Increase insulin secretion | Tolbutamide Glibenclamide Glipizide | 2 | Commercial | Hypoglicemia Weight gain | [220,221] |
| PPARγ agonists | Increase tissues sensibility to insulin action | Rosiglitazone * Pioglitazone * | 2 | Commercial | Fluid retention Weight gain Trauma-related fractures | [222,223] |
| Alpha glucosidase inhibitors | Slow the carbohydrate absorption | Acarbose | 2 | Commercial | Diarrhea Nausea Abdominal pain | [224] |
| Class | Mecanism of Action | Drugs | Current State | Side Effects | Reference |
|---|---|---|---|---|---|
| Biguanides | Reduce aminotransferase levels Increase insulin sensitivity | Metformin * | Under studies | Abdominal discomfort Diarrhea Nausea | [285] |
| PPARy agonist | Increase FFA uptake, hepatic lipogenesis and insulin sensitivity | Rosiglitazone * Pioglitazone * | Under studies | Increase risk of CVD development, congestive heart failure, bladder cancer and bone loss | [283,286] |
| GLP-1 receptor agonists | Delay fibrosis progression | Liraglutide * Exenatide * | Under studies | Transient nausea Vomiting | [287,288,289] |
| SGLT2 inhibitors | Reduce fatty liver content Improve levels of serum liver enzymes | Canagliflozin * Dapagliflozin * Empagliflozin * | Under studies | Urinary tract and genital infections Decrease in blood pressure Weight gain | [290] |
| FXR receptor agonist | Regulate hepatic metabolism of bile and cholesterol | Obeticholic acid | Under studies | Pruritus Dyslipidemia Fatigue Headache Gastrointestinal side effect | [291,292,293,294,295] |
| Statins | Decrease hepatic FFA, steatosis, hepatic fibrosis and inflammatory markers | Atorvastatin Fluvastatin Lovastatin Pitavastatin Pravastatin Rosuvastatin | Under studies | Muscle pain (myalgia) Creatine phosphokinase elevation | [296,297] |
| Polyunsaturated fatty acids | Improve overall symptoms Decrease TG and alanine transaminase levels | Eicosapentaenoic acid Docosahexaenoic acid | Under studies | Unknown | [298] |
| Lipid lowering | Inhibit cholesterol and phytosterol absorption | Ezetimibe | Under studies | Hepatotoxicity Severe cholestatic hepatitis Acute autoimmune hepatitis | [300,301] |
| Stearoyl-CoA desaturase inhibitors | Improve hepatic lipid accumulation | Aramchol | Under studies | Unknown | [302,303] |
| Acetyl-CoA carboxylase inhibitors | Reduce DNL and FA content | GS0976 | Under studies | Unknown | [305,306] |
| Antioxidants | Decrease aminotransferases levels Improve inflammation, steatosis, ballooning and steatohepatitis | Vitamin E | Under studies | Increase blood pressure and heart failure risk | [307,308,309,310,311] |
| Increase glutathione in hepatocytes | N-acetylcysteine | Under studies | Nausea Vomiting Diarrhea Transient skin rash Flushing Epigastric pain Constipation | [312] | |
| Induce anti-inflammatory, cytoprotective, antiapoptotic and anti-steatogenic actions | Betaine | Under studies | Gastrointestinal side effects | [313] | |
| PPAR-α/δ agonists | Induce anti-inflammatory and antifibrotic effects Improve insulin sensitivity and hepatic function | Elafibranor | Under studies | Congestive heart failure Peripheral edema Bone fractures Weight gain | [314,315,316] |
| Xanthine derivatives | Improve ALT levels, steatosis, inflammation and fibrosis | Pentoxifylline | Under studies | Unknown | [317] |
| Probiotic therapy | Improve hepatic histology, inflammation and biochemical markers | - | Under studies | Unknown | [318,319] |
| Hyperimmune bovine colostrum | Improve glycemic control Reduce liver exposure to LPS and gut bacterial bioproducts | IMM-124E | Under studies | Unknown | [320] |
| Class | Mecanism of Action | Drugs | Current State | Side Effects | Reference |
|---|---|---|---|---|---|
