
Analysis of the Gut Mycobiome in Adult Patients with Type 1 and Type 2 Diabetes Using Next-Generation Sequencing (NGS) with Increased Sensitivity—Pilot Study

 , , ,
, , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Materials
2.3. Library Preparation
2.4. Next-Generation Sequencing
2.5. Bioinformatic and Statistical Analysis
3. Results
3.1. Characteristics of the Study Population
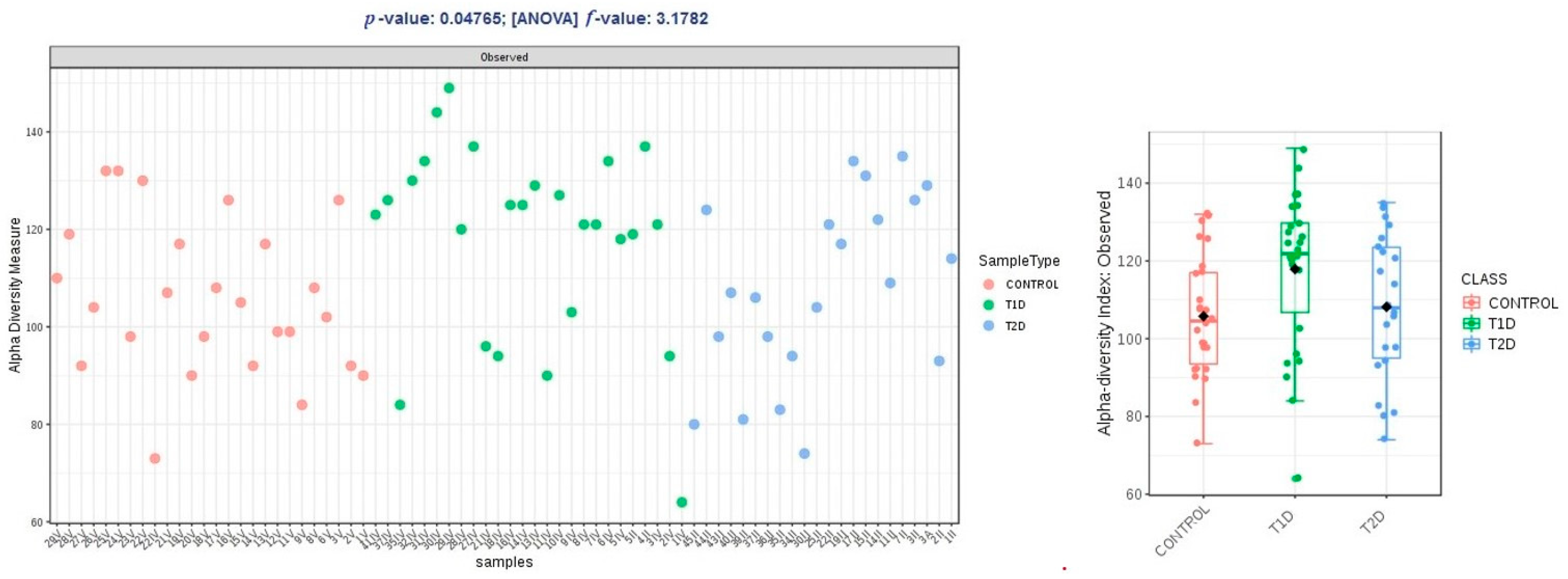
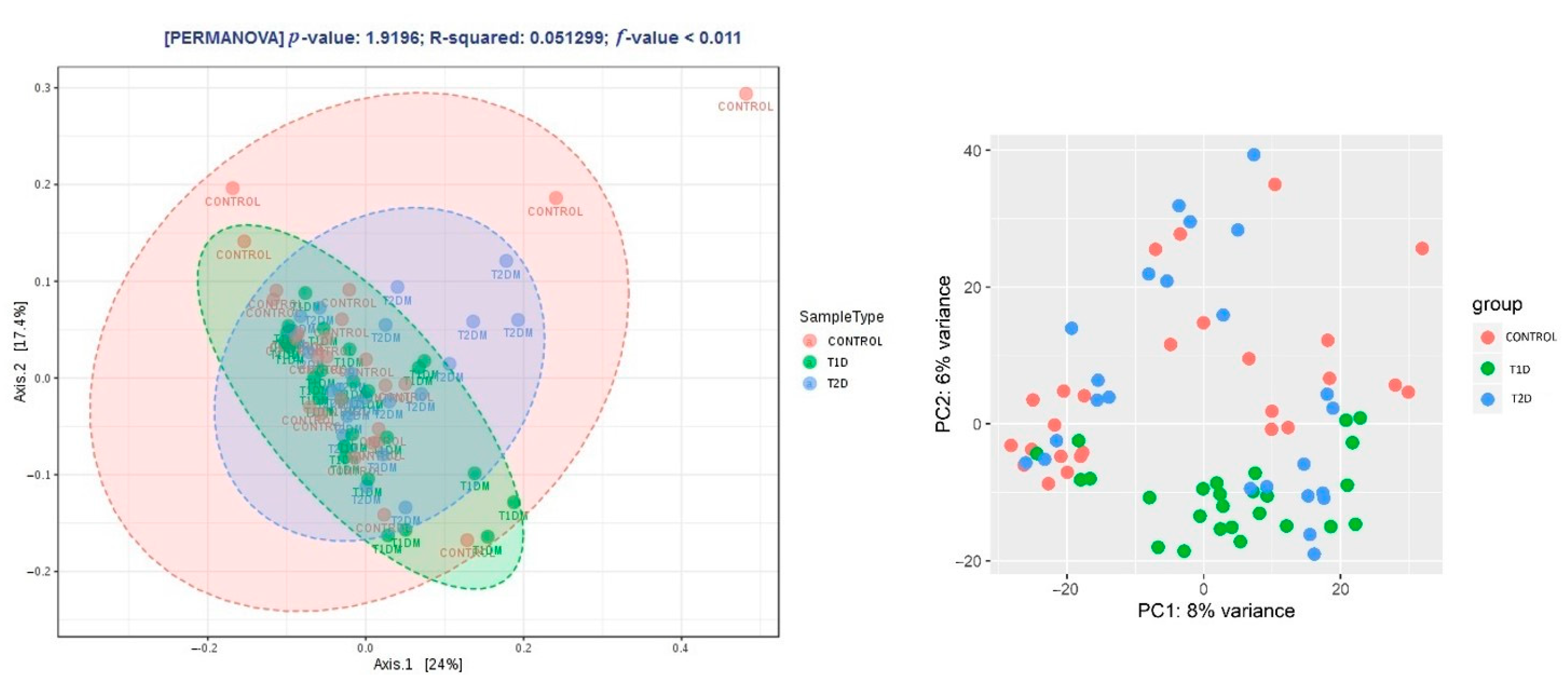
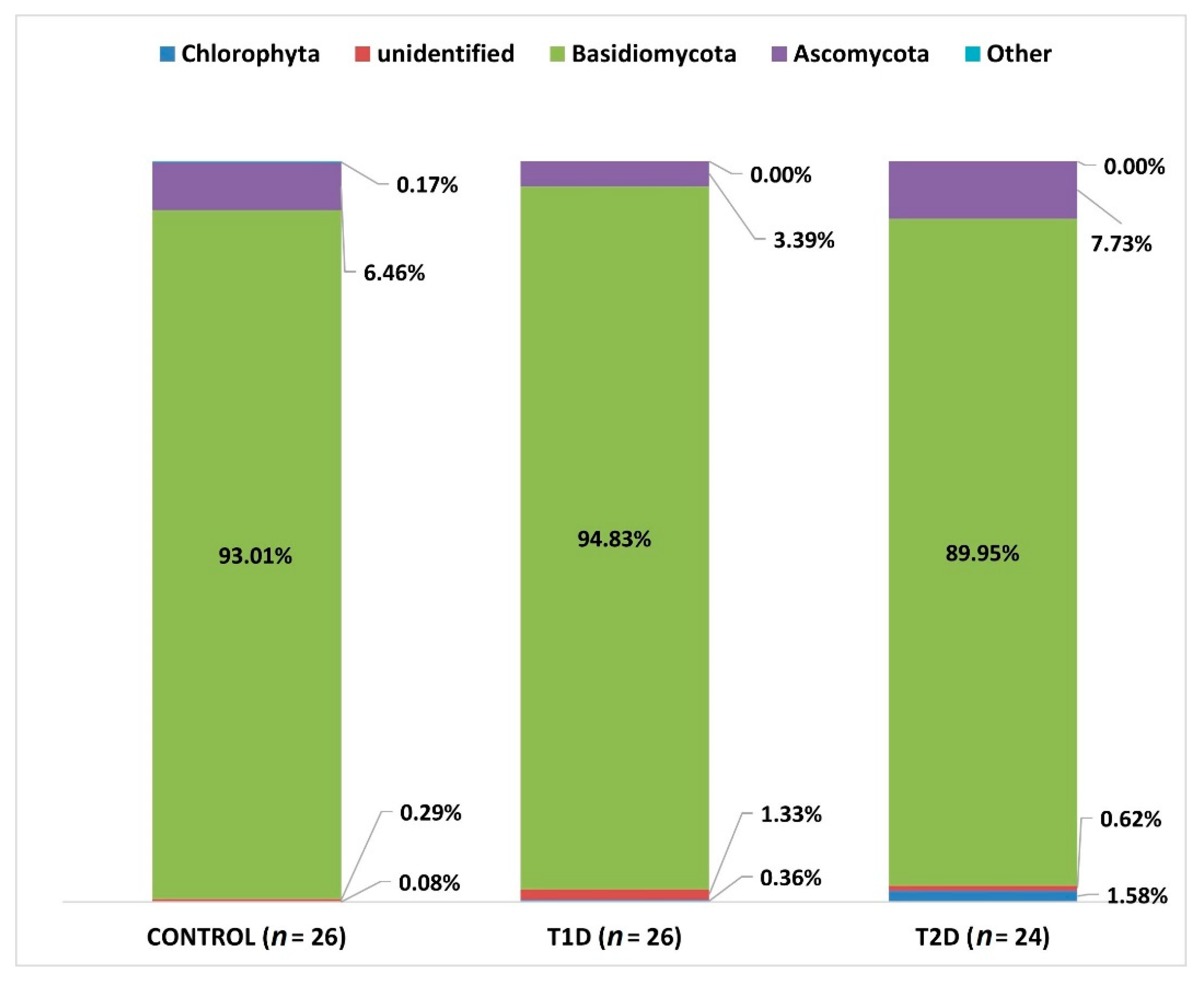
3.2. Metagenomic Sequencing
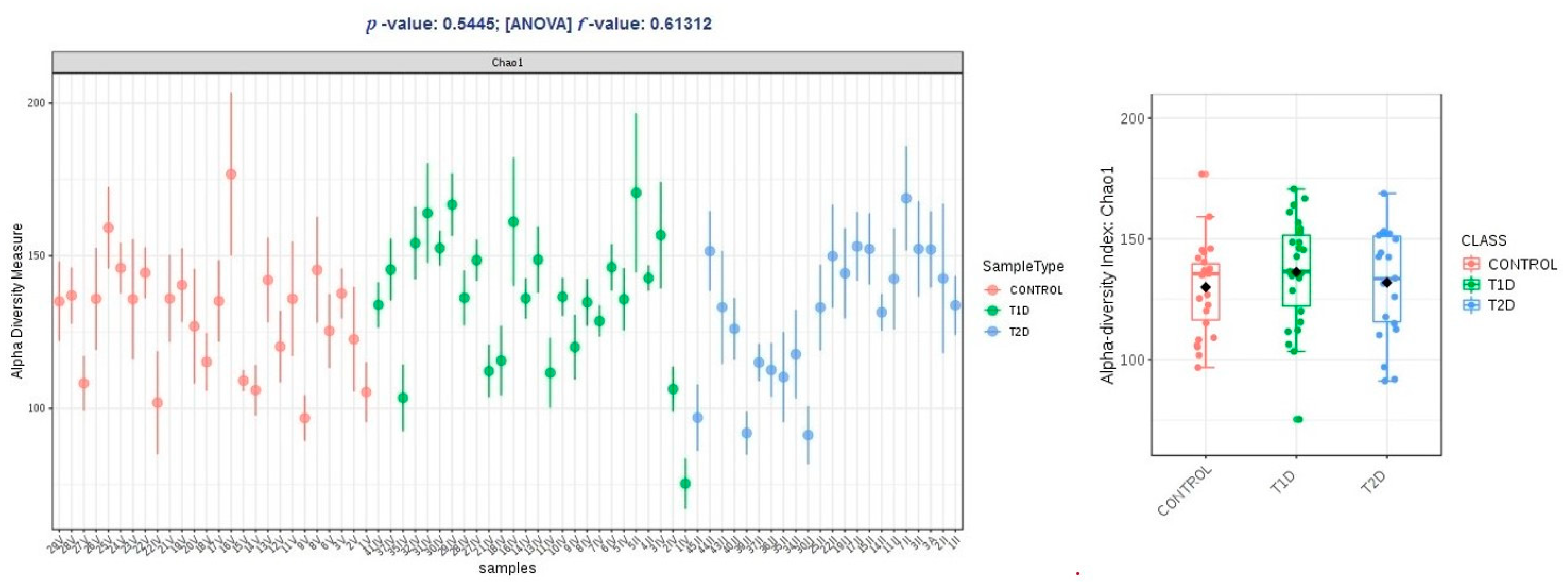
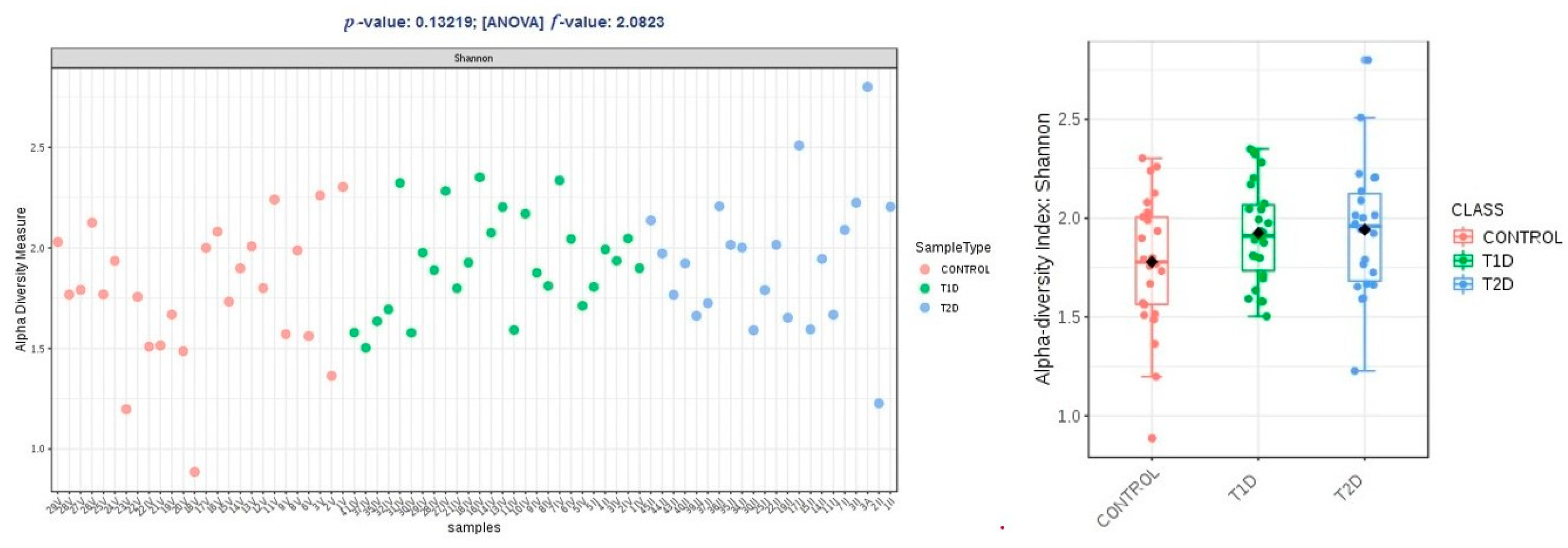
3.3. Correlation Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- International Diabetes Federation. IDF Diabetes Atlas—2019; International Diabetes Federation: Brussels, Belgium, 2019; ISBN 9782930229812. [Google Scholar]
- Bonnefond, A.; Froguel, P. Rare and common genetic events in type 2 diabetes: What should biologists know? Cell Metab. 2015, 21, 357–368. [Google Scholar] [CrossRef]
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef]
- Semenkovich, C.F.; Danska, J.; Darsow, T.; Dunne, J.L.; Huttenhower, C.; Insel, R.A.; McElvaine, A.T.; Ratner, R.E.; Shuldiner, A.R.; Blaser, M.J. American diabetes association and JDRF research symposium: Diabetes and the microbiome. Diabetes 2015, 64, 3967–3977. [Google Scholar] [CrossRef]
