
Phosphate, Microbiota and CKD

 ,
, 
Abstract
1. Chronic Kidney Disease (CKD) Concept and Global Impact
2. CKD-Mineral and Bone Disorder (CKD-MBD)
3. Phosphate and CKD
3.1. Diet
3.2. Phosphate Binders
4. The Microbiota and Biological Impact on CKD
4.1. Microbiota: Concept and Broad Classification
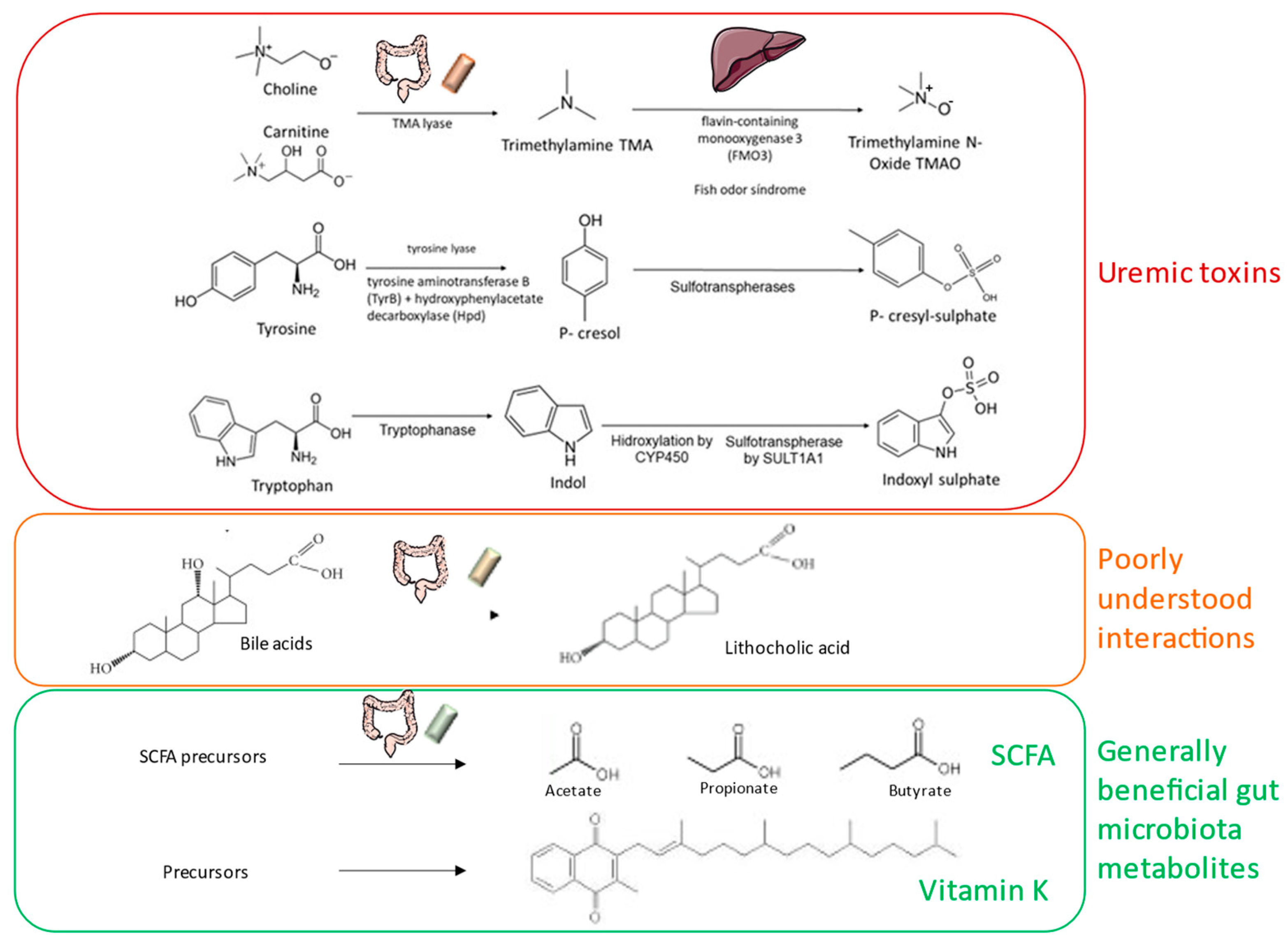
4.2. Bioactive Molecules Released by the Microbiota
4.3. Uremic Toxin Precursors Released by the Microbiota
5. Impact of Dietary Phosphate on Gut Microbiota
6. Impact of Phosphate Binders on Gut Microbiota
7. Impact of Gut Microbiota on CKD-MBD
8. New Tools: Phosphate-Accumulating Organisms
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Perez-Gomez, M.V.; Bartsch, L.-A.; Castillo-Rodriguez, E.; Fernandez-Prado, R.; Fernandez-Fernandez, B.; Martin-Cleary, C.; Gracia-Iguacel, C.; Ortiz, A. Clarifying the Concept of Chronic Kidney Disease for Non-Nephrologists. Clin. Kidney J. 2019, 12, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A. The Unaccomplished Mission of Reducing Mortality in Patients on Kidney Replacement Therapy. Clin. Kidney J. 2020, 13, 948–951. [Google Scholar] [CrossRef] [PubMed]
- Foreman, K.J.; Marquez, N.; Dolgert, A.; Fukutaki, K.; Fullman, N.; McGaughey, M.; Pletcher, M.A.; Smith, A.E.; Tang, K.; Yuan, C.-W.; et al. Forecasting Life Expectancy, Years of Life Lost, and All-Cause and Cause-Specific Mortality for 250 Causes of Death: Reference and Alternative Scenarios for 2016-40 for 195 Countries and Territories. Lancet 2018, 392, 2052–2090. [Google Scholar] [CrossRef]
- Ortiz, A.; Sanchez-Niño, M.D.; Crespo-Barrio, M.; De-Sequera-Ortiz, P.; Fernández-Giráldez, E.; García-Maset, R.; Macía-Heras, M.; Pérez-Fontán, M.; Rodríguez-Portillo, M.; Salgueira-Lazo, M.; et al. The Spanish Society of Nephrology (SENEFRO) Commentary to the Spain GBD 2016 Report: Keeping Chronic Kidney Disease out of Sight of Health Authorities Will Only Magnify the Problem. Nefrologia 2019, 39, 29–34. [Google Scholar] [CrossRef]
- ERA-EDTA Council. ERACODA Working Group Chronic Kidney Disease is a Key Risk Factor for Severe COVID-19: A Call to Action by the ERA-EDTA. Nephrol. Dial. Transplant. 2021, 36, 87–94. [Google Scholar] [CrossRef]
- Sanchez-Niño, M.D.; Fernandez-Fernandez, B.; Ortiz, A. Klotho, the Elusive Kidney-Derived Anti-Ageing Factor. Clin. Kidney J. 2020, 13, 125–127. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Van Laecke, S.; Glorieux, G.; Verbeke, F.; Castillo-Rodriguez, E.; Ortiz, A. Deleting Death and Dialysis: Conservative Care of Cardio-Vascular Risk and Kidney Function Loss in Chronic Kidney Disease (CKD). Toxins 2018, 10, 237. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Prado, R.; Esteras, R.; Perez-Gomez, M.V.; Gracia-Iguacel, C.; Gonzalez-Parra, E.; Sanz, A.B.; Ortiz, A.; Sanchez-Niño, M.D. Nutrients Turned into Toxins: Microbiota Modulation of Nutrient Properties in Chronic Kidney Disease. Nutrients 2017, 9, 489. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Parra, E.; Tuñón, J.; Egido, J.; Ortiz, A. Phosphate: A Stealthier Killer than Previously Thought? Cardiovasc. Pathol. 2012, 21, 372–381. [Google Scholar] [CrossRef]
- Castillo-Rodriguez, E.; Fernandez-Prado, R.; Esteras, R.; Perez-Gomez, M.V.; Gracia-Iguacel, C.; Fernandez-Fernandez, B.; Kanbay, M.; Tejedor, A.; Lazaro, A.; Ruiz-Ortega, M.; et al. Impact of Altered Intestinal Microbiota on Chronic Kidney Disease Progression. Toxins 2018, 10, 300. [Google Scholar] [CrossRef]
- Fuller, D.S.; Dluzniewski, P.J.; Cooper, K.; Bradbury, B.D.; Robinson, B.M.; Tentori, F. Combinations of Mineral and Bone Disorder Markers and Risk of Death and Hospitalizations in the International Dialysis Outcomes and Practice Patterns Study. Clin. Kidney J. 2020, 13, 1056–1062. [Google Scholar] [CrossRef] [PubMed]
- Iseri, K.; Dai, L.; Chen, Z.; Qureshi, A.R.; Brismar, T.B.; Stenvinkel, P.; Lindholm, B. Bone Mineral Density and Mortality in End-Stage Renal Disease Patients. Clin. Kidney J. 2020, 13, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Update Work Group KDIGO 2017 Clinical Practice Guideline Update for the Diagnosis, Evaluation, Prevention, and Treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kidney Int. Suppl. 2017, 7, 1–59. [CrossRef] [PubMed]
- Fernández-Fernández, B.; Valiño-Rivas, L.; Sánchez-Niño, M.D.; Ortiz, A. Albuminuria Downregulation of the Anti-Aging Factor Klotho: The Missing Link Potentially Explaining the Association of Pathological Albuminuria with Premature Death. Adv. Ther. 2020, 37, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Fernandez, B.; Izquierdo, M.C.; Valiño-Rivas, L.; Nastou, D.; Sanz, A.B.; Ortiz, A.; Sanchez-Niño, M.D. Albumin Downregulates Klotho in Tubular Cells. Nephrol. Dial. Transplant. 2018, 33, 1712–1722. [Google Scholar] [CrossRef] [PubMed]
- Kuro-o, M. The Klotho Proteins in Health and Disease. Nat. Rev. Nephrol. 2019, 15, 27–44. [Google Scholar] [CrossRef]
- Kalantar-Zadeh, K.; Gutekunst, L.; Mehrotra, R.; Kovesdy, C.P.; Bross, R.; Shinaberger, C.S.; Noori, N.; Hirschberg, R.; Benner, D.; Nissenson, A.R.; et al. Understanding Sources of Dietary Phosphorus in the Treatment of Patients with Chronic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 519–530. [Google Scholar] [CrossRef]
- Block, G.A.; Hulbert-Shearon, T.E.; Levin, N.W.; Port, F.K. Association of Serum Phosphorus and Calcium x Phosphate Product with Mortality Risk in Chronic Hemodialysis Patients: A National Study. Am. J. Kidney Dis. 1998, 31, 607–617. [Google Scholar] [CrossRef]
- Grabner, A.; Mazzaferro, S.; Cianciolo, G.; Krick, S.; Capelli, I.; Rotondi, S.; Ronco, C.; La Manna, G.; Faul, C. Fibroblast Growth Factor 23: Mineral Metabolism and Beyond. Contrib. Nephrol. 2017, 190, 83–95. [Google Scholar] [CrossRef]
- Sanchez-Niño, M.D.; Aguilera-Correa, J.-J.; Politei, J.; Esteban, J.; Requena, T.; Ortiz, A. Unraveling the Drivers and Consequences of Gut Microbiota Disruption in Fabry Disease: The Lyso-Gb3 Link. Future Microbiol. 2020, 15, 227–231. [Google Scholar] [CrossRef] [PubMed]
- 2015–2020 Dietary Guidelines for Americans. Available online: https://health.gov/sites/default/files/2019-09/2015-2020_Dietary_Guidelines.pdf (accessed on 9 February 2021).
