
DeltaF508 CFTR Hetero- and Homozygous Paediatric Patients with Cystic Fibrosis Do Not Differ with Regard to Nutritional Status

 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
3. Results
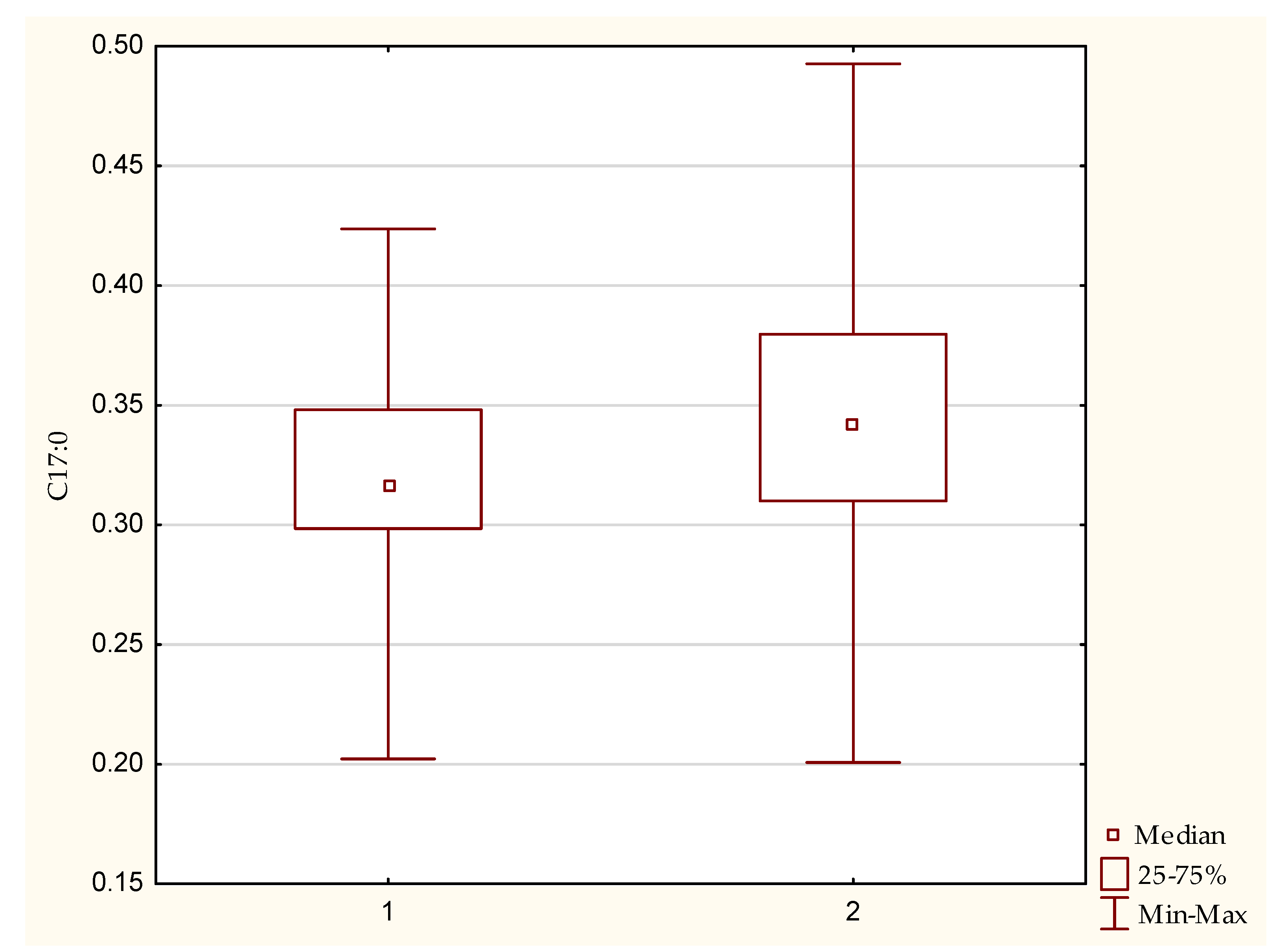
3.1. Fatty Acid Profile
3.2. Vitamins
3.3. Anthropometric Measurements
3.4. Correlation Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
| Parameters | CF Patients n = 75 | Healthy Controls n = 33 | p |
|---|---|---|---|
| Mean age (months) (range) | 110.3 ± 64.6 (9–216) | 116 ± 65 (9–216) | p = 0.71 |
| Anthropometric Measurements | |||
| Mean FFM%-anthropometric | 77.53 ± 6.54 | 68.77 ± 9.4 | p = 0.00 |
| Mean FM%-anthropometric | 22.47 ± 6.54 | 31.23 ± 9.4 | p = 0.00 |
| Mean FFM%-BIA | 81.20 ± 10.93 | 80.77 ± 8.06 | p = 0.08 |
| Mean FM%-BIA | 17.6 ± 6.04 | 19.23 ± 8.06 | p = 0.08 |
| Mean BMI | 16.8 ± 2.2 | 19 ± 4.00 | p = 0.02 |
| BMI Percentile | |||
| <3 | 8% | 3.03% | - |
| 3–15 | 12% | 3.03% | - |
| 15–50 | 45.3% | 36.4% | - |
| 50–85 | 24% | 24.24% | - |
| 85–97 | 9% | 18.18% | - |
| ≥97 | 2.7% | 15.15% | - |
| Fat-Soluble Vitamins Mean | |||
| A (μg/mL) | 0.8 ± 0.2 | 1.0 ± 0 | p = 0.09 |
| 25-OH-D3 (ng/mL) | 13.0 ± 3.3 | 24.0 ± 5.0 | p = 0.0 |