- Davis-Richardson, A.G.; Triplett, E.W. A model for the role of gut bacteria in the development of autoimmunity for type 1 diabetes. Diabetologia 2015, 58, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- Gurung, M.; Li, Z.; You, H.; Rodrigues, R.; Jump, D.B.; Morgun, A.; Shulzhenko, N. Role of gut microbiota in type 2 diabetes pathophysiology. EBioMedicine 2020, 51, 102590. [Google Scholar] [CrossRef]
- Salamon, D.; Sroka-Oleksiak, A.; Kapusta, P.; Szopa, M.; Mrozińska, S.; Ludwig-Słomczyńska, A.H.; Wołkow, P.P.; Bulanda, M.; Klupa, T.; Małecki, M.T.; et al. Characteristics of gut microbiota in adult patients with type 1 and type 2 diabetes based on next-generation sequencing of the 16S rRNA gene fragment. Pol. Arch. Intern. Med. 2018, 128, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Sroka-Oleksiak, A.; Młodzińska, A.; Bulanda, M.; Salamon, D.; Major, P.; Stanek, M.; Gosiewski, T. Metagenomic Analysis of Duodenal Microbiota Reveals a Potential Biomarker of Dysbiosis in the Course of Obesity and Type 2 Diabetes: A Pilot Study. J. Clin. Med. 2020, 9, 369. [Google Scholar] [CrossRef] [PubMed]