- Institute of Medicine (US) Standing Committee on the Scientific Evaluation of Dietary Reference Intakes. Dietary Reference Intakes for Calcium, Phosphorus, Magnesium, Vitamin D, and Fluoride; The National Academies Collection: Reports funded by National Institutes of Health; National Academies Press (US): Washington, DC, USA, 1997; ISBN 978-0-309-06350-0.
- Kemi, V.E.; Rita, H.J.; Kärkkäinen, M.U.M.; Viljakainen, H.T.; Laaksonen, M.M.; Outila, T.A.; Lamberg-Allardt, C.J.E. Habitual High Phosphorus Intakes and Foods with Phosphate Additives Negatively Affect Serum Parathyroid Hormone Concentration: A Cross-Sectional Study on Healthy Premenopausal Women. Public Health Nutr. 2009, 12, 1885–1892. [Google Scholar] [CrossRef] [PubMed]
- De Seigneux, S.; Courbebaisse, M.; Rutkowski, J.M.; Wilhelm-Bals, A.; Metzger, M.; Khodo, S.N.; Hasler, U.; Chehade, H.; Dizin, E.; Daryadel, A.; et al. Proteinuria Increases Plasma Phosphate by Altering Its Tubular Handling. J. Am. Soc. Nephrol. 2015, 26, 1608–1618. [Google Scholar] [CrossRef] [PubMed]
- Barreto, F.C.; Barreto, D.V.; Massy, Z.A.; Drüeke, T.B. Strategies for Phosphate Control in Patients with CKD. Kidney Int. Rep. 2019, 4, 1043–1056. [Google Scholar] [CrossRef] [PubMed]
- Bover, J.; Cozzolino, M. Small Steps towards the Potential of “preventive” Treatment of Early Phosphate Loading in Chronic Kidney Disease Patients. Clin. Kidney J. 2019, 12, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Bouma-de Krijger, A.; van Ittersum, F.J.; Hoekstra, T.; Ter Wee, P.M.; Vervloet, M.G. Short-Term Effects of Sevelamer-Carbonate on Fibroblast Growth Factor 23 and Pulse Wave Velocity in Patients with Normophosphataemic Chronic Kidney Disease Stage 3. Clin. Kidney J. 2019, 12, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Van Camp, Y.P.M.; Vrijens, B.; Abraham, I.; Van Rompaey, B.; Elseviers, M.M. Adherence to Phosphate Binders in Hemodialysis Patients: Prevalence and Determinants. J. Nephrol. 2014, 27, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.; Sanchez-Niño, M.D. The Demise of Calcium-Based Phosphate Binders. Lancet 2013, 382, 1232–1234. [Google Scholar] [CrossRef]
- De Solis, A.J.; González-Pacheco, F.R.; Deudero, J.J.P.; Neria, F.; Albalate, M.; Petkov, V.; Susanibar, L.; Fernandez-Sanchez, R.; Calabia, O.; Ortiz, A.; et al. Alkalinization Potentiates Vascular Calcium Deposition in an Uremic Milieu. J. Nephrol. 2009, 22, 647–653. [Google Scholar] [PubMed]
- Bover Sanjuán, J.; Navarro-González, J.F.; Arenas, M.D.; Torregrosa, J.-V.; Tamargo Menéndez, J.; de Francisco, A.L.M.; González-Parra, E.; Lloret Cora, M.J.; Sánchez Álvarez, J.E.; Martín-Malo, A.; et al. Pharmacological Interactions of Phosphate Binders. Nefrologia 2018, 38, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Slatopolsky, E.; Weerts, C.; Lopez-Hilker, S.; Norwood, K.; Zink, M.; Windus, D.; Delmez, J. Calcium Carbonate as a Phosphate Binder in Patients with Chronic Renal Failure Undergoing Dialysis. N. Engl. J. Med. 1986, 315, 157–161. [Google Scholar] [CrossRef]
- Bover, J.; Ureña-Torres, P.; Mateu, S.; DaSilva, I.; Gràcia, S.; Sánchez-Baya, M.; Arana, C.; Fayos, L.; Guirado, L.; Cozzolino, M. Evidence in Chronic Kidney Disease–Mineral and Bone Disorder Guidelines: Is It Time to Treat or Time to Wait? Clin. Kidney J. 2020, 13, 513–521. [Google Scholar] [CrossRef] [PubMed]
- De Schutter, T.M.; Behets, G.J.; Geryl, H.; Peter, M.E.; Steppan, S.; Gundlach, K.; Passlick-Deetjen, J.; D’Haese, P.C.; Neven, E. Effect of a Magnesium-Based Phosphate Binder on Medial Calcification in a Rat Model of Uremia. Kidney Int. 2013, 83, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Osorio, L.; Zambrano, D.P.; Gracia-Iguacel, C.; Rojas-Rivera, J.; Ortiz, A.; Egido, J.; González Parra, E. Use of Sevelamer in Chronic Kidney Disease: Beyond Phosphorus Control. Nefrologia 2015, 35, 207–217. [Google Scholar] [CrossRef][Green Version]
- Biruete, A.; Hill Gallant, K.M.; Lindemann, S.R.; Wiese, G.N.; Chen, N.X.; Moe, S.M. Phosphate Binders and Nonphosphate Effects in the Gastrointestinal Tract. J. Ren. Nutr. 2020, 30, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.P.; Perianayagam, M.C.; Jaber, B.L. Sevelamer Hydrochloride Use and Circulating Endotoxin in Hemodialysis Patients: A Pilot Cross-Sectional Study. J. Ren. Nutr. 2009, 19, 432–438. [Google Scholar] [CrossRef]
- Perianayagam, M.C.; Jaber, B.L. Endotoxin-Binding Affinity of Sevelamer Hydrochloride. Am. J. Nephrol. 2008, 28, 802–807. [Google Scholar] [CrossRef]
- Dai, L.; Meijers, B.K.; Bammens, B.; de Loor, H.; Schurgers, L.J.; Qureshi, A.R.; Stenvinkel, P.; Evenepoel, P. Sevelamer Use in End-Stage Kidney Disease (ESKD) Patients Associates with Poor Vitamin K Status and High Levels of Gut-Derived Uremic Toxins: A Drug-Bug Interaction? Toxins 2020, 12, 351. [Google Scholar] [CrossRef] [PubMed]
- Fusaro, M.; Cozzolino, M.; Plebani, M.; Iervasi, G.; Ketteler, M.; Gallieni, M.; Aghi, A.; Locatelli, F.; Cunningham, J.; Salam, S.; et al. Sevelamer Use, Vitamin K Levels, Vascular Calcifications, and Vertebral Fractures in Hemodialysis Patients: Results from the VIKI Study. J. Bone Miner. Res. 2020. [Google Scholar] [CrossRef]
- Bennis, Y.; Cluet, Y.; Titeca-Beauport, D.; El Esper, N.; Ureña, P.; Bodeau, S.; Combe, C.; Dussol, B.; Fouque, D.; Choukroun, G.; et al. The Effect of Sevelamer on Serum Levels of Gut-Derived Uremic Toxins: Results from In Vitro Experiments and A Multicenter, Double-Blind, Placebo-Controlled, Randomized Clinical Trial. Toxins 2019, 11, 279. [Google Scholar] [CrossRef]
- Brandenburg, V.M.; Schlieper, G.; Heussen, N.; Holzmann, S.; Busch, B.; Evenepoel, P.; Vanholder, R.; Meijers, B.; Meert, N.; Fassbender, W.J.; et al. Serological Cardiovascular and Mortality Risk Predictors in Dialysis Patients Receiving Sevelamer: A Prospective Study. Nephrol. Dial. Transplant. 2010, 25, 2672–2679. [Google Scholar] [CrossRef]
- Ohno, I.; Yamaguchi, Y.; Saikawa, H.; Uetake, D.; Hikita, M.; Okabe, H.; Ichida, K.; Hosoya, T. Sevelamer Decreases Serum Uric Acid Concentration through Adsorption of Uric Acid in Maintenance Hemodialysis Patients. Intern. Med. 2009, 48, 415–420. [Google Scholar] [CrossRef]
- Phan, O.; Ivanovski, O.; Nguyen-Khoa, T.; Mothu, N.; Angulo, J.; Westenfeld, R.; Ketteler, M.; Meert, N.; Maizel, J.; Nikolov, I.G.; et al. Sevelamer Prevents Uremia-Enhanced Atherosclerosis Progression in Apolipoprotein E-Deficient Mice. Circulation 2005, 112, 2875–2882. [Google Scholar] [CrossRef] [PubMed]