| E (μg/mL) | 6.1 ± 3.4 | 10.0 ± 2.0 | p = 0.0 |
| K1 (ng/mL)2 | 0.4 ± 0.2 | 1.0 ± 0.0 | p = 0.03 |
| Select Data on Fatty Acid Profiles in Erythrocyte Membranes: Acids with Statistically Significant Differences | |||
| Saturated | |||
| Myristic acid (C14:0) | 0.6 ± 0.2 | 0.0 ± 0 | p = 0.0 |
| Lignoceric acid (C24:0) | 4.0 ± 0.6 | 4.0 ± 0 | p = 0.001 |
| Behenic acid (C22:0) | 1.6 ± 0.3 | 2.0 ± 0 | p = 0.001 |
| Monounsaturated | |||
| Palmitoleic acid (C16:1 n-7) | 0.4 ± 0.2 | 0.0 ± 0 | p = 0.0 |
| Heptadecenoic acid (C17:1) | 0.1 ± 0 | 0.0 ± 0 | p = 0.04 |
| Vaccenic acid (C18:1 n-7 cis) | 1.0 ± 0.2 | 1.0 ± 0 | p = 0.038 |
| Erucic acid (C22:1) | 0.2 ± 0.1 | 0.0 ± 0 | p = 0.009 |
| PUFA: Omega n-6 | |||
| Gamma-linolenic acid (18:3 n-6) | 0.1 ± 0.0 | 0.0 ± 0 | p = 0.00 |
| Dihomo-gamma-linolenic acid (20:3 n-6) | 1.7 ± 0.4 | 1.0 ± 0 | p = 0.003 |
| Linoleic acid (18:2 n-6) | 7.9 ± 1.5 | 9.0 ± 1 | p = 0.0 |
| PUFA: Omega n-3 | |||
| Hexadecatrienoic acid (16:3 n-3) | 0.1 ± 0.1 | 0.0 ± 1 | p = 0.024 |
References
- O’Sullivan, B.P.; Freedman, S.D. Cystic fibrosis. Lancet 2009, 373, 1891–1904. [Google Scholar] [CrossRef]
- Cystic Fibrosis Mutation Database. Available online: www.genet.sickkids.on.ca (accessed on 5 January 2021).