- Underhill, D.M.; Iliev, I.D. The mycobiota: Interactions between commensal fungi and the host immune system. Nat. Rev. Immunol. 2014, 14, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Ruszkowski, J.; Kaźmierczak-Siedlecka, K.; Witkowski, J.M.; Dębska-Ślizień, A. Mycobiota of the human gastrointestinal tract. Postepy Hig. Med. Dosw. 2020, 74, 301–313. [Google Scholar] [CrossRef]
- Chin, V.K.; Yong, V.C.; Chong, P.P.; Amin Nordin, S.; Basir, R.; Abdullah, M. Mycobiome in the Gut: A Multiperspective Review. Mediators Inflamm. 2020, 2020, 9560684. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, D.; Yu, B.; He, J.; Zheng, P.; Mao, X.; Yu, J.; Luo, J.; Tian, G.; Huang, Z.; et al. Fungi in Gastrointestinal Tracts of Human and Mice: From Community to Functions. Microb. Ecol. 2018, 75, 821–829. [Google Scholar] [CrossRef]
- Nash, A.K.; Auchtung, T.A.; Wong, M.C.; Smith, D.P.; Gesell, J.R.; Ross, M.C.; Stewart, C.J.; Metcalf, G.A.; Muzny, D.M.; Gibbs, R.A.; et al. The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome 2017, 5, 153. [Google Scholar] [CrossRef]