- Riccio, E.; Sabbatini, M.; Bruzzese, D.; Grumetto, L.; Marchetiello, C.; Amicone, M.; Andreucci, M.; Guida, B.; Passaretti, D.; Russo, G.; et al. Plasma P-Cresol Lowering Effect of Sevelamer in Non-Dialysis CKD Patients: Evidence from a Randomized Controlled Trial. Clin. Exp. Nephrol. 2018, 22, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Parra, E.; Gonzalez-Casaus, M.L.; Galán, A.; Martinez-Calero, A.; Navas, V.; Rodriguez, M.; Ortiz, A. Lanthanum Carbonate Reduces FGF23 in Chronic Kidney Disease Stage 3 Patients. Nephrol. Dial. Transplant. 2011, 26, 2567–2571. [Google Scholar] [CrossRef] [PubMed]
- Nastou, D.; Fernández-Fernández, B.; Elewa, U.; González-Espinoza, L.; González-Parra, E.; Sanchez-Niño, M.D.; Ortiz, A. Next-Generation Phosphate Binders: Focus on Iron-Based Binders. Drugs 2014, 74, 863–877. [Google Scholar] [CrossRef] [PubMed]
- Rahbar Saadat, Y.; Niknafs, B.; Hosseiniyan Khatibi, S.M.; Ardalan, M.; Majdi, H.; Bahmanpoor, Z.; Abediazar, S.; Zununi Vahed, S. Gut Microbiota; an Overlooked Effect of Phosphate Binders. Eur. J. Pharmacol. 2020, 868, 172892. [Google Scholar] [CrossRef]
- Pascale, A.; Marchesi, N.; Marelli, C.; Coppola, A.; Luzi, L.; Govoni, S.; Giustina, A.; Gazzaruso, C. Microbiota and Metabolic Diseases. Endocrine 2018, 61, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.R.; Pop, M.; DeBoy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic Analysis of the Human Distal Gut Microbiome. Science 2006, 312, 1355–1359. [Google Scholar] [CrossRef] [PubMed]
- Natividad, J.M.M.; Verdu, E.F. Modulation of Intestinal Barrier by Intestinal Microbiota: Pathological and Therapeutic Implications. Pharmacol. Res. 2013, 69, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Gensollen, T.; Iyer, S.S.; Kasper, D.L.; Blumberg, R.S. How Colonization by Microbiota in Early Life Shapes the Immune System. Science 2016, 352, 539–544. [Google Scholar] [CrossRef]
- Long, S.L.; Gahan, C.G.M.; Joyce, S.A. Interactions between Gut Bacteria and Bile in Health and Disease. Mol. Asp. Med. 2017, 56, 54–65. [Google Scholar] [CrossRef]
- Weiss, G.A.; Hennet, T. Mechanisms and Consequences of Intestinal Dysbiosis. Cell. Mol. Life Sci. 2017, 74, 2959–2977. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the Immune System. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef]
- Jalanka-Tuovinen, J.; Salonen, A.; Nikkilä, J.; Immonen, O.; Kekkonen, R.; Lahti, L.; Palva, A.; Vos, W.M. de Intestinal Microbiota in Healthy Adults: Temporal Analysis Reveals Individual and Common Core and Relation to Intestinal Symptoms. PLoS ONE 2011, 6, e23035. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Ley, R.E.; Volchkov, P.Y.; Stranges, P.B.; Avanesyan, L.; Stonebraker, A.C.; Hu, C.; Wong, F.S.; Szot, G.L.; Bluestone, J.A.; et al. Innate Immunity and Intestinal Microbiota in the Development of Type 1 Diabetes. Nature 2008, 455, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [PubMed]
- Flint, H.J.; Scott, K.P.; Louis, P.; Duncan, S.H. The Role of the Gut Microbiota in Nutrition and Health. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.I.; Kapila, R. Dietary Metabolites Derived from Gut Microbiota: Critical Modulators of Epigenetic Changes in Mammals. Nutr. Rev. 2017, 75, 374–389. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.W. Histone-Deacetylase Inhibitors: Novel Drugs for the Treatment of Cancer. Nat. Rev. Drug Discov. 2002, 1, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Thangaraju, M.; Prasad, P.D.; Martin, P.M.; Lambert, N.A.; Boettger, T.; Offermanns, S.; Ganapathy, V. Blockade of Dendritic Cell Development by Bacterial Fermentation Products Butyrate and Propionate through a Transporter (Slc5a8)-Dependent Inhibition of Histone Deacetylases. J. Biol. Chem. 2010, 285, 27601–27608. [Google Scholar] [CrossRef]
- Cummings, J.H.; Pomare, E.W.; Branch, W.J.; Naylor, C.P.; Macfarlane, G.T. Short Chain Fatty Acids in Human Large Intestine, Portal, Hepatic and Venous Blood. Gut 1987, 28, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Frost, G.; Sleeth, M.L.; Sahuri-Arisoylu, M.; Lizarbe, B.; Cerdan, S.; Brody, L.; Anastasovska, J.; Ghourab, S.; Hankir, M.; Zhang, S.; et al. The Short-Chain Fatty Acid Acetate Reduces Appetite via a Central Homeostatic Mechanism. Nat. Commun. 2014, 5, 3611. [Google Scholar] [CrossRef] [PubMed]
- Kimura, I.; Inoue, D.; Maeda, T.; Hara, T.; Ichimura, A.; Miyauchi, S.; Kobayashi, M.; Hirasawa, A.; Tsujimoto, G. Short-Chain Fatty Acids and Ketones Directly Regulate Sympathetic Nervous System via G Protein-Coupled Receptor 41 (GPR41). Proc. Natl. Acad. Sci. USA 2011, 108, 8030–8035. [Google Scholar] [CrossRef]
- González Hernández, M.A.; Canfora, E.E.; Jocken, J.W.E.; Blaak, E.E. The Short-Chain Fatty Acid Acetate in Body Weight Control and Insulin Sensitivity. Nutrients 2019, 11, 1943. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Moreno, J.M.; Fontecha-Barriuso, M.; Martín-Sánchez, D.; Sánchez-Niño, M.D.; Ruiz-Ortega, M.; Sanz, A.B.; Ortiz, A. The Contribution of Histone Crotonylation to Tissue Health and Disease: Focus on Kidney Health. Front. Pharmacol. 2020, 11, 393. [Google Scholar] [CrossRef]
- Fontecha-Barriuso, M.; Martin-Sanchez, D.; Ruiz-Andres, O.; Poveda, J.; Sanchez-Niño, M.D.; Valiño-Rivas, L.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. Targeting Epigenetic DNA and Histone Modifications to Treat Kidney Disease. Nephrol. Dial. Transplant. 2018, 33, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Andres, O.; Sanchez-Niño, M.D.; Cannata-Ortiz, P.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A.; Sanz, A.B. Histone Lysine Crotonylation during Acute Kidney Injury in Mice. Dis. Model Mech. 2016, 9, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.A.; Izquierdo, M.C.; Sanchez-Niño, M.D.; Suárez-Alvarez, B.; Lopez-Larrea, C.; Jakubowski, A.; Blanco, J.; Ramirez, R.; Selgas, R.; Ruiz-Ortega, M.; et al. The Inflammatory Cytokines TWEAK and TNFα Reduce Renal Klotho Expression through NFκB. J. Am. Soc. Nephrol. 2011, 22, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jiang, C.; Liu, G.; Wang, P.; Shi, M.; Yang, M.; Zhong, Z.; Ding, S.; Li, Y.; Liu, B.; et al. Sodium Butyrate Protects against Oxidative Stress in Human Nucleus Pulposus Cells via Elevating PPARγ-Regulated Klotho Expression. Int. Immunopharmacol. 2020, 85, 106657. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Andres, O.; Suarez-Alvarez, B.; Sánchez-Ramos, C.; Monsalve, M.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A.; Sanz, A.B. The Inflammatory Cytokine TWEAK Decreases PGC-1α Expression and Mitochondrial Function in Acute Kidney Injury. Kidney Int. 2016, 89, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Fontecha-Barriuso, M.; Martín-Sánchez, D.; Martinez-Moreno, J.M.; Carrasco, S.; Ruiz-Andrés, O.; Monsalve, M.; Sanchez-Ramos, C.; Gómez, M.J.; Ruiz-Ortega, M.; Sánchez-Niño, M.D.; et al. PGC-1α Deficiency Causes Spontaneous Kidney Inflammation and Increases the Severity of Nephrotoxic AKI. J. Pathol. 2019, 249, 65–78. [Google Scholar] [CrossRef]
