
Decreased Efficiency of Very-Low-Density Lipoprotein Lipolysis Is Linked to Both Hypertriglyceridemia and Hypercholesterolemia, but It Can Be Counteracted by High-Density Lipoprotein

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Groups
2.2. VLDL and HDL Isolation
2.3. VLDL Lipolysis Study
2.4. Biochemical Analysis and Electrophoresis
2.5. Statistical Analysis
3. Results
3.1. Lipid Profile and VLDL and HDL Composition
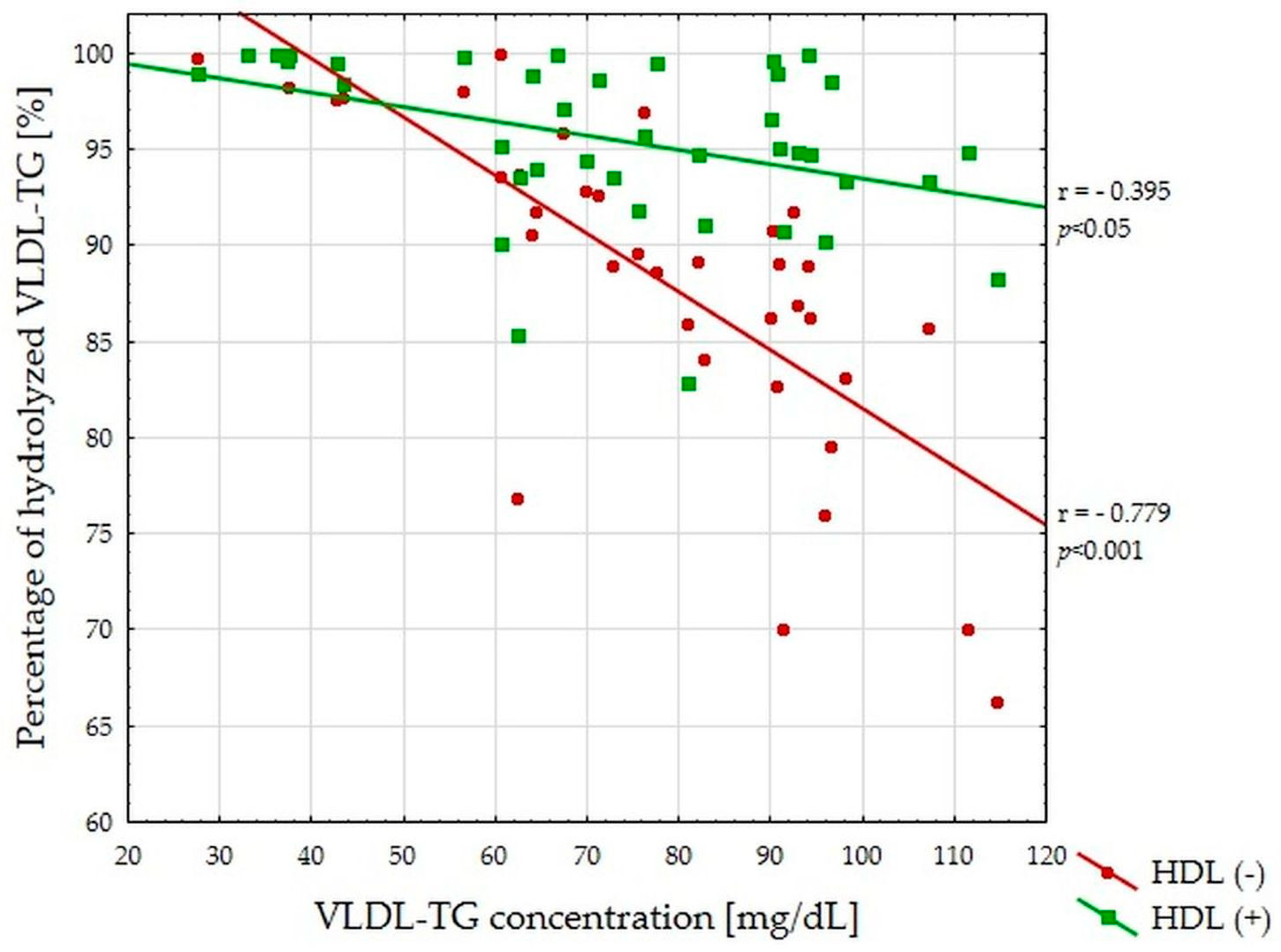
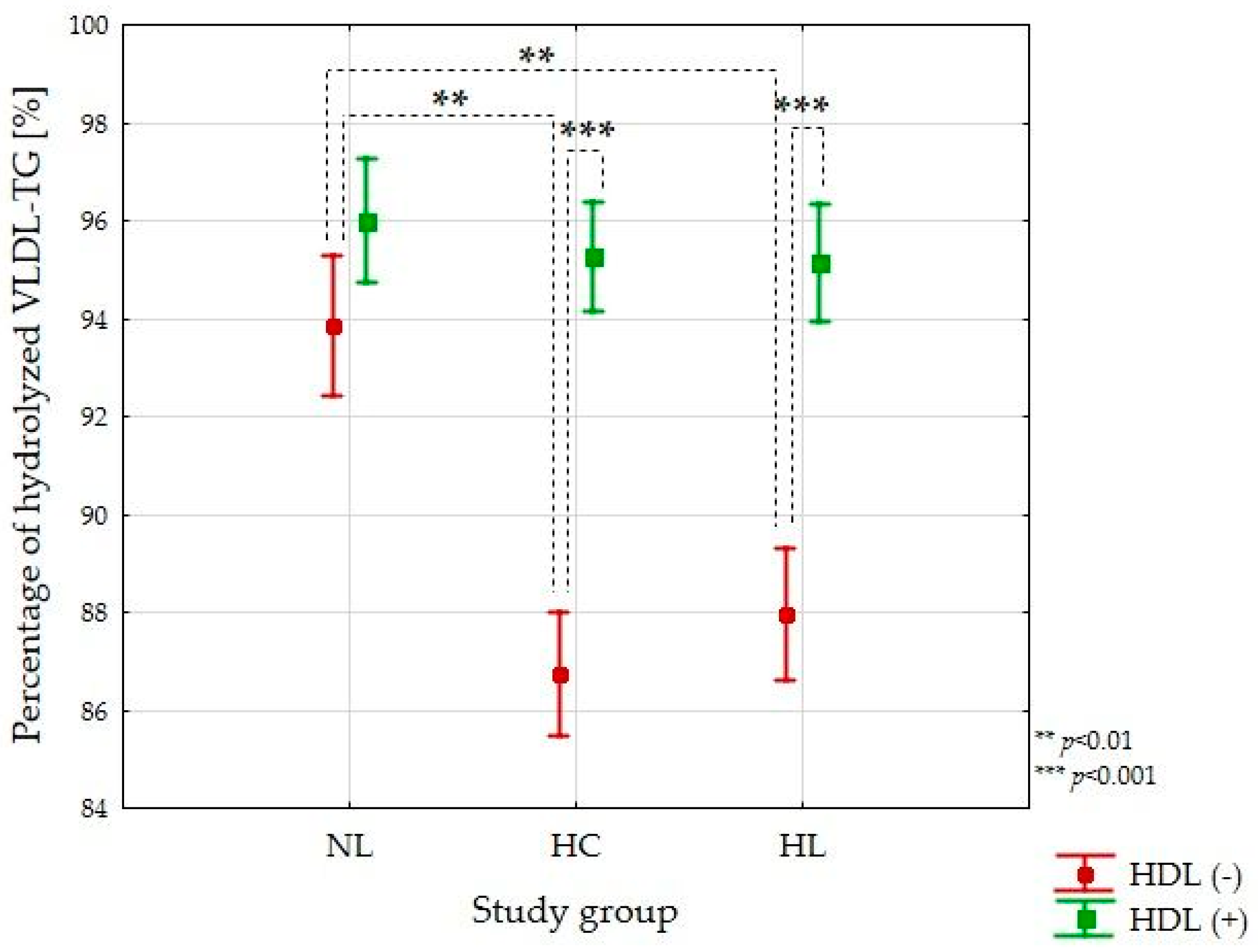
3.2. VLDL-TG Lipolysis Efficiency Study
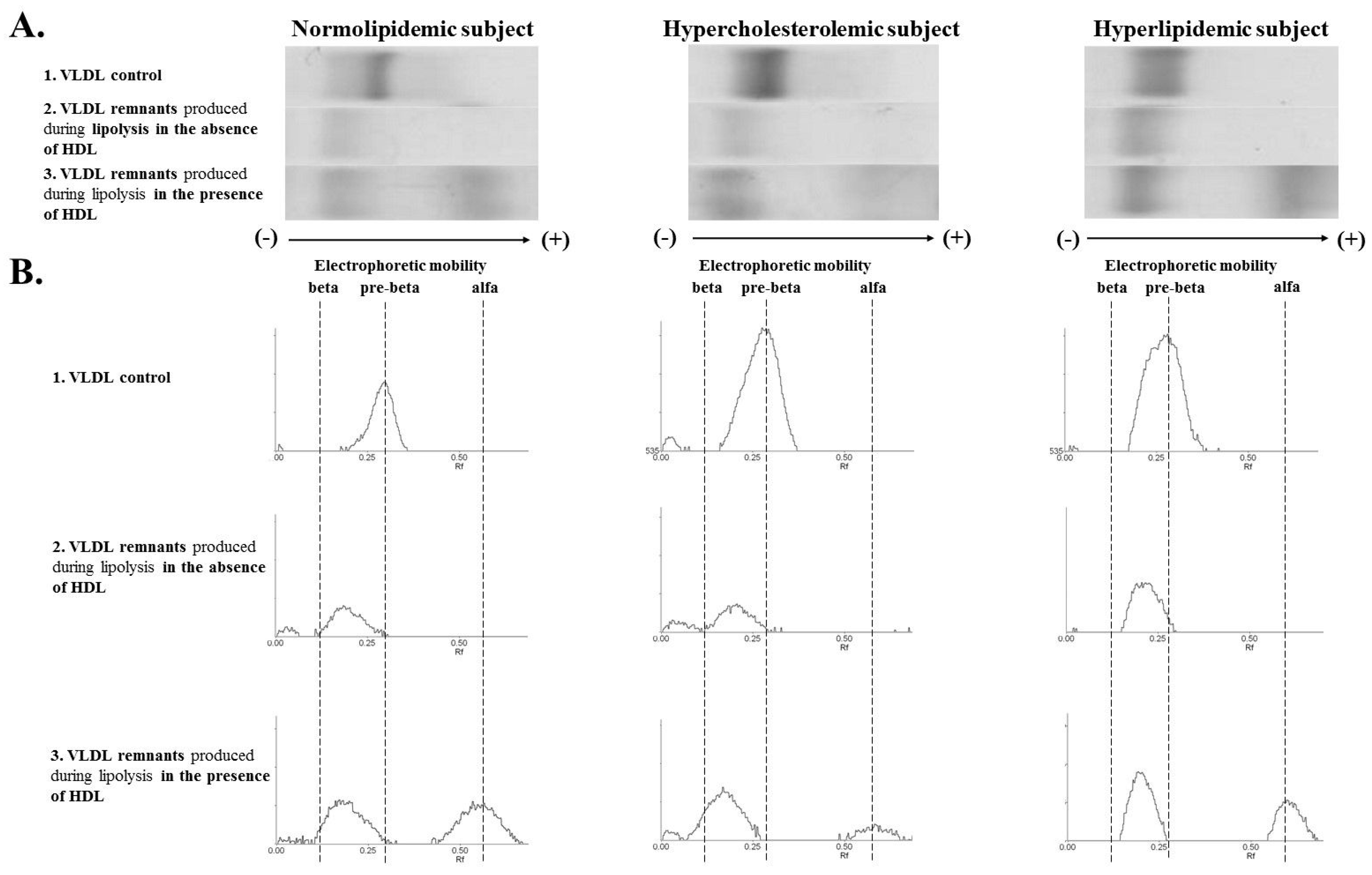
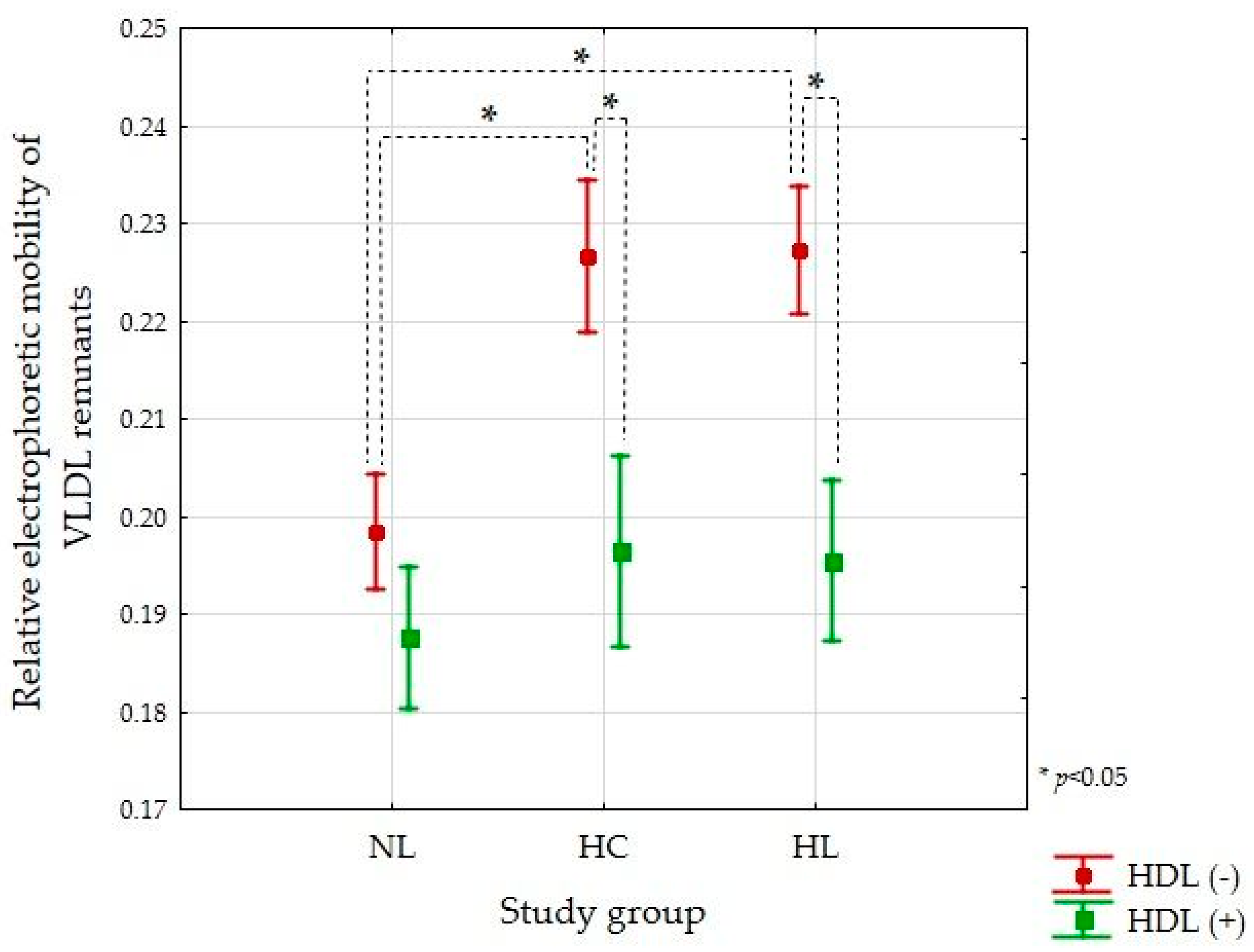
3.3. VLDL Remnants Mobility Produced during Lipolysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jagannathan, R.; Patel, S.A.; Ali, M.K.; Narayan, K.M.V. Global Updates on Cardiovascular Disease Mortality Trends and Attribution of Traditional Risk Factors. Curr. Diab. Rep. 2019, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baignet, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Delgado, V. 2019 ESC / EAS Guidelines for the Management of Dyslipidaemias: Lipid Modification to Reduce Cardiovascular Risk the Task Force for the Management of Dyslipidaemias of the the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur. Hear. J. 2020, 41, 111–188. [Google Scholar] [CrossRef] [PubMed]
- Arca, M.; Veronesi, C.; D’Erasmo, L.; Borghi, C.; Colivicchi, F.; De Ferrari, G.M.; Desideri, G.; Pontremoli, R.; Temporelli, P.L.; Perrone, V.; et al. Association of Hypertriglyceridemia with All-Cause Mortality and Atherosclerotic Cardiovascular Events in a Low-Risk Italian Population: The TG-REAL Retrospective Cohort Analysis. J. Am. Heart Assoc. 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Nordestgaard, B.G. Triglyceride-Rich Lipoproteins and Atherosclerotic Cardiovascular Disease. New Insights from Epidemiology, Genetics, and Biology. Circ. Res. 2016, 118, 547–563. [Google Scholar] [CrossRef]