- Welsh, M.J.; Smith, A.E. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 1993, 73, 1251–1254. [Google Scholar] [CrossRef]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 2016, 27, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Ratjen, F.; Bell, S.C.; Rowe, S.M.; Goss, C.H.; Quittner, A.L.; Bush, A. Cystic fibrosis. Nat. Rev. Dis. Primers 2015, 1, 15010. [Google Scholar] [CrossRef] [PubMed]
- De Boeck, K.; Amaral, M.D. Progress in therapies for cystic fibrosis. Lancet Respir. Med. 2016, 4, 662–674. [Google Scholar] [CrossRef]
- McKone, E.F.; Goss, C.H.; Aitken, M.L. CFTR genotype as a predictor of prognosis in cystic fibrosis. Chest 2006, 130, 1441–1447. [Google Scholar] [CrossRef]
- Sobczyńska-Tomaszewska, A.; Ołtarzewski, M.; Czerska, K.; Wertheim-Tysarowska, K.; Sands, D.; Walkowiak, J.; Bal, J.; Mazurczak, T. Newborn screening for cystic fibrosis: Polish 4 years’ experience with CFTR sequencing strategy. Eur. J. Hum. Genet. 2013, 21, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Castellani, C.; Cuppens, H.; Macek, M.; Cassiman, J.J.; Kerem, E.; Durie, P.; Tullis, E.; Assael, B.M.; Bombieri, C.; Brown, A.; et al. Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice. J. Cyst. Fibros. 2008, 7, 179–196. [Google Scholar] [CrossRef]
- Couper, R.T.; Corey, M.; Moore, D.J.; Fisher, L.J.; Forstner, G.G.; Durie, P.R. Decline of exocrine pancreatic function in cystic fibrosis patients with pancreatic sufficiency. Pediatr. Res. 1992, 32, 179–182. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dodge, J.A.; Turck, D. Cystic fibrosis: Nutritional consequences and management. Best Pract. Res. Clin. Gastroenterol. 2006, 20, 531–546. [Google Scholar] [CrossRef]
- Pencharz, P.B.; Durie, P.R. Pathogenesis of malnutrition in cystic fibrosis, and its treatment. Clin. Nutr. 2000, 19, 387–394. [Google Scholar] [CrossRef]
- Umławska, W.; Susanne, C. Growth and nutritional status in children and adolescents with cystic fibrosis. Ann. Hum. Biol. 2008, 35, 145–153. [Google Scholar] [CrossRef]
- Sands, D.; Umławska, W.; Zielińska, A. A cross-sectional study of growth, nutritional status and body proportions in children and adolescents at a medical center specializing in the treatment of cystic fibrosis in Poland. Arch. Med. Sci. 2015, 11, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Kerem, E.; Conway, S.; Elborn, S.; Heijerman, H.; Committee, C. Standards of care for patients with cystic fibrosis: A European consensus. J. Cyst. Fibros. 2005, 4, 7–26. [Google Scholar] [CrossRef] [PubMed]
- Kröger, J.; Jacobs, S.; Jansen, E.H.; Fritsche, A.; Boeing, H.; Schulze, M.B. Erythrocyte membrane fatty acid fluidity and risk of type 2 diabetes in the EPIC-Potsdam study. Diabetologia 2015, 58, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Staff, M.C.L. Test Catalog. Available online: https://www.mayocliniclabs.com/test-catalog/index.html (accessed on 5 January 2021).