- Hoffmann, C.; Dollive, S.; Grunberg, S.; Chen, J.; Li, H.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Archaea and Fungi of the Human Gut Microbiome: Correlations with Diet and Bacterial Residents. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Kowalska-Duplaga, K.; Krawczyk, A.; Sroka-Oleksiak, A.; Salamon, D.; Wedrychowicz, A.; Fyderek, K.; Gosiewski, T. Dependence of colonization of the large intestine by candida on the treatment of Crohn’s disease. Polish J. Microbiol. 2019, 68, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Bhute, S.S.; Suryavanshi, M.V.; Joshi, S.M.; Yajnik, C.S.; Shouche, Y.S.; Ghaskadbi, S.S. Gut microbial diversity assessment of Indian type-2-diabetics reveals alterations in eubacteria, archaea, and eukaryotes. Front. Microbiol. 2017, 8, 214. [Google Scholar] [CrossRef] [PubMed]
- Gosiewski, T.; Salamon, D.; Szopa, M.; Sroka, A.; Malecki, M.T.; Bulanda, M. Quantitative evaluation of fungi of the genus Candida in the feces of adult patients with type 1 and 2 diabetes—A pilot study. Gut Pathog. 2014, 6. [Google Scholar] [CrossRef]
- Fungal Sequencing and Classification with the ITS Metagenomics Protocol. Available online: https://www.illumina.com/content/dam/illumina-marketing/documents/products/appnotes/its-metagenomics-app-note-1270-2018-001-web.pdf (accessed on 24 March 2021).