- Meldrum, D.R.; Morris, M.A.; Gambone, J.C. Obesity Pandemic: Causes, Consequences, and Solutions—But Do We Have the Will? Fertil. Steril. 2017, 107, 833–839. [Google Scholar] [CrossRef]
- Peng, J.; Luo, F.; Ruan, G.; Peng, R.; Li, X. Hypertriglyceridemia and Atherosclerosis. Lipids Health Dis. 2017, 16, 1–12. [Google Scholar] [CrossRef]
- Ooi, E.M.M.; Barrett, P.H.R.; Chan, D.C.; Watts, G.F. Apolipoprotein C-III: Understanding an Emerging Cardiovascular Risk Factor. Clin. Sci. 2008, 114, 611–624. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Varbo, A. Triglycerides and Cardiovascular Disease. Lancet 2014, 384, 626–635. [Google Scholar] [CrossRef]
- Björnson, E.; Adiels, M.; Taskinen, M.-R.; Borén, J. Kinetics of Plasma Triglycerides in Abdominal Obesity. Curr. Opin. Lipidol. 2016, 28, 11–18. [Google Scholar] [CrossRef]
- Shin, H.K.; Kim, Y.K.; Kim, K.Y.; Lee, J.H.; Hong, K.W. Remnant Lipoprotein Particles Induce Apoptosis in Endothelial Cells by NAD(P)H Oxidase-Mediated Production of Superoxide and Cytokines via Lectin-like Oxidized Low-Density Lipoprotein Receptor-1 Activation: Prevention by Cilostazol. Circulation 2004, 109, 1022–1028. [Google Scholar] [CrossRef]
- Wang, L.; Gill, R.; Pedersen, T.L.; Higgins, L.J.; Newman, J.W.; Rutledge, J.C. Triglyceride-Rich Lipoprotein Lipolysis Releases Neutral and Oxidized FFAs That Induce Endothelial Cell Inflammation. J. Lipid Res. 2009, 50, 204–213. [Google Scholar] [CrossRef]
- Rached, F.H.; Chapman, M.J.; Kontush, A. HDL Particle Subpopulations: Focus on Biological Function. BioFactors 2015, 41, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Hernáez, Á.; Soria-Florido, M.T.; Schröder, H.; Ros, E.; Pintó, X.; Estruch, R.; Salas-Salvadó, J.; Corella, D.; Arós, F.; Serra-Majem, L.; et al. Role of HDL Function and LDL Atherogenicity on Cardiovascular Risk: A Comprehensive Examination. PLoS ONE 2019, 14, 1–15. [Google Scholar] [CrossRef]