- Kerem, E.; Corey, M.; Kerem, B.S.; Rommens, J.; Markiewicz, D.; Levison, H.; Tsui, L.C.; Durie, P. The relation between genotype and phenotype in cystic fibrosis--analysis of the most common mutation (delta F508). N. Engl. J. Med. 1990, 323, 1517–1522. [Google Scholar] [CrossRef]
- Boas, S.R.; Hageman, J.R.; Ho, L.T.; Liveris, M. Very high-dose ergocalciferol is effective for correcting vitamin D deficiency in children and young adults with cystic fibrosis. J. Cyst. Fibros. 2009, 8, 270–272. [Google Scholar] [CrossRef] [PubMed]
- Rana, M.; Wong-See, D.; Katz, T.; Gaskin, K.; Whitehead, B.; Jaffe, A.; Coakley, J.; Lochhead, A. Fat-soluble vitamin deficiency in children and adolescents with cystic fibrosis. J. Clin. Pathol. 2014, 67, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Rusińska, A.; Płudowski, P.; Walczak, M.; Borszewska-Kornacka, M.K.; Bossowski, A.; Chlebna-Sokół, D.; Czech-Kowalska, J.; Dobrzańska, A.; Franek, E.; Helwich, E.; et al. Vitamin D Supplementation Guidelines for General Population and Groups at Risk of Vitamin D Deficiency in Poland-Recommendations of the Polish Society of Pediatric Endocrinology and Diabetes and the Expert Panel With Participation of National Specialist Consultants and Representatives of Scientific Societies-2018 Update. Front. Endocrinol. 2018, 9, 246. [Google Scholar]
- Voo, V.T.F.; Stankovich, J.; O’Brien, T.J.; Butzkueven, H.; Monif, M. Vitamin D status in an Australian patient population: A large retrospective case series focusing on factors associated with variations in serum 25(OH)D. BMJ Open 2020, 10, e032567. [Google Scholar] [CrossRef] [PubMed]
- Daley, T.; Hughan, K.; Rayas, M.; Kelly, A.; Tangpricha, V. Vitamin D deficiency and its treatment in cystic fibrosis. J. Cyst. Fibros. 2019, 18 (Suppl. S2), S66–S73. [Google Scholar] [CrossRef] [PubMed]
- Wani, W.A.; Nazir, M.; Bhat, J.I.; Malik, E.U.; Ahmad, Q.I.; Charoo, B.A.; Ali, S.W. Vitamin D status correlates with the markers of cystic fibrosis-related pulmonary disease. Pediatr. Neonatol. 2019, 60, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.M.; Mischler, E.H.; Engle, M.J.; Brown, D.J.; Lau, S.M. Fatty acid abnormalities in cystic fibrosis. Pediatr. Res. 1985, 19, 104–109. [Google Scholar] [CrossRef]
- Kuo, P.T.; Huang, N.N.; Bassett, D.R. The fatty acid composition of the serum chylomicrons and adipose tissue of children with cystic fibrosis of the pancreas. J. Pediatr. 1962, 60, 394–403. [Google Scholar] [CrossRef]
- Batal, I.; Ericsoussi, M.B.; Cluette-Brown, J.E.; O’Sullivan, B.P.; Freedman, S.D.; Savaille, J.E.; Laposata, M. Potential utility of plasma fatty acid analysis in the diagnosis of cystic fibrosis. Clin. Chem. 2007, 53, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Coste, T.C.; Armand, M.; Lebacq, J.; Lebecque, P.; Wallemacq, P.; Leal, T. An overview of monitoring and supplementation of omega 3 fatty acids in cystic fibrosis. Clin. Biochem. 2007, 40, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Roulet, M.; Frascarolo, P.; Rappaz, I.; Pilet, M. Essential fatty acid deficiency in well nourished young cystic fibrosis patients. Eur. J. Pediatr. 1997, 156, 952–956. [Google Scholar] [CrossRef]