- Nilsson, R.H.; Anslan, S.; Bahram, M.; Wurzbacher, C.; Baldrian, P.; Tedersoo, L. Mycobiome diversity: High-throughput sequencing and identification of fungi. Nat. Rev. Microbiol. 2019, 17, 95–109. [Google Scholar] [CrossRef]
- Toju, H.; Tanabe, A.S.; Yamamoto, S.; Sato, H. High-coverage ITS primers for the DNA-based identification of ascomycetes and basidiomycetes in environmental samples. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Gosiewski, T.; Szała, L.; Pietrzyk, A.; Brzychczy-Włoch, M.; Heczko, P.B.; Bulanda, M. Comparison of methods for isolation of bacterial and fungal DNA from human blood. Curr. Microbiol. 2014, 68, 149–155. [Google Scholar] [CrossRef] [PubMed]
- 16S Metagenomic Sequencing Library Preparation. Available online: https://support.illumina.com/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf (accessed on 24 March 2021).
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Kõljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.S.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson-Palme, J.; Callaghan, T.M.; et al. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Faul, F.; Erdfelder, E.; Lang, A.G.; Buchner, A. G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods 2007, 39, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Kukwa, M.; Kosecka, M.; Guzow-Krzemińska, B. One Name—One Fungus: The Influence of Photosynthetic Partners on the Taxonomy and Systematics of Lichenized Fungi. Acta Soc. Bot. Pol. 2020, 89. [Google Scholar] [CrossRef]
- Nowakowska, D.; Kurnatowska, A.; Stray-Pedersen, B.; Wilczyński, J. Species distribution and influence of glycemic control on fungal infections in pregnant women with diabetes. J. Infect. 2004, 48, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Siljander, H.; Honkanen, J.; Knip, M. Microbiome and type 1 diabetes. EBioMedicine 2019, 46, 512–521. [Google Scholar] [CrossRef]
- Jayasudha, R.; Das, T.; Kalyana Chakravarthy, S.; Sai Prashanthi, G.; Bhargava, A.; Tyagi, M.; Rani, P.K.; Pappuru, R.R.; Shivaji, S. Gut mycobiomes are altered in people with type 2 Diabetes Mellitus and Diabetic Retinopathy. PLoS ONE 2020, 15, e0243077. [Google Scholar] [CrossRef]
- Al Bataineh, M.T.; Dash, N.R.; Lassen, P.B.; Banimfreg, B.H.; Nada, A.M.; Belda, E.; Clément, K. Revealing links between gut microbiome and its fungal community in Type 2 Diabetes Mellitus among Emirati subjects: A pilot study. Sci. Rep. 2020, 10, 9624. [Google Scholar] [CrossRef]