- Li, Y.J.; Chen, X.; Kwan, T.K.; Loh, Y.W.; Singer, J.; Liu, Y.; Ma, J.; Tan, J.; Macia, L.; Mackay, C.R.; et al. Dietary Fiber Protects against Diabetic Nephropathy through Short-Chain Fatty Acid-Mediated Activation of G Protein-Coupled Receptors GPR43 and GPR109A. J. Am. Soc. Nephrol. 2020, 31, 1267–1281. [Google Scholar] [CrossRef] [PubMed]
- Makki, K.; Deehan, E.C.; Walter, J.; Bäckhed, F. The Impact of Dietary Fiber on Gut Microbiota in Host Health and Disease. Cell Host Microbe 2018, 23, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Trompette, A.; Gollwitzer, E.S.; Yadava, K.; Sichelstiel, A.K.; Sprenger, N.; Ngom-Bru, C.; Blanchard, C.; Junt, T.; Nicod, L.P.; Harris, N.L.; et al. Gut Microbiota Metabolism of Dietary Fiber Influences Allergic Airway Disease and Hematopoiesis. Nat. Med. 2014, 20, 159–166. [Google Scholar] [CrossRef]
- Ragsdale, S.W.; Pierce, E. Acetogenesis and the Wood-Ljungdahl Pathway of CO2 Fixation. Biochim. Biophys. Acta 2008, 1784, 1873–1898. [Google Scholar] [CrossRef]
- Hetzel, M.; Brock, M.; Selmer, T.; Pierik, A.J.; Golding, B.T.; Buckel, W. Acryloyl-CoA Reductase from Clostridium Propionicum. An Enzyme Complex of Propionyl-CoA Dehydrogenase and Electron-Transferring Flavoprotein. Eur. J. Biochem. 2003, 270, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.P.; Martin, J.C.; Campbell, G.; Mayer, C.-D.; Flint, H.J. Whole-Genome Transcription Profiling Reveals Genes up-Regulated by Growth on Fucose in the Human Gut Bacterium “Roseburia Inulinivorans". J. Bacteriol. 2006, 188, 4340–4349. [Google Scholar] [CrossRef] [PubMed]
- Duncan, S.H.; Barcenilla, A.; Stewart, C.S.; Pryde, S.E.; Flint, H.J. Acetate Utilization and Butyryl Coenzyme A (CoA):Acetate-CoA Transferase in Butyrate-Producing Bacteria from the Human Large Intestine. Appl. Environ. Microbiol. 2002, 68, 5186–5190. [Google Scholar] [CrossRef] [PubMed]
- Pryde, S.E.; Duncan, S.H.; Hold, G.L.; Stewart, C.S.; Flint, H.J. The Microbiology of Butyrate Formation in the Human Colon. FEMS Microbiol. Lett. 2002, 217, 133–139. [Google Scholar] [CrossRef]
- Kimura, I.; Miyamoto, J.; Ohue-Kitano, R.; Watanabe, K.; Yamada, T.; Onuki, M.; Aoki, R.; Isobe, Y.; Kashihara, D.; Inoue, D.; et al. Maternal Gut Microbiota in Pregnancy Influences Offspring Metabolic Phenotype in Mice. Science 2020, 367. [Google Scholar] [CrossRef]
- Meijers, B.K.I.; De Loor, H.; Bammens, B.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. P-Cresyl Sulfate and Indoxyl Sulfate in Hemodialysis Patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1932–1938. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Sato, T.; Nomoto, K.; Tsuji, H. Identification of Phenol- and p-Cresol-Producing Intestinal Bacteria by Using Media Supplemented with Tyrosine and Its Metabolites. FEMS Microbiol. Ecol. 2018, 94. [Google Scholar] [CrossRef] [PubMed]
- Pignanelli, M.; Just, C.; Bogiatzi, C.; Dinculescu, V.; Gloor, G.B.; Allen-Vercoe, E.; Reid, G.; Urquhart, B.L.; Ruetz, K.N.; Velenosi, T.J.; et al. Mediterranean Diet Score: Associations with Metabolic Products of the Intestinal Microbiome, Carotid Plaque Burden, and Renal Function. Nutrients 2018, 10, 779. [Google Scholar] [CrossRef] [PubMed]
- Gryp, T.; De Paepe, K.; Vanholder, R.; Kerckhof, F.-M.; Van Biesen, W.; Van de Wiele, T.; Verbeke, F.; Speeckaert, M.; Joossens, M.; Couttenye, M.M.; et al. Gut Microbiota Generation of Protein-Bound Uremic Toxins and Related Metabolites Is Not Altered at Different Stages of Chronic Kidney Disease. Kidney Int. 2020, 97, 1230–1242. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, G.; Gryp, T.; Perna, A. Gut-Derived Metabolites and Their Role in Immune Dysfunction in Chronic Kidney Disease. Toxins 2020, 12, 245. [Google Scholar] [CrossRef]
- Tang, W.H.W.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal Microbial Metabolism of Phosphatidylcholine and Cardiovascular Risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Moraes, C.; Fouque, D.; Amaral, A.C.F.; Mafra, D. Trimethylamine N-Oxide From Gut Microbiota in Chronic Kidney Disease Patients: Focus on Diet. J. Ren. Nutr. 2015, 25, 459–465. [Google Scholar] [CrossRef] [PubMed]
- El-Deeb, O.S.; Atef, M.M.; Hafez, Y.M. The Interplay between Microbiota-Dependent Metabolite Trimethylamine N-Oxide, Transforming Growth Factor β/SMAD Signaling and Inflammasome Activation in Chronic Kidney Disease Patients: A New Mechanistic Perspective. J. Cell. Biochem. 2019, 120, 14476–14485. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Y.; Yang, P.; Liu, X.; Lu, L.; Chen, Y.; Zhong, X.; Li, Z.; Liu, H.; Ou, C.; et al. Trimethylamine-N-Oxide Promotes Vascular Calcification Through Activation of NLRP3 (Nucleotide-Binding Domain, Leucine-Rich-Containing Family, Pyrin Domain-Containing-3) Inflammasome and NF-ΚB (Nuclear Factor ΚB) Signals. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 751–765. [Google Scholar] [CrossRef]
- Ardura, J.A.; Sanz, A.B.; Ortiz, A.; Esbrit, P. Parathyroid Hormone-Related Protein Protects Renal Tubuloepithelial Cells from Apoptosis by Activating Transcription Factor Runx2. Kidney Int. 2013, 83, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Gessner, A.; König, J.; Fromm, M.F. Contribution of Multidrug and Toxin Extrusion Protein 1 (MATE1) to Renal Secretion of Trimethylamine-N-Oxide (TMAO). Sci. Rep. 2018, 8, 6659. [Google Scholar] [CrossRef] [PubMed]
- Teft, W.A.; Morse, B.L.; Leake, B.F.; Wilson, A.; Mansell, S.E.; Hegele, R.A.; Ho, R.H.; Kim, R.B. Identification and Characterization of Trimethylamine-N-Oxide Uptake and Efflux Transporters. Mol. Pharm. 2017, 14, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Buffa, J.A.; Roberts, A.B.; Sangwan, N.; Skye, S.M.; Li, L.; Ho, K.J.; Varga, J.; DiDonato, J.A.; Tang, W.H.W.; et al. Targeted Inhibition of Gut Microbial Trimethylamine N-Oxide Production Reduces Renal Tubulointerstitial Fibrosis and Functional Impairment in a Murine Model of Chronic Kidney Disease. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1239–1255. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-J.; Chen, H.-H.; Pan, C.-F.; Chuang, C.-K.; Wang, T.-J.; Sun, F.-J.; Wu, C.-J. P-Cresylsulfate and Indoxyl Sulfate Level at Different Stages of Chronic Kidney Disease. J. Clin. Lab. Anal. 2011, 25, 191–197. [Google Scholar] [CrossRef]
- Wang, C.-P.; Lu, L.-F.; Yu, T.-H.; Hung, W.-C.; Chiu, C.-A.; Chung, F.-M.; Hsu, C.-C.; Lu, Y.-C.; Lee, Y.-J.; Houng, J.-Y. Associations among Chronic Kidney Disease, High Total p-Cresylsulfate and Major Adverse Cardiac Events. J. Nephrol. 2013, 26, 111–118. [Google Scholar] [CrossRef]