- Strandvik, B.; Gronowitz, E.; Enlund, F.; Martinsson, T.; Wahlström, J. Essential fatty acid deficiency in relation to genotype in patients with cystic fibrosis. J. Pediatr. 2001, 139, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Coste, T.C.; Deumer, G.; Reychler, G.; Lebecque, P.; Wallemacq, P.; Leal, T. Influence of pancreatic status and sex on polyunsaturated fatty acid profiles in cystic fibrosis. Clin. Chem. 2008, 54, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.G.; Fitzgerald, D.A.; Garg, M.L. Hypothesis: Vitamin E complements polyunsaturated fatty acids in essential fatty acid deficiency in cystic fibrosis. J. Am. Coll. Nutr. 2003, 22, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Underwood, B.A.; Denning, C.R.; Navab, M. Polyunsaturated fatty acids and tocopherol levels in patients with cystic fibrosis. Ann. N. Y. Acad. Sci. 1972, 203, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, M.H.; Ott, P.; Michaelsen, K.F.; Porsgaard, T.; Jensen, F.; Lanng, S. Long-chain PUFA in granulocytes, mononuclear cells, and RBC in patients with cystic fibrosis: Relation to liver disease. J. Pediatr. Gastroenterol. Nutr. 2012, 55, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Christophe, A.B.; Warwick, W.J.; Holman, R.T. Serum fatty acid profiles in cystic fibrosis patients and their parents. Lipids 1994, 29, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Al-Turkmani, M.R.; Freedman, S.D.; Laposata, M. Fatty acid alterations and n-3 fatty acid supplementation in cystic fibrosis. Prostaglandins Leukot. Essent. Fat. Acids 2007, 77, 309–318. [Google Scholar] [CrossRef]
- Turck, D.; Braegger, C.P.; Colombo, C.; Declercq, D.; Morton, A.; Pancheva, R.; Robberecht, E.; Stern, M.; Strandvik, B.; Wolfe, S.; et al. ESPEN-ESPGHAN-ECFS guidelines on nutrition care for infants, children, and adults with cystic fibrosis. Clin. Nutr. 2016, 35, 557–577. [Google Scholar] [CrossRef] [PubMed]
- Watson, H.; Stackhouse, C. Omega-3 fatty acid supplementation for cystic fibrosis. Cochrane Database Syst. Rev. 2020, 4, CD002201. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, M.R.; Mondick, J.; Barrett, J.S.; Wilson, M.; Stallings, V.A.; Schall, J.I. Malabsorption blood test: Assessing fat absorption in patients with cystic fibrosis and pancreatic insufficiency. J. Clin. Pharmacol. 2015, 55, 854–865. [Google Scholar] [CrossRef]
- Jenkins, B.; West, J.A.; Koulman, A. A review of odd-chain fatty acid metabolism and the role of pentadecanoic Acid (c15:0) and heptadecanoic Acid (c17:0) in health and disease. Molecules 2015, 20, 2425–2444. [Google Scholar] [CrossRef] [PubMed]
- Pfeuffer, M.; Jaudszus, A. Pentadecanoic and Heptadecanoic Acids: Multifaceted Odd-Chain Fatty Acids. Adv. Nutr. 2016, 7, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Fuhrman, B.J.; Barba, M.; Krogh, V.; Micheli, A.; Pala, V.; Lauria, R.; Chajes, V.; Riboli, E.; Sieri, S.; Berrino, F.; et al. Erythrocyte membrane phospholipid composition as a biomarker of dietary fat. Ann. Nutr. Metab. 2006, 50, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Bhura-Bandali, F.N.; Suh, M.; Man, S.F.; Clandinin, M.T. The deltaF508 mutation in the cystic fibrosis transmembrane conductance regulator alters control of essential fatty acid utilization in epithelial cells. J. Nutr. 2000, 130, 2870–2875. [Google Scholar] [CrossRef] [PubMed]