- Zago, V.; Gorzalczany, S.; Lucero, D.; Taira, C.; Schreier, L. Role of HDL in Neutralizing the VLDL Effect on Endothelial Dysfunction. Microvasc. Res. 2013, 89, 153–158. [Google Scholar] [CrossRef]
- Ćwiklińska, A.; Cackowska, M.; Wieczorek, E.; Król, E.; Kowalski, R.; Kuchta, A.; Kortas-Stempak, B.; Gliwińska, A.; Dąbkowski, K.; Zielińska, J.; et al. Progression of Chronic Kidney Disease Affects HDL Impact on Lipoprotein Lipase (LPL)-Mediated VLDL Lipolysis Efficiency. Kidney Blood Press. Res. 2018, 43, 970–978. [Google Scholar] [CrossRef] [PubMed]
- Kolovou, G.D.; Anagnostopoulou, K.K.; Pilatis, N.D.; Iraklianou, S.; Hoursalas, I.S.; Liberi, S.; Pavlidis, A.N.; Dritsas, A.; Mikhailidis, D.P.; Cokkinos, D.V. Heterozygote Men with Familial Hypercholesterolaemia May Have an Abnormal Triglyceride Response Post-Prandially. Evidence for Another Predictor of Vascular Risk in Familial Hypercholesterolaemia. Int. J. Clin. Pract. 2005, 59, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Pavlidis, A.N.; Kolovou, G.D.; Anagnostopoulou, K.K.; Petrou, P.C.; Cokkinos, D.V. Postprandial Metabolic Heterogeneity in Men with Primary Dyslipidaemia. Arch. Med. Sci. 2010, 6, 879–886. [Google Scholar] [CrossRef]
- Chan, D.C.; Pang, J.; Hugh, R.B.; Sullivan, D.R.; Burnett, J.R.; Van Bockxmeer, F.M.; Watts, G.F. ω-3 Fatty Acid Ethyl Esters Diminish Postprandial Lipemia in Familial Hypercholesterolemia. J. Clin. Endocrinol. Metab. 2016, 101, 3732–3739. [Google Scholar] [CrossRef]
- McEneny, J.; Loughrey, C.M.; McNamee, P.T.; Trimble, E.R.; Young, I.S. Susceptibility of VLDL to Oxidation in Patients on Regular Haemodialysis. Atherosclerosis 1997, 129, 215–220. [Google Scholar] [CrossRef]