- Strati, F.; Di Paola, M.; Stefanini, I.; Albanese, D.; Rizzetto, L.; Lionetti, P.; Calabrò, A.; Jousson, O.; Donati, C.; Cavalieri, D.; et al. Age and gender affect the composition of fungal population of the human gastrointestinal tract. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Mar Rodríguez, M.; Pérez, D.; Javier Chaves, F.; Esteve, E.; Marin-Garcia, P.; Xifra, G.; Vendrell, J.; Jové, M.; Pamplona, R.; Ricart, W.; et al. Obesity changes the human gut mycobiome. Sci. Rep. 2015, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hamad, I.; Ranque, S.; Azhar, E.I.; Yasir, M.; Jiman-Fatani, A.A.; Tissot-Dupont, H.; Raoult, D.; Bittar, F. Culturomics and Amplicon-based Metagenomic Approaches for the Study of Fungal Population in Human Gut Microbiota. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Spatz, M.; Richard, M.L. Overview of the Potential Role of Malassezia in Gut Health and Disease. Front. Cell. Infect. Microbiol. 2020, 10, 201. [Google Scholar] [CrossRef] [PubMed]
- Fryssouli, V.; Zervakis, G.; Polemis, E.; Typas, M.A. A global meta-analysis of ITS rDNA sequences from material belonging to the genus Ganoderma (Basidiomycota, Polyporales) including new data from selected taxa. MycoKeys 2020, 75, 71–143. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.C.; Xi, R.J.; Li, Y.; Wang, D.M.; Yao, Y.J. The species identity of the widely cultivated ganoderma, “G. lucidum” (ling-zhi), in China. PLoS ONE 2012, 7, 40857. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Obin, M.S.; Zhao, L. The gut microbiota, obesity and insulin resistance. Mol. Aspects Med. 2013, 34, 39–58. [Google Scholar] [CrossRef]
- Albuquerque, R.C.M.F.; Brandão, A.B.P.; De Abreu, I.C.M.E.; Ferreira, F.G.; Santos, L.B.; Moreira, L.N.; Taddei, C.R.; Aimbire, F.; Cunha, T.S. Saccharomyces boulardii Tht 500101 changes gut microbiota and ameliorates hyperglycaemia, dyslipidaemia, and liver inflammation in streptozotocin-diabetic mice. Benef. Microbes 2019, 10, 901–912. [Google Scholar] [CrossRef]
- Juntachai, W.; Oura, T.; Murayama, S.Y.; Kajiwara, S. The lipolytic enzymes activities of Malassezia species. Med. Mycol. 2009, 47, 477–484. [Google Scholar] [CrossRef]
- Aykut, B.; Pushalkar, S.; Chen, R.; Li, Q.; Abengozar, R.; Kim, J.I.; Shadaloey, S.A.; Wu, D.; Preiss, P.; Verma, N.; et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature 2019, 574, 264–267. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