- Jakobik, V.; Burus, I.; Decsi, T. Fatty acid composition of erythrocyte membrane lipids in healthy subjects from birth to young adulthood. Eur. J. Pediatr. 2009, 168, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Laryea, M.; Cieslicki, P.; Diekmann, E.; Wendel, U. Age-dependent fatty acid composition of erythrocyte membrane phospholipids in healthy children. Z. Für Ernährungswissenschaft 1990, 29, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Mickle, J.E.; Cutting, G.R. Genotype-phenotype relationships in cystic fibrosis. Med. Clin. N. Am. 2000, 84, 597–607. [Google Scholar] [CrossRef]
- Johansen, H.K.; Nir, M.; Høiby, N.; Koch, C.; Schwartz, M. Severity of cystic fibrosis in patients homozygous and heterozygous for delta F508 mutation. Lancet 1991, 337, 631–634. [Google Scholar] [CrossRef]

| Type of Mutation | Number of Patients | Percentage |
|---|---|---|
| dele2,3(21 kb) | 5 | 14.7 |
| 1717–1G > A | 3 | 8.82 |
| 3659 delC | 2 | 5.88 |
| 3600 + 2 insT | 2 | 5.88 |
| N1303K | 2 | 5.88 |
| 3849 + 10 kbC > T | 2 | 5.88 |
| 4374 + 1G > T | 1 | 2.94 |
| 3171insC | 1 | 2.94 |
| IVS2 + 1G > T | 1 | 2.94 |
| N1282X | 1 | 2.94 |
| E92K | 1 | 2.94 |
| R1158X | 1 | 2.94 |
| 2184insA | 1 | 2.94 |
| K710X | 1 | 2.94 |
| dup16,17a | 1 | 2.94 |
| G542X | 1 | 2.94 |
| 1078delT | 1 | 2.94 |
| M952T | 1 | 2.94 |
| 2184insA | 1 | 2.94 |
| Y1092X | 1 | 2.94 |
| 296 + 1G > T | 1 | 2.94 |
| IVS-5T(TG)11 | 1 | 2.94 |
| 3272–26A-G | 1 | 2.94 |
| 2143delT | 1 | 2.94 |
| Clinical and Demographic Features | Homozygotes | Heterozygotes | p |
|---|---|---|---|
| Total number | 30 | 34 | - |
| Mean age (months) (range) | 116 (10–198) | 92.1 (8–216) | 0.1 |
| Sex: females n (%) | 16 (53.3) | 15 (44.1) | 0.96 |
| Sex: males n (%) | 14 (46.7) | 19 (55.9) | 0.96 |
| Mean stool elastaze-1 (μg/g) 1 (range) | 86.7 (15–200) | 147.3 (16–652) | 0.37 |
| Pseudomonas aeruginosa-colonised patients n (%) | 6 (20) | 8 (23.5) | 0.96 |
| Fatty Acid (%) | Homozygous | Heterozygous | p | |
|---|---|---|---|---|
| Saturated | C 14:0 | 0.58 | 0.58 | 0.95 |
| C 15:0 | 0.17 | 0.16 | 0.94 | |
| C 16:0 | 25.12 | 25.08 | 0.85 | |
| C 17:0 | 0.34 | 0.32 | 0.04 | |
| C 18:0 | 15.45 | 15.95 | 0.55 | |
| C 20:0 | 0.49 | 0.50 | 0.81 | |
| C 22:0 | 1.61 | 1.58 | 0.82 | |
| C 24:0 | 4.01 | 4.03 | 0.94 | |
| Unsaturated | C16:1 n-7 cis | 0.38 | 0.34 | 0.39 |
| C17:1 | 0.09 | 0.08 | 0.27 | |
| C18:1 n-9 trans | 0.16 | 0.15 | 0.68 | |
| C18:1 n-7 trans | 0.20 | 0.20 | 0.56 | |
| C18:1 n-9 cis | 12.03 | 11.87 | 0.55 | |
| C18:1 n-7 cis | 0.98 | 0.94 | 0.61 | |
| C20:1 n-9 cis | 0.35 | 0.62 | 0.51 | |
| C22:1 | 0.24 | 0.20 | 0.48 | |
| C22:1 n-9 cis | 0.23 | 0.25 | 0.24 | |
| C24:1 n-9 | 5.36 | 5.22 | 0.45 | |
| Polyunsaturated omega n-3 | C16:3 n-3 | 0.09 | 0.10 | 0.53 |
| C18:3 n-3 | 0.21 | 0.20 | 0.67 | |
| C20:5 n-3 | 1.46 | 1.36 | 0.34 | |
| C22:5 n-3 | 2.31 | 2.24 | 0.95 | |
| C22:6 n-3 | 3.69 | 3.57 | 0.81 | |
| Polyunsaturated omega n-6 | C18:2 n-6 cis | 7.83 | 7.95 | 0.88 |
| C18:3 n-6 | 0.08 | 0.07 | 0.10 | |
| C20:2 | 0.26 | 0.24 | 0.58 | |
| C20:3 n-6 | 1.70 | 1.62 | 0.98 | |