- McPherson, P.A.C.; Young, I.S.; McKibben, B.; McEneny, J. High Density Lipoprotein Subfractions: Isolation, Composition, and Their Duplicitous Role in Oxidation. J. Lipid Res. 2007, 48, 86–95. [Google Scholar] [CrossRef]
- Ćwiklińska, A.; Kortas-Stempak, B.; Gliwińska, A.; Pacanis, A.; Kuchta, A.; Wróblewska, M. Interaction between VLDL and Phosphatidylcholine Liposomes Generates New γ-LpE-like Particles. Lipids 2014, 49, 143–153. [Google Scholar] [CrossRef]
- Couch, S.C.; Isasi, C.R.; Karmally, W.; Blaner, W.S.; Starc, T.J.; Kaluski, D.; Deckelbaum, R.J.; Ginsberg, H.N.; Shea, S.; Berglund, L. Predictors of Postprandial Triacylglycerol Response in Children: The Columbia University Biomarkers Study. Am. J. Clin. Nutr. 2000, 72, 1119–1127. [Google Scholar] [CrossRef]
- O’Meara, N.M.; Lewis, G.F.; Cabana, V.G.; Iverius, P.H.; Getz, G.S.; Polonsky, K.S. Role of Basal Triglyceride and High Density Lipoprotein in Determination of Postprandial Lipid and Lipoprotein Responses. J. Clin. Endocrinol. Metab. 1992, 75, 465–471. [Google Scholar] [CrossRef]
- Van Barlingen, H.H.; Kock, L.A.; De Man, F.H.; Erkelens, D.W.; De Bruin, T.W. In Vitro Lipolysis of Human VLDL: Effect of Different VLDL Compositions in Normolipidemia, Familial Combined Hyperlipidemia and Familial Hypertriglyceridemia. Atherosclerosis 1996, 121, 75–84. [Google Scholar] [CrossRef]
- Chung, B.H.; Segrest, J.P. Resistance of a Very Low Density Lipoprotein Subpopulation from Familial Dysbetalipoproteinemia to in Vitro Lipolytic Conversion to the Low Density Lipoprotein Density Fraction. J. Lipid Res. 1983, 24, 1148–1159. [Google Scholar] [CrossRef]