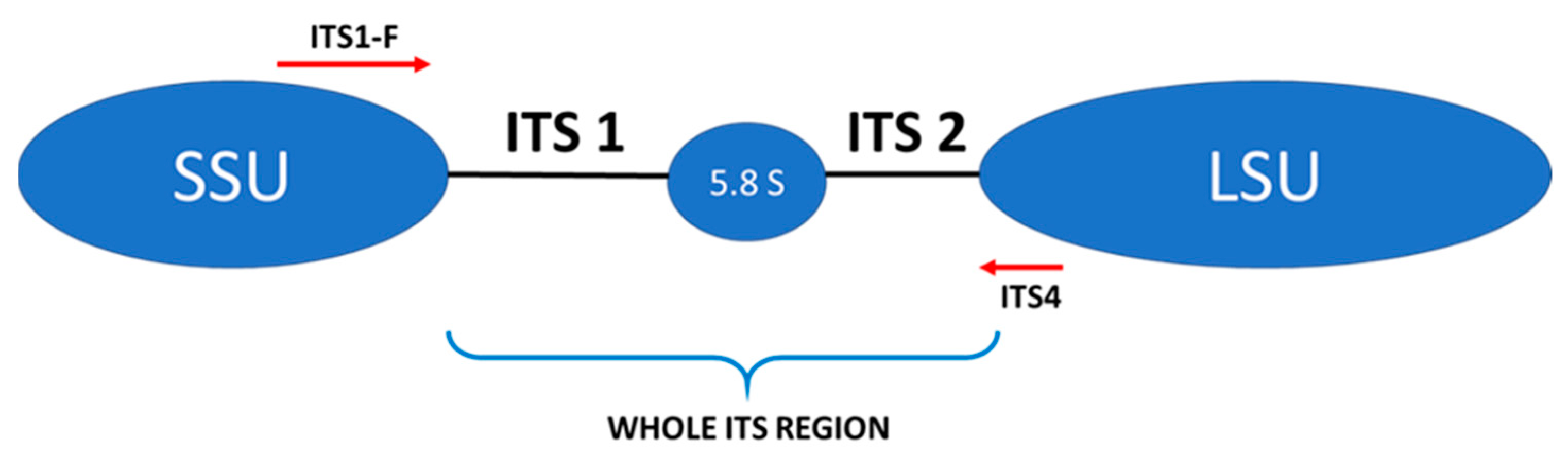
| Primer Sequence 5′→3′ | Reaction Mixture | Thermal Amplification Program | |
|---|---|---|---|
| External primers a F: AAATGCGATAAGTAATGTGAATTGCAGAATT R: TTACTAGGGGAATCCTTGTTAGTTTCT | Water Kapa c Primer F (10 µM) Primer R (10 µM) DNA | 2.0 μL 5.0 μL 0.5 μL 0.5 μL 2.0 μL |  |
| Internal primers b ITS1-F(F): CTGGTCATTTAGAAGTAA ITS4 (R): TCCTCCGCTTATTGTATGC | Water Kapa c Primer F (10 µM) Primer R (10 µM) DNA | 9.5 μL 12.5 μL 0.5 μL 0.5 μL 2.0 μL |  |
| Parameters | CONTROL (n = 26) | T1D (n = 26) | T2D (n = 24) | p-Value |
|---|---|---|---|---|
| F:M | 19:7 | 20:6 | 9:15 | - |
| Age, years | 36 (31−46.5) | 33(30−47) | 56 (56.25−62.75) | <0.001 a |
| BMI, kg/m2 | 23.1 (22.2−24.6) | 22.2 (20.3−25) | 27.2 (25−28.7) | <0.001 a |
| HbA1c, % | 5.35(5.2−5.5) | 7.95(6.77−9.65) | 7.1(6.41−8.56) | <0.001 b |
| Total cholesterol, mmol/L | 5.2(4.92−5.75) | 5.0 (4.12−5.42) | 4.82 (4.04−5.9) | 0.458 |
| HDL-C, mmol/L | 1.8 (1.5−1.9) | 1.6 (1.42−2.0) | 1.08 (0.87−1.2) | <0.001 a |
| LDL-C, mmol/L | 3.15 (2.72−3.55) | 2.7 (2.3−3.25) | 2.94 (2.49−3.77) | 0.215 |
| TGs mmol/L | 0.8 (0.69−1.19) | 0.8 (0.65−1.35) | 1.72 (1.4−2.29) | 0.274 |
| ALT, U/L | 17 (13.2−19.85) | 14 (11.2−19.5) | 24.5 (20.5−35) | <0.001 a |
| Creatinine, μmol/L | 60 (56−66) | 58 (55−68) | 59 (56−65) | <0.39 |
| eGFR (MDRD), mL/min/1.73 m2 | 115.3 (118.6−110.8) | 118.7 (121.3−111.25) | 108.2 (110.5−103.9) | 0.06 |
| Duration of diabetes, years | - | 15.5 (5.5−22.75) | 5.5 (2.25−10) | 0.004 c,* |
| Taxonomic Level | CONTROL | T1D | T2D | |||
|---|---|---|---|---|---|---|
| No. of Classified Reads | Percent of Reads | No. of Reads | Percent of Classified Reads | No. of Reads | Percent of Classified Reads | |
| Kingdom | 2,896,459 | 96.92 | 4,142,146 | 96.56 | 2,782,278 | 95.83 |
| Phylum | 2,880,932 | 99.48 | 4,118,805 | 99.35 | 2,765,589 | 99.33 |
| Class | 2,862,193 | 99.33 | 4,079,130 | 99.04 | 2,732,493 | 98.66 |
| Order | 2,813,641 | 98.23 | 4,021,210 | 98.69 | 2,700,384 | 98.75 |
| Family | 2,784,972 | 99.07 | 3,976,863 | 99.01 | 2,670,820 | 98.80 |
| Genus | 2,751,978 | 98.91 | 3,887,741 | 98.14 | 2,652,556 | 99.31 |
| Species | 2,492,288 | 91.98 | 3,684,955 | 94.59 | 2,381,592 | 90.98 |
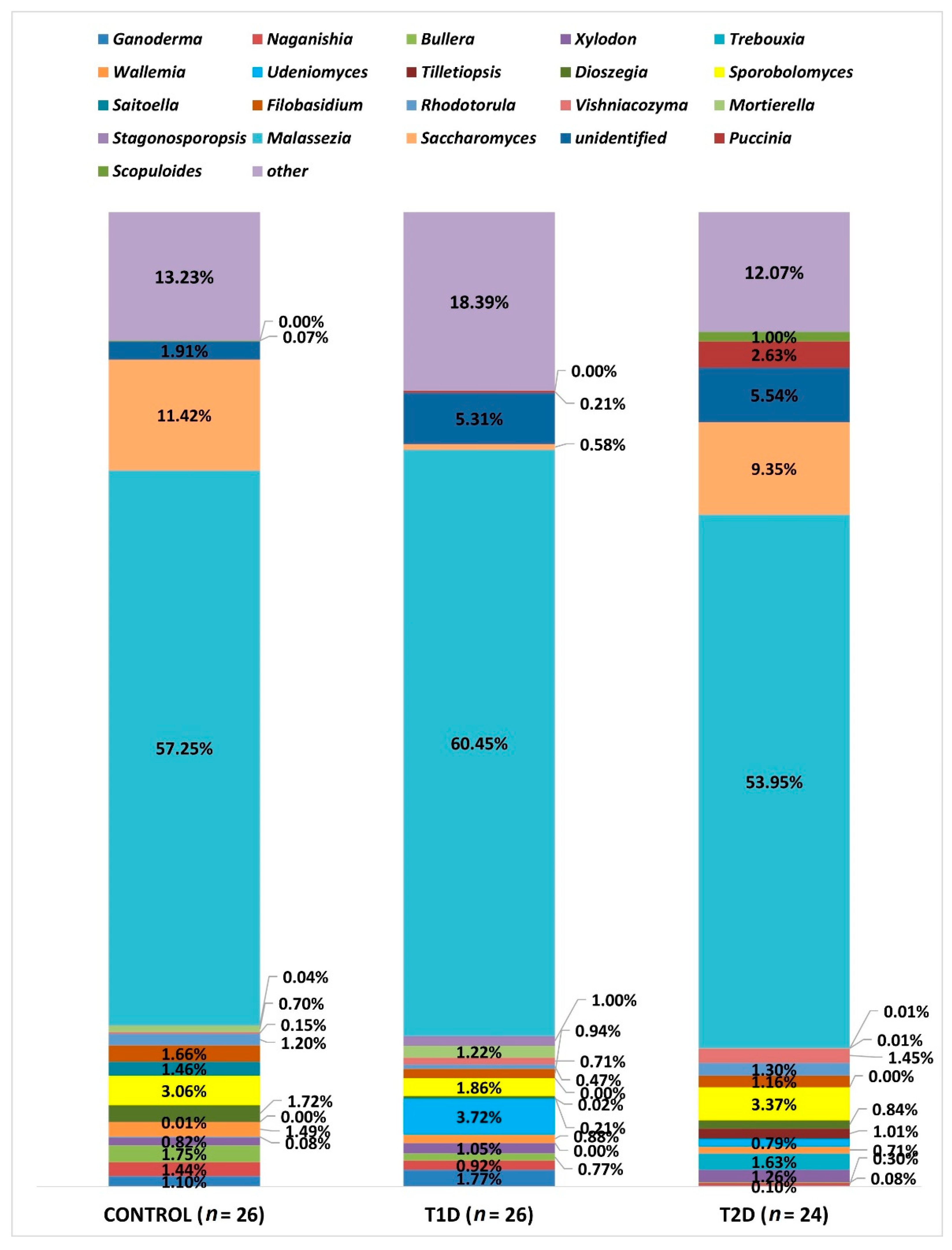
| Genus | Difference Between Groups (Relative Percentage) | Adjusted p-Value |