- Liabeuf, S.; Barreto, D.V.; Barreto, F.C.; Meert, N.; Glorieux, G.; Schepers, E.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; et al. Free P-Cresylsulphate Is a Predictor of Mortality in Patients at Different Stages of Chronic Kidney Disease. Nephrol. Dial. Transplant. 2010, 25, 1183–1191. [Google Scholar] [CrossRef]
- Viaene, L.; Annaert, P.; de Loor, H.; Poesen, R.; Evenepoel, P.; Meijers, B. Albumin Is the Main Plasma Binding Protein for Indoxyl Sulfate and P-Cresyl Sulfate. Biopharm. Drug Dispos. 2013, 34, 165–175. [Google Scholar] [CrossRef]
- Bosch-Panadero, E.; Mas, S.; Civantos, E.; Abaigar, P.; Camarero, V.; Ruiz-Priego, A.; Ortiz, A.; Egido, J.; González-Parra, E. Bisphenol A Is an Exogenous Toxin That Promotes Mitochondrial Injury and Death in Tubular Cells. Environ. Toxicol. 2018, 33, 325–332. [Google Scholar] [CrossRef]
- Brocca, A.; Virzì, G.M.; de Cal, M.; Cantaluppi, V.; Ronco, C. Cytotoxic Effects of P-Cresol in Renal Epithelial Tubular Cells. Blood Purif. 2013, 36, 219–225. [Google Scholar] [CrossRef]
- Lin, H.-H.; Huang, C.-C.; Lin, T.-Y.; Lin, C.-Y. P-Cresol Mediates Autophagic Cell Death in Renal Proximal Tubular Cells. Toxicol. Lett. 2015, 234, 20–29. [Google Scholar] [CrossRef]
- Vanholder, R.; Bammens, B.; de Loor, H.; Glorieux, G.; Meijers, B.; Schepers, E.; Massy, Z.; Evenepoel, P. Warning: The Unfortunate End of p-Cresol as a Uraemic Toxin. Nephrol. Dial. Transplant. 2011, 26, 1464–1467. [Google Scholar] [CrossRef] [PubMed]
- Koppe, L.; Alix, P.M.; Croze, M.L.; Chambert, S.; Vanholder, R.; Glorieux, G.; Fouque, D.; Soulage, C.O. P-Cresyl Glucuronide Is a Major Metabolite of p-Cresol in Mouse: In Contrast to p-Cresyl Sulphate, p-Cresyl Glucuronide Fails to Promote Insulin Resistance. Nephrol. Dial. Transplant. 2017, 32, 2000–2009. [Google Scholar] [CrossRef]
- Poveda, J.; Sanchez-Niño, M.D.; Glorieux, G.; Sanz, A.B.; Egido, J.; Vanholder, R.; Ortiz, A. P-Cresyl Sulphate Has pro-Inflammatory and Cytotoxic Actions on Human Proximal Tubular Epithelial Cells. Nephrol. Dial. Transplant. 2014, 29, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Bolati, D.; Higashiyama, Y.; Nishijima, F.; Shimizu, K.; Niwa, T. Indoxyl Sulfate Upregulates Renal Expression of MCP-1 via Production of ROS and Activation of NF-ΚB, P53, ERK, and JNK in Proximal Tubular Cells. Life Sci. 2012, 90, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.-Y.; Hsu, H.-H.; Wu, M.-S. P-Cresol Sulfate and Indoxyl Sulfate Induce Similar Cellular Inflammatory Gene Expressions in Cultured Proximal Renal Tubular Cells. Nephrol. Dial. Transplant. 2013, 28, 70–78. [Google Scholar] [CrossRef]
- Sun, C.-Y.; Chang, S.-C.; Wu, M.-S. Uremic Toxins Induce Kidney Fibrosis by Activating Intrarenal Renin-Angiotensin-Aldosterone System Associated Epithelial-to-Mesenchymal Transition. PLoS ONE 2012, 7, e34026. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Choi, H.I.; Bae, E.H.; Ma, S.K.; Kim, S.W. Paricalcitol Attenuates Indoxyl Sulfate-Induced Apoptosis through the Inhibition of MAPK, Akt, and NF-KB Activation in HK-2 Cells. Korean J. Intern. Med. 2019, 34, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-J.; Chang, C.-H.; Sun, M.-F.; Hsu, S.-F.; Weng, C.-S. DPP-4 Inhibitor Attenuates Toxic Effects of Indoxyl Sulfate on Kidney Tubular Cells. PLoS ONE 2014, 9, e93447. [Google Scholar] [CrossRef]
- Wang, L.; Gao, Z.; Wang, L.; Gao, Y. Upregulation of Nuclear Factor-ΚB Activity Mediates CYP24 Expression and Reactive Oxygen Species Production in Indoxyl Sulfate-Induced Chronic Kidney Disease. Nephrology 2016, 21, 774–781. [Google Scholar] [CrossRef]
- Ellis, R.J.; Small, D.M.; Ng, K.L.; Vesey, D.A.; Vitetta, L.; Francis, R.S.; Gobe, G.C.; Morais, C. Indoxyl Sulfate Induces Apoptosis and Hypertrophy in Human Kidney Proximal Tubular Cells. Toxicol. Pathol. 2018, 46, 449–459. [Google Scholar] [CrossRef]
- Saito, S.; Shimizu, H.; Yisireyili, M.; Nishijima, F.; Enomoto, A.; Niwa, T. Indoxyl Sulfate-Induced Activation of (pro)Renin Receptor Is Involved in Expression of TGF-Β1 and α-Smooth Muscle Actin in Proximal Tubular Cells. Endocrinology 2014, 155, 1899–1907. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Yisireyili, M.; Shimizu, H.; Ng, H.-Y.; Niwa, T. Indoxyl Sulfate Upregulates Prorenin Expression via Nuclear Factor-ΚB P65, Signal Transducer and Activator of Transcription 3, and Reactive Oxygen Species in Proximal Tubular Cells. J. Ren. Nutr. 2015, 25, 145–148. [Google Scholar] [CrossRef]
- Milanesi, S.; Garibaldi, S.; Saio, M.; Ghigliotti, G.; Picciotto, D.; Ameri, P.; Garibotto, G.; Barisione, C.; Verzola, D. Indoxyl Sulfate Induces Renal Fibroblast Activation through a Targetable Heat Shock Protein 90-Dependent Pathway. Oxid. Med. Cell. Longev. 2019, 2019, 2050183. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Yisireyili, M.; Nishijima, F.; Niwa, T. Indoxyl Sulfate Enhances P53-TGF-Β1-Smad3 Pathway in Proximal Tubular Cells. Am. J. Nephrol. 2013, 37, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.-Y.; Young, G.-H.; Hsieh, Y.-T.; Chen, Y.-H.; Wu, M.-S.; Wu, V.-C.; Lee, J.-H.; Lee, C.-C. Protein-Bound Uremic Toxins Induce Tissue Remodeling by Targeting the EGF Receptor. J. Am. Soc. Nephrol. 2015, 26, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Edamatsu, T.; Fujieda, A.; Itoh, Y. Phenyl Sulfate, Indoxyl Sulfate and p-Cresyl Sulfate Decrease Glutathione Level to Render Cells Vulnerable to Oxidative Stress in Renal Tubular Cells. PLoS ONE 2018, 13, e0193342. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.-Y.; Cheng, M.-L.; Pan, H.-C.; Lee, J.-H.; Lee, C.-C. Protein-Bound Uremic Toxins Impaired Mitochondrial Dynamics and Functions. Oncotarget 2017, 8, 77722–77733. [Google Scholar] [CrossRef]
- Vila Cuenca, M.; van Bezu, J.; Beelen, R.H.J.; Vervloet, M.G.; Hordijk, P.L. Stabilization of Cell-Cell Junctions by Active Vitamin D Ameliorates Uraemia-Induced Loss of Human Endothelial Barrier Function. Nephrol. Dial. Transplant. 2019, 34, 252–264. [Google Scholar] [CrossRef]
- Chen, S.-C.; Huang, S.-Y.; Wu, C.-C.; Hsu, C.-F. P-Cresylsulfate, the Protein-Bound Uremic Toxin, Increased Endothelial Permeability Partly Mediated by Src-Induced Phosphorylation of VE-Cadherin. Toxins 2020, 12, 62. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.-H.; Wang, C.-P.; Yu, T.-H.; Tai, P.-Y.; Liang, S.-S.; Hung, W.-C.; Wu, C.-C.; Huang, S.-H.; Lee, Y.-J.; Chen, S.-C. Protein-Bounded Uremic Toxin p-Cresylsulfate Induces Vascular Permeability Alternations. Histochem. Cell Biol. 2018, 149, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-C.; Tomino, Y.; Lu, K.-C. Impacts of Indoxyl Sulfate and P-Cresol Sulfate on Chronic Kidney Disease and Mitigating Effects of AST-120. Toxins 2018, 10, 367. [Google Scholar] [CrossRef] [PubMed]