- Tiihonen, K.; Rautonen, N.; Alhoniemi, E.; Ahotupa, M.; Stowell, J.; Vasankari, T. Postprandial Triglyceride Response in Normolipidemic, Hyperlipidemic and Obese Subjects—The Influence of Polydextrose, a Non-Digestible Carbohydrate. Nutr. J. 2015, 14, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cortés, B.; Núñez, I.; Cofán, M.; Gilabert, R.; Pérez-Heras, A.; Casals, E.; Deulofeu, R.; Ros, E. Acute Effects of High-Fat Meals Enriched with Walnuts or Olive Oil on Postprandial Endothelial Function. J. Am. Coll. Cardiol. 2006, 48, 1666–1671. [Google Scholar] [CrossRef] [PubMed]
- Cabezas, M.C.; De Bruin, T.W.A.; Westerveld, H.E.; Meijer, E.; Erkelens, D.W. Delayed Chylomicron Remnant Clearance in Subjects with Heterozygous Familial Hypercholesterolaemia. J. Intern. Med. 1998, 244, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Packard, C.J.; Boren, J.; Taskinen, M.R. Causes and Consequences of Hypertriglyceridemia. Front. Endocrinol. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wolska, A.; Dunbar, R.L.; Freeman, L.A.; Ueda, M.; Amar, M.J.; Sviridov, D.O. Apolipoprotein C-II: New Findings Related to Genetics, Biochemistry, and Role in Triglyceride Metabolism. Atherosclerosis 2017, 267, 549–562. [Google Scholar] [CrossRef]
- Dominiczak, M.H.; Caslake, M.J. Review Article Apolipoproteins: Metabolic Role and Clinical Biochemistry Applications. Ann. Clin. Biochem. 2011, 48, 498–515. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.C.; Lee, Y.T.; Chen, M.F. Effect of N-3 Fatty Acids on the Composition and Binding Properties of Lipoproteins in Hypertriglyceridemic Patients. Am. J. Clin. Nutr. 2000, 71, 28–35. [Google Scholar] [CrossRef]
- Chan, D.C.; Nguyen, M.N.; Watts, G.F.; Barrett, P.H.R. Plasma Apolipoprotein C-III Transport in Centrally Obese Men: Associations with Very Low-Density Lipoprotein Apolipoprotein B and High-Density Lipoprotein Apolipoprotein A-I Metabolism. J. Clin. Endocrinol. Metab. 2008, 93, 557–564. [Google Scholar] [CrossRef]