|---|---|---|
| Saccharomyces | Control (11.42%) vs. T1D (0.58%) T1D (0.58%) vs. T2D (9.35%) | <0.001 <0.0001 |
| Dioszegia | Control (1.72%) vs. T1D (0.21%) | 0.005 |
| Xylodon | Control (0.81%) vs. T1D (1.05%) | 0.005 |
| Mortierella | Control (0.7%) vs. T1D (1.22%) | 0.008 |
| Naganishia | Control (1.44%) vs. T2D (0.3%) T1D (0.92%) vs. T2D (0.3%) | <0.0001 <0.0001 |
| Udeniomyces | Control (0.01%) vs. T1D(3.72%) Control (0.01%) vs. T2D (0.79%) T1D (3.72%) vs. T2D (0.79%) | <0.0001 <0.0001 0.007 |
| Bullera | Control (1.75%) vs. T2D (0.08%) T1D (0.77%) vs. T2D (0.08%) | <0.0001 <0.001 |
| Tilletiopsis | Control (0.002%) vs. T2D (1.01%) T1D (0.02%) vs. T2D (1.01%) | <0.0001 <0.0001 |
| Saitoella | Control (1.46%) vs. T2D (0.002%) | <0.001 |
| Ganoderma | Control (1.1%) vs. T2D (0.1%) T1D (1.77%) vs. T2D (0.1%) | 0.013 0.02 |
| Vishniacozyma | Control (0.15%) vs. T2D (1.45%) | 0.04 |
| Wallemia | Control (1.49%) vs. T2D (0.71%) | 0.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salamon, D.; Sroka-Oleksiak, A.; Gurgul, A.; Arent, Z.; Szopa, M.; Bulanda, M.; Małecki, M.T.; Gosiewski, T. Analysis of the Gut Mycobiome in Adult Patients with Type 1 and Type 2 Diabetes Using Next-Generation Sequencing (NGS) with Increased Sensitivity—Pilot Study. Nutrients 2021, 13, 1066. https://doi.org/10.3390/nu13041066
Salamon D, Sroka-Oleksiak A, Gurgul A, Arent Z, Szopa M, Bulanda M, Małecki MT, Gosiewski T. Analysis of the Gut Mycobiome in Adult Patients with Type 1 and Type 2 Diabetes Using Next-Generation Sequencing (NGS) with Increased Sensitivity—Pilot Study. Nutrients. 2021; 13(4):1066. https://doi.org/10.3390/nu13041066
Chicago/Turabian StyleSalamon, Dominika, Agnieszka Sroka-Oleksiak, Artur Gurgul, Zbigniew Arent, Magdalena Szopa, Małgorzata Bulanda, Maciej T. Małecki, and Tomasz Gosiewski. 2021. "Analysis of the Gut Mycobiome in Adult Patients with Type 1 and Type 2 Diabetes Using Next-Generation Sequencing (NGS) with Increased Sensitivity—Pilot Study" Nutrients 13, no. 4: 1066. https://doi.org/10.3390/nu13041066
APA StyleSalamon, D., Sroka-Oleksiak, A., Gurgul, A., Arent, Z., Szopa, M., Bulanda, M., Małecki, M. T., & Gosiewski, T. (2021). Analysis of the Gut Mycobiome in Adult Patients with Type 1 and Type 2 Diabetes Using Next-Generation Sequencing (NGS) with Increased Sensitivity—Pilot Study. Nutrients, 13(4), 1066. https://doi.org/10.3390/nu13041066