- Schulman, G.; Berl, T.; Beck, G.J.; Remuzzi, G.; Ritz, E.; Arita, K.; Kato, A.; Shimizu, M. Randomized Placebo-Controlled EPPIC Trials of AST-120 in CKD. J. Am. Soc. Nephrol. 2015, 26, 1732–1746. [Google Scholar] [CrossRef]
- Saito, H.; Yoshimura, M.; Saigo, C.; Komori, M.; Nomura, Y.; Yamamoto, Y.; Sagata, M.; Wakida, A.; Chuman, E.; Nishi, K.; et al. Hepatic Sulfotransferase as a Nephropreventing Target by Suppression of the Uremic Toxin Indoxyl Sulfate Accumulation in Ischemic Acute Kidney Injury. Toxicol. Sci. 2014, 141, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.-T.; Lin, S.-H. Uremic Vascular Calcification: The Pathogenic Roles and Gastrointestinal Decontamination of Uremic Toxins. Toxins 2020, 12, 812. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-C.; Wu, C.-C.; Lim, P.-S.; Chien, S.-W.; Hou, Y.-C.; Zheng, C.-M.; Shyu, J.-F.; Lin, Y.-F.; Lu, K.-C. Effect of Uremic Toxin-Indoxyl Sulfate on the Skeletal System. Clin. Chim. Acta 2018, 484, 197–206. [Google Scholar] [CrossRef]
- Watanabe, K.; Tominari, T.; Hirata, M.; Matsumoto, C.; Hirata, J.; Murphy, G.; Nagase, H.; Miyaura, C.; Inada, M. Indoxyl Sulfate, a Uremic Toxin in Chronic Kidney Disease, Suppresses Both Bone Formation and Bone Resorption. FEBS Open Bio 2017, 7, 1178–1185. [Google Scholar] [CrossRef] [PubMed]
- Liabeuf, S.; Villain, C.; Massy, Z.A. Protein-Bound Toxins: Has the Cinderella of Uraemic Toxins Turned into a Princess? Clin. Sci. 2016, 130, 2209–2216. [Google Scholar] [CrossRef] [PubMed]
- Lau, W.L.; Chang, Y.; Vaziri, N.D. The Consequences of Altered Microbiota in Immune-Related Chronic Kidney Disease. Nephrol. Dial. Transplant. 2020. [Google Scholar] [CrossRef] [PubMed]
- Gryp, T.; Huys, G.R.B.; Joossens, M.; Van Biesen, W.; Glorieux, G.; Vaneechoutte, M. Isolation and Quantification of Uremic Toxin Precursor-Generating Gut Bacteria in Chronic Kidney Disease Patients. Int. J. Mol. Sci. 2020, 21, 1986. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.-Y.; Lin, C.-J.; Pan, H.-C.; Lee, C.-C.; Lu, S.-C.; Hsieh, Y.-T.; Huang, S.-Y.; Huang, H.-Y. Clinical Association between the Metabolite of Healthy Gut Microbiota, 3-Indolepropionic Acid and Chronic Kidney Disease. Clin. Nutr. 2019, 38, 2945–2948. [Google Scholar] [CrossRef]
- Dodd, D.; Spitzer, M.H.; Van Treuren, W.; Merrill, B.D.; Hryckowian, A.J.; Higginbottom, S.K.; Le, A.; Cowan, T.M.; Nolan, G.P.; Fischbach, M.A.; et al. A Gut Bacterial Pathway Metabolizes Aromatic Amino Acids into Nine Circulating Metabolites. Nature 2017, 551, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Marzocco, S.; Fazeli, G.; Di Micco, L.; Autore, G.; Adesso, S.; Dal Piaz, F.; Heidland, A.; Di Iorio, B. Supplementation of Short-Chain Fatty Acid, Sodium Propionate, in Patients on Maintenance Hemodialysis: Beneficial Effects on Inflammatory Parameters and Gut-Derived Uremic Toxins, A Pilot Study (PLAN Study). J. Clin. Med. 2018, 7, 315. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Tian, H.; Wang, Y.; Shen, Y.; Zhu, Q.; Ding, F. Effect of Ionic Strength, PH and Chemical Displacers on the Percentage Protein Binding of Protein-Bound Uremic Toxins. Blood Purif. 2019, 47, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.-H.; Park, D.; Kim, Y.J.; Lee, I.; Kim, S.; Oh, C.-T.; Kim, J.-Y.; Yang, J.; Jo, S.-K. Lactobacillus Salivarius BP121 Prevents Cisplatin-induced Acute Kidney Injury by Inhibition of Uremic Toxins Such as Indoxyl Sulfate and P-cresol Sulfate via Alleviating Dysbiosis. Int. J. Mol. Med. 2020, 45, 1130–1140. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, Q.; Henning, S.M.; Zhong, J.; Hsu, M.; Lee, R.; Long, J.; Chan, B.; Nagami, G.T.; Heber, D.; et al. Effects of Prebiotic Fiber Xylooligosaccharide in Adenine-Induced Nephropathy in Mice. Mol. Nutr. Food Res. 2018, e1800014. [Google Scholar] [CrossRef]
- Plata, C.; Cruz, C.; Cervantes, L.G.; Ramírez, V. The Gut Microbiota and Its Relationship with Chronic Kidney Disease. Int. Urol. Nephrol. 2019, 51, 2209–2226. [Google Scholar] [CrossRef] [PubMed]
- Eidi, F.; Poor-Reza Gholi, F.; Ostadrahimi, A.; Dalili, N.; Samadian, F.; Barzegari, A. Effect of Lactobacillus Rhamnosus on Serum Uremic Toxins (Phenol and P-Cresol) in Hemodialysis Patients: A Double Blind Randomized Clinical Trial. Clin. Nutr. ESPEN 2018, 28, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Wei, K.-Y.; Gritter, M.; Vogt, L.; de Borst, M.H.; Rotmans, J.I.; Hoorn, E.J. Dietary Potassium and the Kidney: Lifesaving Physiology. Clin. Kidney J. 2020, 13, 952–968. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, P.; Ruilope, L.M.; Cupisti, A.; Ketteler, M.; Wheeler, D.C.; Pignot, M.; Cornea, G.; Schulmann, T.; Lund, L.H. Recurrent Hyperkalaemia Management and Use of Renin-Angiotensin-Aldosterone System Inhibitors: A European Multi-National Targeted Chart Review. Clin. Kidney J. 2020, 13, 714–719. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.R.; Lazo, M.; Appel, L.J.; Gutiérrez, O.M.; Grams, M.E. High Dietary Phosphorus Intake Is Associated with All-Cause Mortality: Results from NHANES III123. Am. J. Clin. Nutr. 2014, 99, 320–327. [Google Scholar] [CrossRef]
- Skrypnik, K.; Suliburska, J. Association between the Gut Microbiota and Mineral Metabolism. J. Sci. Food Agric. 2018, 98, 2449–2460. [Google Scholar] [CrossRef]
- Trautvetter, U.; Camarinha-Silva, A.; Jahreis, G.; Lorkowski, S.; Glei, M. High Phosphorus Intake and Gut-Related Parameters—Results of a Randomized Placebo-Controlled Human Intervention Study. Nutr. J. 2018, 17, 23. [Google Scholar] [CrossRef] [PubMed]
- Martel, J.; Wu, C.-Y.; Peng, H.-H.; Young, J.D. Mineralo-Organic Nanoparticles in Health and Disease: An Overview of Recent Findings. Nanomedicine 2018, 13, 1787–1793. [Google Scholar] [CrossRef] [PubMed]
- The Facts about Your Favorite Beverages (U.S.)|Phosphorus. Available online: https://www.pepsicobeveragefacts.com/home/phosphorus (accessed on 9 February 2021).
- Zaborin, A.; Smith, D.; Garfield, K.; Quensen, J.; Shakhsheer, B.; Kade, M.; Tirrell, M.; Tiedje, J.; Gilbert, J.A.; Zaborina, O.; et al. Membership and Behavior of Ultra-Low-Diversity Pathogen Communities Present in the Gut of Humans during Prolonged Critical Illness. mBio 2014, 5, e01361. [Google Scholar] [CrossRef] [PubMed]
- Ponsuksili, S.; Reyer, H.; Hadlich, F.; Weber, F.; Trakooljul, N.; Oster, M.; Siengdee, P.; Muráni, E.; Rodehutscord, M.; Camarinha-Silva, A.; et al. Identification of the Key Molecular Drivers of Phosphorus Utilization Based on Host MiRNA-MRNA and Gut Microbiome Interactions. Int. J. Mol. Sci. 2020, 21, 2818. [Google Scholar] [CrossRef] [PubMed]