- Ooi, E.M.; Chan, D.C.; Hodson, L.; Adiels, M.; Boren, J.; Karpe, F.; Fielding, B.A.; Watts, G.F.; Barrett, P.H.R. Triglyceride-Rich Lipoprotein Metabolism in Women: Roles of apoC-II and apoC-III. Eur. J. Clin. Investig. 2016, 46, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Kei, A.A.; Filippatos, T.D.; Tsimihodimos, V.; Elisaf, M.S. A Review of the Role of Apolipoprotein C-II in Lipoprotein Metabolism and Cardiovascular Disease. Metabolism 2012, 61, 906–921. [Google Scholar] [CrossRef] [PubMed]
- Batal, R.; Tremblay, M.; Barrett, P.H.R.; Jacques, H.; Fredenrich, A.; Mamer, O.; Davignon, J.; Cohn, J.S. Plasma Kinetics of apoC-III and apoE in Normolipidemic and Hypertriglyceridemic Subjects. J. Lipid Res. 2000, 41, 706–718. [Google Scholar] [CrossRef]
- Tomiyasu, K.; Walsh, B.W.; Ikewaki, K.; Judge, H.; Sacks, F.M. Differential Metabolism of Human VLDL according to Content of apoE and apoC-III. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1494–1500. [Google Scholar] [CrossRef]
- Mendivil, C.O.; Zheng, C.; Furtado, J.; Lel, J.; Sacks, F.M. Metabolism of Very-Low-Density Lipoprotein and Low-Density Lipoprotein Containing Apolipoprotein C-III and Not Other Small Apolipoproteins. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 239–245. [Google Scholar] [CrossRef]
- Ćwiklińska, A.; Gliwińska, A.; Senderowska, Z.; Kortas-Stempak, B.; Kuchta, A.; Dąbkowski, K.; Jankowski, M. Impact of Phosphatidylcholine Liposomes on the Compositional Changes of VLDL during Lipoprotein Lipase (LPL)-Mediated Lipolysis. Chem. Phys. Lipids 2016, 195, 63–70. [Google Scholar] [CrossRef]
- Chung, B.H.; Dashti, N. Lipolytic Remnants of Human VLDL Produced in Vitro: Effect of HDL Levels in the Lipolysis Mixtures on the apoCs to apoE Ratio and Metabolic Properties of VLDL Core Remnants. J. Lipid Res. 2000, 41, 285–297. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Normolipidemic (NL) Group | Hypercholesterolemic (HC) Group | Hyperlipidemic (HL) Group | p-Value |