| GLP-1 receptor agonists | Reduce abdominal visceral fat and systolic blood pressure Improve endothelial and myocardial function | Liraglutide * Exenatide * | Under studies | Transient nausea Vomiting | [363,372,373] |
| DPP4 inhibitors | Degrade GLP-1 | Sitagliptin * Vildagliptin * Saxagliptin * Linagliptin * Alogliptin * | Under studies | Hypoglicemia Loss of consciousness Gastrointestinal side effects | [374,375] |
| SGLT2 inhibitors | Promote glucose reabsorption Decrease blood glucose concentrationImprove insulin sensitivity Reduce glucose toxicity and blood pressure Induce nephroprotection | Empagliflozin * | Under studies | Urinary tract and genital infections Decrease in blood pressure Weight gain | [213,376] |
| AMPK activators | Induce NO production Suppress 26S mediated GTP-cyclohydrolase degradation | Metformin * | Under studies | Abdominal discomfort Diarrhea Nausea | [381,382,383] |
| Reduce isquemia, reperfusion and myocardial infarction | TZDs | Under studies | Increase risk of CVD development, congestive heart failure, bladder cancer and bone loss | [384] | |
| Lp(a)and LPL-C modulators | Reduce Lp(a), LDL-c, apo B-100, sdLDL and TG levels Raise HDL levels | Niacin | Under studies | Hepatic toxicity Myopathy Blurred vision Nausea Vomiting | [387,388,389] |
| Decrease Lp(a) levels | ASA | Under studies | GI upset Nausea | [390] | |
| Reduce LDL-c levels | Evolocumab Alirocumab | Under studies | Nasopharyngitis Injection site pain Arthralgia Back pain | [386,391] | |
| Inhibit apo(a) synthesis and LP(a) secretion | ASOs | Under studies | Unknown | [392,393] | |
| LPL inhibitors | Suppress LPL activityReduce TG level s Increase HDL-C levels | ANGPTLs | Under studies | Unknown | [395,396,397,398,399,400,401,402] |
| Mitochondrial therapies | Decrease ROS production | MitoQ1 | Under studies | Unknown | [407] |
| Reduce oxidative stress and inflammation | Statins ACE inhibitors AT-1 receptor blocker | Under studies | Unknown | [409,410,411,412] | |
| RNA-based therapies | Reduce myocardial infarct size Improve cardiac function | miR-146a | Under studies | Unknown | [422] |
| Prevent cardiomyocyte apoptosis Promote autophagy | miR-99a | Under studies | Unknown | [423] | |
| Ameliorate cardiac fibrosis and ventricular dysfunction | miR-433 | Under studies | Unknown | [424] | |
| Attenuate atherosclerosis | miR-100 | Under studies | Unknown | [426] | |
| Promote cardiac repair Reduce infarct size Preserve cardiac function | miR-199a-3p miR-590-3p | Under studies | Unknown | [427] | |
| Endocannabinoids | Induce hypotensive and vascular effects | Anandamide 2-arachidonoylglycerol | Under studies | Neuropsychiatric side effects | [429,430,431,432,433,434] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerra, J.V.S.; Dias, M.M.G.; Brilhante, A.J.V.C.; Terra, M.F.; García-Arévalo, M.; Figueira, A.C.M. Multifactorial Basis and Therapeutic Strategies in Metabolism-Related Diseases. Nutrients 2021, 13, 2830. https://doi.org/10.3390/nu13082830
Guerra JVS, Dias MMG, Brilhante AJVC, Terra MF, García-Arévalo M, Figueira ACM. Multifactorial Basis and Therapeutic Strategies in Metabolism-Related Diseases. Nutrients. 2021; 13(8):2830. https://doi.org/10.3390/nu13082830
Chicago/Turabian StyleGuerra, João V. S., Marieli M. G. Dias, Anna J. V. C. Brilhante, Maiara F. Terra, Marta García-Arévalo, and Ana Carolina M. Figueira. 2021. "Multifactorial Basis and Therapeutic Strategies in Metabolism-Related Diseases" Nutrients 13, no. 8: 2830. https://doi.org/10.3390/nu13082830
APA StyleGuerra, J. V. S., Dias, M. M. G., Brilhante, A. J. V. C., Terra, M. F., García-Arévalo, M., & Figueira, A. C. M. (2021). Multifactorial Basis and Therapeutic Strategies in Metabolism-Related Diseases. Nutrients, 13(8), 2830. https://doi.org/10.3390/nu13082830