- Heyer, C.M.E.; Weiss, E.; Schmucker, S.; Rodehutscord, M.; Hoelzle, L.E.; Mosenthin, R.; Stefanski, V. The Impact of Phosphorus on the Immune System and the Intestinal Microbiota with Special Focus on the Pig. Nutr. Res. Rev. 2015, 28, 67–82. [Google Scholar] [CrossRef] [PubMed]
- Heyer, C.M.E.; Schmucker, S.; Burbach, K.; Weiss, E.; Eklund, M.; Aumiller, T.; Capezzone, F.; Steuber, J.; Rodehutscord, M.; Hoelzle, L.E.; et al. Phytate Degradation, Intestinal Microbiota, Microbial Metabolites and Immune Values Are Changed in Growing Pigs Fed Diets with Varying Calcium–Phosphorus Concentration and Fermentable Substrates. J. Anim. Physiol. Anim. Nutr. 2019, 103, 1185–1197. [Google Scholar] [CrossRef]
- Metzler-Zebeli, B.U.; Zijlstra, R.T.; Mosenthin, R.; Gänzle, M.G. Dietary Calcium Phosphate Content and Oat β-Glucan Influence Gastrointestinal Microbiota, Butyrate-Producing Bacteria and Butyrate Fermentation in Weaned Pigs. FEMS Microbiol. Ecol. 2011, 75, 402–413. [Google Scholar] [CrossRef]
- Metzler-Zebeli, B.U.; Vahjen, W.; Baumgärtel, T.; Rodehutscord, M.; Mosenthin, R. Ileal Microbiota of Growing Pigs Fed Different Dietary Calcium Phosphate Levels and Phytase Content and Subjected to Ileal Pectin Infusion. J. Anim. Sci. 2010, 88, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.-Y.; Xu, C.-M.; Xia, M.; Zhu, H.-Q.; Chen, Y.-Q. Relationship between Gut Microbiota and Phosphorus Metabolism in Hemodialysis Patients: A Preliminary Exploration. Chin. Med. J. 2018, 131, 2792–2799. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; DeSantis, T.Z.; Ni, Z.; Nguyen, T.-H.; Andersen, G.L. Chronic Kidney Disease Alters Intestinal Microbial Flora. Kidney Int. 2013, 83, 308–315. [Google Scholar] [CrossRef]
- Wu, P.-H.; Liu, P.-Y.; Chiu, Y.-W.; Hung, W.-C.; Lin, Y.-T.; Lin, T.-Y.; Hung, S.-C.; Delicano, R.A.; Kuo, M.-C.; Wu, C.-Y. Comparative Gut Microbiome Differences between Ferric Citrate and Calcium Carbonate Phosphate Binders in Patients with End-Stage Kidney Disease. Microorganisms 2020, 8, 2040. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Luo, Y.; Ranjit, S.; Xie, C.; Libby, A.E.; Orlicky, D.J.; Dvornikov, A.; Wang, X.X.; Myakala, K.; Jones, B.A.; et al. Bile Acid Sequestration Reverses Liver Injury and Prevents Progression of Nonalcoholic Steatohepatitis in Western Diet-Fed Mice. J. Biol. Chem. 2020, 295, 4733–4747. [Google Scholar] [CrossRef] [PubMed]
- Brønden, A.; Mikkelsen, K.; Sonne, D.P.; Hansen, M.; Våben, C.; Gabe, M.N.; Rosenkilde, M.; Tremaroli, V.; Wu, H.; Bäckhed, F.; et al. Glucose-Lowering Effects and Mechanisms of the Bile Acid-Sequestering Resin Sevelamer. Diabetes Obes. Metab. 2018, 20, 1623–1631. [Google Scholar] [CrossRef] [PubMed]
- Lau, W.L.; Vaziri, N.D.; Nunes, A.C.F.; Comeau, A.M.; Langille, M.G.I.; England, W.; Khazaeli, M.; Suematsu, Y.; Phan, J.; Whiteson, K. The Phosphate Binder Ferric Citrate Alters the Gut Microbiome in Rats with Chronic Kidney Disease. J. Pharmacol. Exp. Ther. 2018, 367, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Cantorna, M.T.; Rogers, C.J.; Arora, J. Aligning the Paradoxical Role of Vitamin D in Gastrointestinal Immunity. Trends Endocrinol. Metab. 2019, 30, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Rawat, A.; Alwakeel, M.; Sharif, E.; Al Khodor, S. The Potential Role of Vitamin D Supplementation as a Gut Microbiota Modifier in Healthy Individuals. Sci. Rep. 2020, 10, 21641. [Google Scholar] [CrossRef] [PubMed]
- Sun, J. Vitamin D and Mucosal Immune Function. Curr. Opin. Gastroenterol. 2010, 26, 591–595. [Google Scholar] [CrossRef]
- Agrawal, T.; Gupta, G.K.; Agrawal, D.K. Vitamin D Deficiency Decreases the Expression of VDR and Prohibitin in the Lungs of Mice with Allergic Airway Inflammation. Exp. Mol. Pathol. 2012, 93, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Lagishetty, V.; Misharin, A.V.; Liu, N.Q.; Lisse, T.S.; Chun, R.F.; Ouyang, Y.; McLachlan, S.M.; Adams, J.S.; Hewison, M. Vitamin D Deficiency in Mice Impairs Colonic Antibacterial Activity and Predisposes to Colitis. Endocrinology 2010, 151, 2423–2432. [Google Scholar] [CrossRef]
- Chaudhari, S.N.; Luo, J.N.; Harris, D.A.; Aliakbarian, H.; Yao, L.; Paik, D.; Subramaniam, R.; Adhikari, A.A.; Vernon, A.H.; Kiliç, A.; et al. A Microbial Metabolite Remodels the Gut-Liver Axis Following Bariatric Surgery. Cell Host Microbe 2020. [Google Scholar] [CrossRef]
- Song, X.; Sun, X.; Oh, S.F.; Wu, M.; Zhang, Y.; Zheng, W.; Geva-Zatorsky, N.; Jupp, R.; Mathis, D.; Benoist, C.; et al. Microbial Bile Acid Metabolites Modulate Gut RORγ+ Regulatory T Cell Homeostasis. Nature 2020, 577, 410–415. [Google Scholar] [CrossRef]
- Stacchiotti, V.; Rezzi, S.; Eggersdorfer, M.; Galli, F. Metabolic and Functional Interplay between Gut Microbiota and Fat-Soluble Vitamins. Crit. Rev. Food Sci. Nutr. 2020, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Massy, Z.A.; Drueke, T.B. Gut Microbiota Orchestrates PTH Action in Bone: Role of Butyrate and T Cells. Kidney Int. 2020, 98, 269–272. [Google Scholar] [CrossRef]
- Li, J.-Y.; Yu, M.; Pal, S.; Tyagi, A.M.; Dar, H.; Adams, J.; Weitzmann, M.N.; Jones, R.M.; Pacifici, R. Parathyroid Hormone–Dependent Bone Formation Requires Butyrate Production by Intestinal Microbiota. J. Clin. Investig. 2020, 130, 1767–1781. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Malik Tyagi, A.; Li, J.-Y.; Adams, J.; Denning, T.L.; Weitzmann, M.N.; Jones, R.M.; Pacifici, R. PTH Induces Bone Loss via Microbial-Dependent Expansion of Intestinal TNF+ T Cells and Th17 Cells. Nat. Commun. 2020, 11, 468. [Google Scholar] [CrossRef] [PubMed]
- Lopes, R.D.C.S.O.; Balbino, K.P.; de Paula Jorge, M.; Ribeiro, A.Q.; Martino, H.S.D.; Alfenas, R.D.C.G. Modulation of Intestinal Microbiota, Control of Nitrogen Products and Inflammation by Pre/Probiotics in Chronic Kidney Disease: A Systematic Review. Nutr. Hosp. 2018, 35, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Tarayre, C.; Nguyen, H.-T.; Brognaux, A.; Delepierre, A.; De Clercq, L.; Charlier, R.; Michels, E.; Meers, E.; Delvigne, F. Characterisation of Phosphate Accumulating Organisms and Techniques for Polyphosphate Detection: A Review. Sensors 2016, 16, 797. [Google Scholar] [CrossRef] [PubMed]