|---|---|---|---|---|
| Number | 12 | 15 | 13 | - |
| Age (years) | 31 ± 7 | 40 ±13 | 41 ± 11 | 0.050 a |
| BMI (kg/m2) | 28 ± 5 | 28 ± 5 | 28 ± 4 | 0.935 a |
| Gender (Female/Male) | 8/4 (67%/33%) | 6/9 (40%/60%) | 6/7 (46%/54%) | 0.366 c |
| Hypertension | 1 (8%) | 5 (33%) | 3 (23%) | 0.302 c |
| Smoking | 3 (25%) | 2 (13%) | 3 (23%) | 0.766 c |
| Serum lipids and apolipoproteins | ||||
| Triglycerides (mg/dL) | 81 (72–114) | 101 (75–135) | 164 (151–238) d,e | <0.001 b |
| Total cholesterol (mg/dL) | 180 (169–188) | 215 (197–239) d | 240 (210–258) d | <0.001 b |
| LDL-cholesterol (mg/dL) | 101 ± 8 | 149 ± 35 d | 147 ±33 d | <0.001 a |
| Phospholipids (mg/dL) | 227 (203–249) | 232 (210–254) | 259 (234–291) d,e | 0.007 b |
| Apo B (mg/dL) | 80 ± 13 | 105 ± 16 d | 110 ± 18 d | <0.001 a |
| Apo CII (mg/dL) | 3.53 ± 1.21 | 4.24 ± 1.41 | 5.88 ± 1.43 d,e | <0.001 a |
| Apo CIII (mg/dL) | 8.56 (5.87–12.21) | 10.22 (7.73–11.22) | 16.68 (13.50–18.53) d,e | <0.001 b |
| Apo E (mg/dL) | 3.15 ± 1.03 | 3.79 ± 0.84 | 5.28 ± 1.41 d,e | <0.001 a |
| VLDL lipids and apolipoproteins | ||||
| Triglycerides (mg/dL) | 53 ± 20 | 60 ± 28 | 139 ± 40 d,e | <0.001 a |
| Cholesterol (mg/dL) | 10 (6–11) | 13 (7–18) | 26 (24–32) d,e | <0.001 b |
| Phospholipids (mg/dL) | 16 ± 5 | 22 ± 12 | 46 ± 17 d,e | <0.001 a |
| Apo B (mg/dL) | 4.78 (3.15–6.03) | 6.00 (3.25–7.43) | 10.40 (9.50–14.95) d,e | <0.001 b |
| Apo CII (mg/dL) | 0.94 ± 0.46 | 1.28 ± 0.62 | 2.87 ± 1.21 d,e | <0.001 a |
| Apo CIII (mg/dL) | 2.38 ± 1.01 | 3.01 ± 1.33 | 7.29 ± 3.72 d,e | <0.001 a |
| Apo E (mg/dL) | 0.51 ± 0.17 | 0.55 ± 0.27 | 1.16 ± 0.73 d,e | 0.001 a |
| HDL lipids and apolipoproteins | ||||
| Triglycerides (mg/dL) | 16 (12–19) | 12 (11–15) | 15 (14–21) | 0.157 b |