| C20:4 n-6 | 11.62 | 11.62 | 0.99 | |
| C22:2 | 0.10 | 0.09 | 0.98 | |
| C22:4 n-6 | 2.86 | 2.86 | 0.99 | |
| Vitamin | Mean Serum Levels in Heterozygotes ± SD (Range) | Mean Serum Levels in Homozygotes ± SD (Range) | p | Reference Value 2 |
|---|---|---|---|---|
| A (μg/mL) | 0.78 ± 0.2 (0.47–1.18) | 0.77 ± 0.18 (0.44–1) | 0.73 | 0.11–0.97 |
| 25-OH-D3 (ng/mL) | 12.92 ± 3.41 (8.53–17.91) | 13.14 ± 3.31 (7.18–18.02) | 0.82 | 20–50 |
| E (μg/mL) | 6.32 ± 3.49 (0.75–13.21) | 6.50 ± 3.39 (0.72–11.97) | 0.80 | 3.8–18.4 |
| K1 (ng/mL) 1 | 0.41 ± 0.21 (0.18–0.7) | 0.38 ± 0.2 (0.18–0.67) | 0.38 | 0.10–2.20 |
| Anthropometric Indicators | Homozygotes (n = 30) | Heterozygotes (n = 34) | p |
|---|---|---|---|
| Mean height percentile (range) | 41.72 ± 33.75 (0.2 to 95.5) | 49.24 ± 31.4 (2.6 to 99.8) | 0.3 |
| Mean height Z-score (range) | −0.40 ± 1.28 (−2.96 to 1.69) | 0.07 ± 1.2 (−1.94 to 2.9) | 0.3 |
| Mean weight percentile (%) | 35.74 ± 28.67 (0.2 to 84.9) | 49.65 ± 34.43 (0.2 to 99.2) | 0.11 |
| Mean weight Z-score (range) | −0.58 ± 1.07 (−2.84 to 1.03) | −0.06 ± 1.32 (−2.9 to 2.4) | 0.11 |
| Mean BMI (range) | 16.57 ± 2.12 (13.6 to 21.0) | 16.97 ± 2.3 (13.8 to 21.8) | 0.48 |
| Mean BMI percentile (range) | 36.07 ± 26.09 (0.7 to 86.0) | 48.73 ± 31.48 (0.2 to 97.6) | 0.11 |
| Mean BMI percentile Z-score (range ± SD) | −0.5 ± 0.9 (−2.48 to 1.11) | −0.03 ± 1.1 (−2.82 to 1.97) | 0.11 |
| BMI Percentile | Homozygotes n = 30 | Heterozygotes n = 34 |
|---|---|---|
| <5 | 10% (n = 3) | 5.9% (n = 2) |
| 5–24 | 23.3% (n = 7) | 23.5% (n = 8) |
| 25–84 | 63.3% (n = 19) | 50% (n = 17) |
| 85–94 | 3.3% (n = 1) | 14.7% (n = 5) |
| ≥95 | 0% (n = 0) | 5.9% (n = 2) |
| Correlated Parameters | n | Spearman’s R | p |
|---|---|---|---|
| BMI Percentile with α-linolenic acid (C18:3 n-3) | 32 | −0.37 | 0.04 |
| BMI Percentile with heptadecenoic acid (C17:1) | 32 | 0.48 | 0.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mędza, A.; Kaźmierska, K.; Wielgomas, B.; Konieczna, L.; Olędzka, I.; Szlagatys-Sidorkiewicz, A.; Sznurkowska, K. DeltaF508 CFTR Hetero- and Homozygous Paediatric Patients with Cystic Fibrosis Do Not Differ with Regard to Nutritional Status. Nutrients 2021, 13, 1402. https://doi.org/10.3390/nu13051402
Mędza A, Kaźmierska K, Wielgomas B, Konieczna L, Olędzka I, Szlagatys-Sidorkiewicz A, Sznurkowska K. DeltaF508 CFTR Hetero- and Homozygous Paediatric Patients with Cystic Fibrosis Do Not Differ with Regard to Nutritional Status. Nutrients. 2021; 13(5):1402. https://doi.org/10.3390/nu13051402
Chicago/Turabian StyleMędza, Aleksandra, Katarzyna Kaźmierska, Bartosz Wielgomas, Lucyna Konieczna, Ilona Olędzka, Agnieszka Szlagatys-Sidorkiewicz, and Katarzyna Sznurkowska. 2021. "DeltaF508 CFTR Hetero- and Homozygous Paediatric Patients with Cystic Fibrosis Do Not Differ with Regard to Nutritional Status" Nutrients 13, no. 5: 1402. https://doi.org/10.3390/nu13051402
APA StyleMędza, A., Kaźmierska, K., Wielgomas, B., Konieczna, L., Olędzka, I., Szlagatys-Sidorkiewicz, A., & Sznurkowska, K. (2021). DeltaF508 CFTR Hetero- and Homozygous Paediatric Patients with Cystic Fibrosis Do Not Differ with Regard to Nutritional Status. Nutrients, 13(5), 1402. https://doi.org/10.3390/nu13051402