- Reyes, M.; Borrás, L.; Seco, A.; Ferrer, J. Identification and Quantification of Microbial Populations in Activated Sludge and Anaerobic Digestion Processes. Environ. Technol. 2015, 36, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Weissbrodt, D.G.; Maillard, J.; Brovelli, A.; Chabrelie, A.; May, J.; Holliger, C. Multilevel Correlations in the Biological Phosphorus Removal Process: From Bacterial Enrichment to Conductivity-Based Metabolic Batch Tests and Polyphosphatase Assays. Biotechnol. Bioeng. 2014, 111, 2421–2435. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Quinto, A.; Cerqueda-García, D.; Falcón, L.I.; Gaona, O.; Martínez-Correa, S.; Nieto, J.; G-Santoyo, I. Gut Microbiome in Children from Indigenous and Urban Communities in México: Different Subsistence Models, Different Microbiomes. Microorganisms 2020, 8, 1592. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.W.; Choy, D.; Penniston, K.L.; Lange, D. Inhibition of Urinary Stone Disease by a Multi-Species Bacterial Network Ensures Healthy Oxalate Homeostasis. Kidney Int. 2019, 96, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shen, N.; Oehmen, A.; Zhou, Y. The Impact of Temperature on the Metabolism of Volatile Fatty Acids by Polyphosphate Accumulating Organisms (PAOs). Environ. Res. 2020, 188, 109729. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, J.; Oehmen, A.; Le, C.; Geng, Y.; Zhou, Y. Butyrate Can Support PAOs but Not GAOs in Tropical Climates. Water Res. 2021, 193, 116884. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Drug | Usual Dose 1 | Advantages | Disadvantages | Characteristics |
|---|---|---|---|---|
| Calcium-based binders | ||||
| Calcium carbonate | 500–1250 mg (3–12 tablets) | Effectiveness. Non evidence of influence in gut microbiota. | Hypercalcemia and vascular calcification. Gastrointestinal: constipation, nausea, vomiting. | Election therapy in 1980–1990s. Reduce carboxylation of matrix g-carboxyglutamate protein, a protein that inhibits calcification. |
| Calcium acetate * | 667 mg (6–12 capsules) | |||
| Magnesium-based binders | ||||
| Magnesium carbonate * | 63 mg (2–6 capsules) | Lower calcium overload and vascular calcification. Gastrointestinal tolerability. | Diarrhea. Hypermagnesemia. | Experimental data suggests that magnesium interferes with hydroxyapatite crystal formation. |
| Polymeric binders | ||||
| Sevelamer hydrochloride | 800–1600 mg every 8 h | Nonproducing calcium overload. Improves endothelial functions. Reduces bile salt absorption | High bill burden. Gastrointestinal tolerability. Interference in absorption of fat-soluble vitamins. High costs. | Exchange of carbonate or HCl for Pi. First non-metal phosphate binder. Large cross-linked cationic polymer. |
| Sevelamer carbonate | 800–1600 mg every 8 h | |||
| Bixalomer | 250 mg (6–14 tablets) | Gastrointestinal tolerability. Less water absorption Better fluidity. | Non available. | Amine-functional and non-absorbable polymer. Only in Japan. |
| Metal-based binders (non-iron) | ||||
| Aluminum-based | 640 mg (5–6 tablets) | Gastrointestinal tolerability. | Aluminum intoxication: encephalopathy, osteomalacia, microcytic anemia and premature death | First available binder Use strongly discouraged by KDIGO guidelines. |
| Lanthanum carbonate | 250–1000 mg (3–6 chewable tablets) | Lower pill burden. Gastrointestinal tolerability. | Accumulation in bone in dialysis patients. Low solubility. | First calcium-free chewable phosphate binder. Detaches carbonate and forms a lanthanum-phosphate complex. |
| Metal-based binders (iron) | ||||
| Ferric citrate | 210 mg (4–5 tablets) | Lower pill burden. Improves iron parameters. | Gastrointestinal tolerability: diarrhea, nausea, vomiting. Altered gut microbiota | Forms a non-soluble ferric-phosphate complex. |
| Sucroferric oxyhydroxide | 500 mg (2–6 chewable tablets) | Less gastrointestinal effects than ferric citrate. Less alteration of gut microbiota. Lower pill burden. | Polynuclear chewable iron-based phosphate binder. | |
| The gut microbiota is a source of beneficial bioactive molecules (e.g., SCFA, IPA, vitamin K)) and of uremic toxin precursors that collectively may impact host health including CKD and CKD-MBD. |
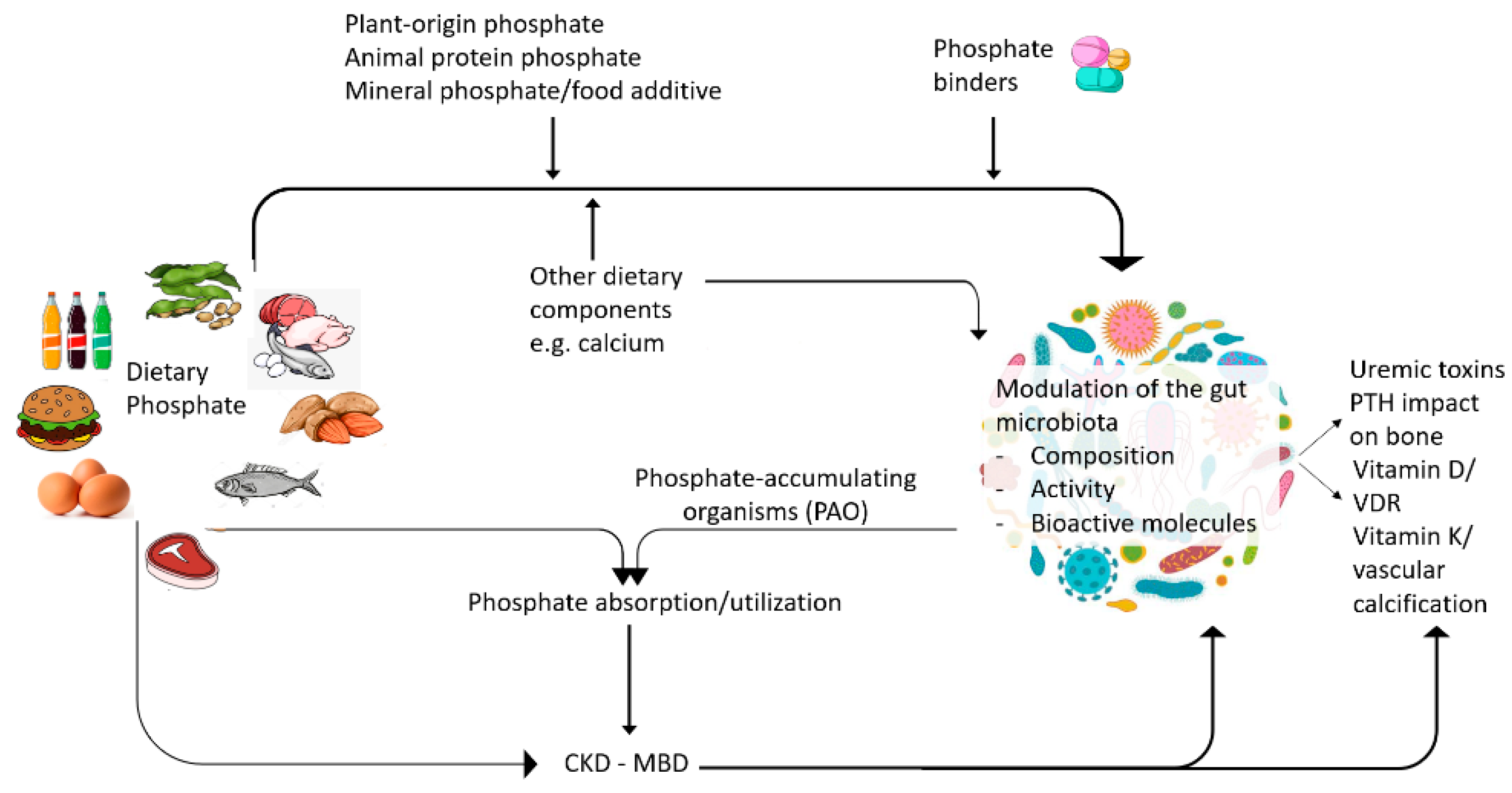
| The source and amount of dietary phosphate and the dietary calcium:phosphate ratio may modify the gut microbiota composition and properties. |
| Treatment for CKD-MBD, including phosphate binders, may influence the gut microbiota composition and properties in a binder-specific manner. |
| The gut microbiota may modulate CKD-MBD through SCFA-mediated modulation of Klotho expression, modulation of vitamin D and PTH activity, thus modulating bone health, serum phosphate and phosphate balance. |
| Phosphorus utilization research in farm animal research explores how to modulate phosphate uptake from the diet. |
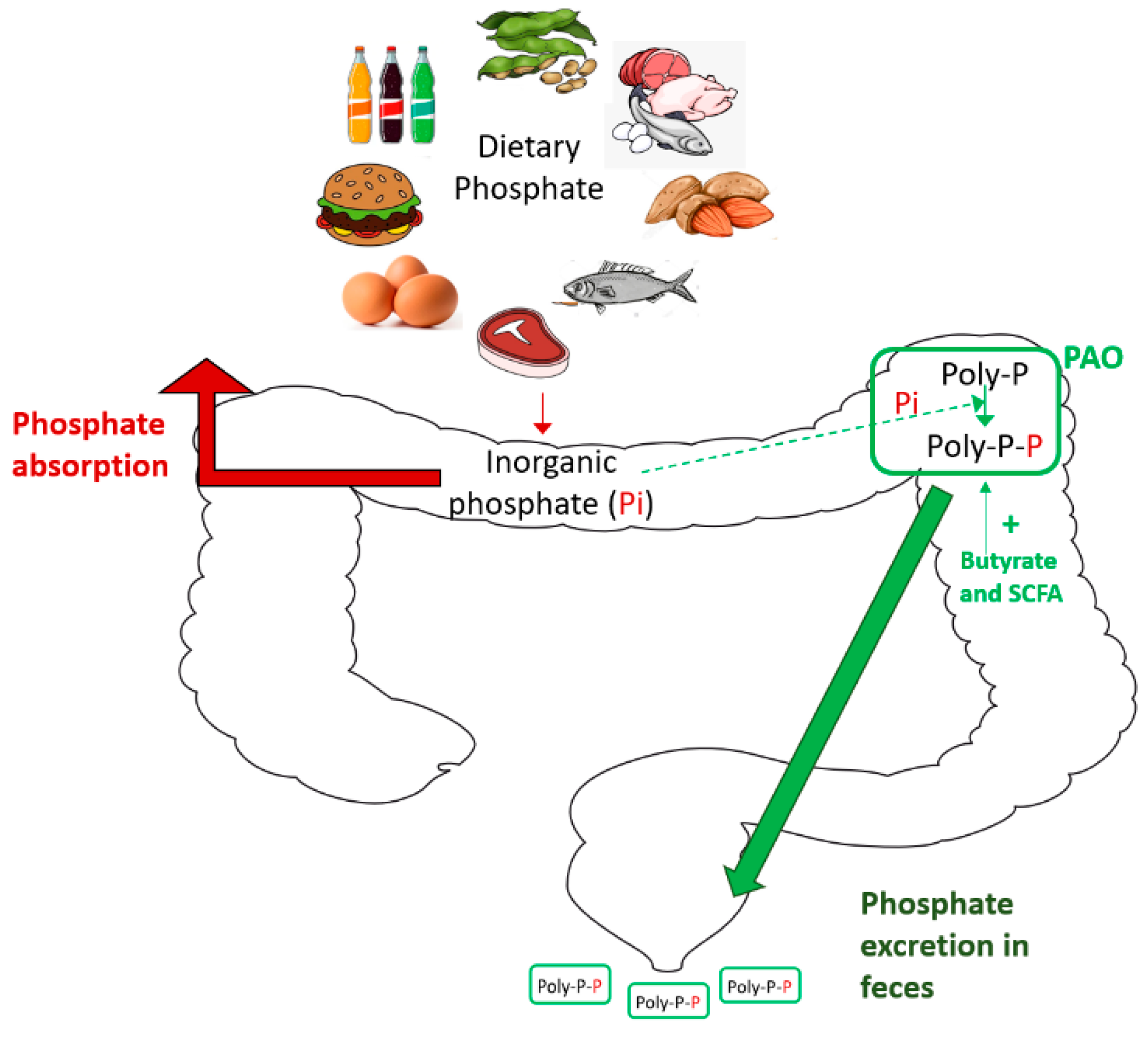
| Phosphate-accumulating organisms (PAOs) are used in wastewater research to remove phosphate for the microenvironment. |
| Findings from phosphorus utilization and PAO research may be applied to prevent dietary phosphate absorption in human CKD. |
| What is the optimal dietary phosphate intake and optimal form of dietary phosphate from the point of view of a healthy microbiota? In the general population? And in CKD patients? |
| What is the optimal phosphate binder from the point of vew of a healthy microbiota? |
| What phosphate binder best promotes the microbiota production of beneficial and bioavailable short chain fatty acids? |
| What is the optimal phosphate binder to decrease uremic toxins production by the gut microbiota? |
| What components of the gut microbiota minimize the adverse consequences of CKD-MBD by modulating vitamin D, PTH or other key host activities? |
| How can we promote and maintain such microbiota? Can dietary interventions, prebiotics, probiotics or symbiotics achieve this? |
| Are there phosphate-accumulating organisms (PAO) in the gut microbiota that can be used to increase the fecal excretion of dietary phosphate? |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Favero, C.; Carriazo, S.; Cuarental, L.; Fernandez-Prado, R.; Gomá-Garcés, E.; Perez-Gomez, M.V.; Ortiz, A.; Fernandez-Fernandez, B.; Sanchez-Niño, M.D. Phosphate, Microbiota and CKD. Nutrients 2021, 13, 1273. https://doi.org/10.3390/nu13041273
Favero C, Carriazo S, Cuarental L, Fernandez-Prado R, Gomá-Garcés E, Perez-Gomez MV, Ortiz A, Fernandez-Fernandez B, Sanchez-Niño MD. Phosphate, Microbiota and CKD. Nutrients. 2021; 13(4):1273. https://doi.org/10.3390/nu13041273
Chicago/Turabian StyleFavero, Chiara, Sol Carriazo, Leticia Cuarental, Raul Fernandez-Prado, Elena Gomá-Garcés, Maria Vanessa Perez-Gomez, Alberto Ortiz, Beatriz Fernandez-Fernandez, and Maria Dolores Sanchez-Niño. 2021. "Phosphate, Microbiota and CKD" Nutrients 13, no. 4: 1273. https://doi.org/10.3390/nu13041273
APA StyleFavero, C., Carriazo, S., Cuarental, L., Fernandez-Prado, R., Gomá-Garcés, E., Perez-Gomez, M. V., Ortiz, A., Fernandez-Fernandez, B., & Sanchez-Niño, M. D. (2021). Phosphate, Microbiota and CKD. Nutrients, 13(4), 1273. https://doi.org/10.3390/nu13041273