| Cholesterol (mg/dL) | 61 ± 15 | 52 ± 11 | 51 ± 15 | 0.122 a |
| Phospholipids (mg/dL) | 164 ± 39 | 133 ± 32 | 136 ± 37 | 0.119 a |
| Apo AI (mg/dL) | 221 ± 48 | 166 ± 29 d | 181 ± 40 d | 0.011 a |
| Apo CII (mg/dL) | 2.13 ± 0.89 | 2.44 ± 0.77 | 2.33 ± 0.68 | 0.652 a |
| Apo CIII (mg/dL) | 6.79 ± 3.56 | 6.36 ± 2.18 | 6.65 ± 2.34 | 0.928 a |
| Apo E (mg/dL) | 0.86 ± 0.27 | 1.05 ± 0.20 | 1.27 ± 0.50 d | 0.045 a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wieczorek, E.; Ćwiklińska, A.; Kuchta, A.; Kortas-Stempak, B.; Gliwińska, A.; Jankowski, M. Decreased Efficiency of Very-Low-Density Lipoprotein Lipolysis Is Linked to Both Hypertriglyceridemia and Hypercholesterolemia, but It Can Be Counteracted by High-Density Lipoprotein. Nutrients 2021, 13, 1224. https://doi.org/10.3390/nu13041224
Wieczorek E, Ćwiklińska A, Kuchta A, Kortas-Stempak B, Gliwińska A, Jankowski M. Decreased Efficiency of Very-Low-Density Lipoprotein Lipolysis Is Linked to Both Hypertriglyceridemia and Hypercholesterolemia, but It Can Be Counteracted by High-Density Lipoprotein. Nutrients. 2021; 13(4):1224. https://doi.org/10.3390/nu13041224
Chicago/Turabian StyleWieczorek, Ewa, Agnieszka Ćwiklińska, Agnieszka Kuchta, Barbara Kortas-Stempak, Anna Gliwińska, and Maciej Jankowski. 2021. "Decreased Efficiency of Very-Low-Density Lipoprotein Lipolysis Is Linked to Both Hypertriglyceridemia and Hypercholesterolemia, but It Can Be Counteracted by High-Density Lipoprotein" Nutrients 13, no. 4: 1224. https://doi.org/10.3390/nu13041224
APA StyleWieczorek, E., Ćwiklińska, A., Kuchta, A., Kortas-Stempak, B., Gliwińska, A., & Jankowski, M. (2021). Decreased Efficiency of Very-Low-Density Lipoprotein Lipolysis Is Linked to Both Hypertriglyceridemia and Hypercholesterolemia, but It Can Be Counteracted by High-Density Lipoprotein. Nutrients, 13(4), 1224. https://doi.org/10.3390/nu13041224