
Wait-and-See Approach or Gluten-Free Diet Administration—The Rational Management of Potential Coeliac Disease

Abstract
1. Introduction
2. Epidemiological Data
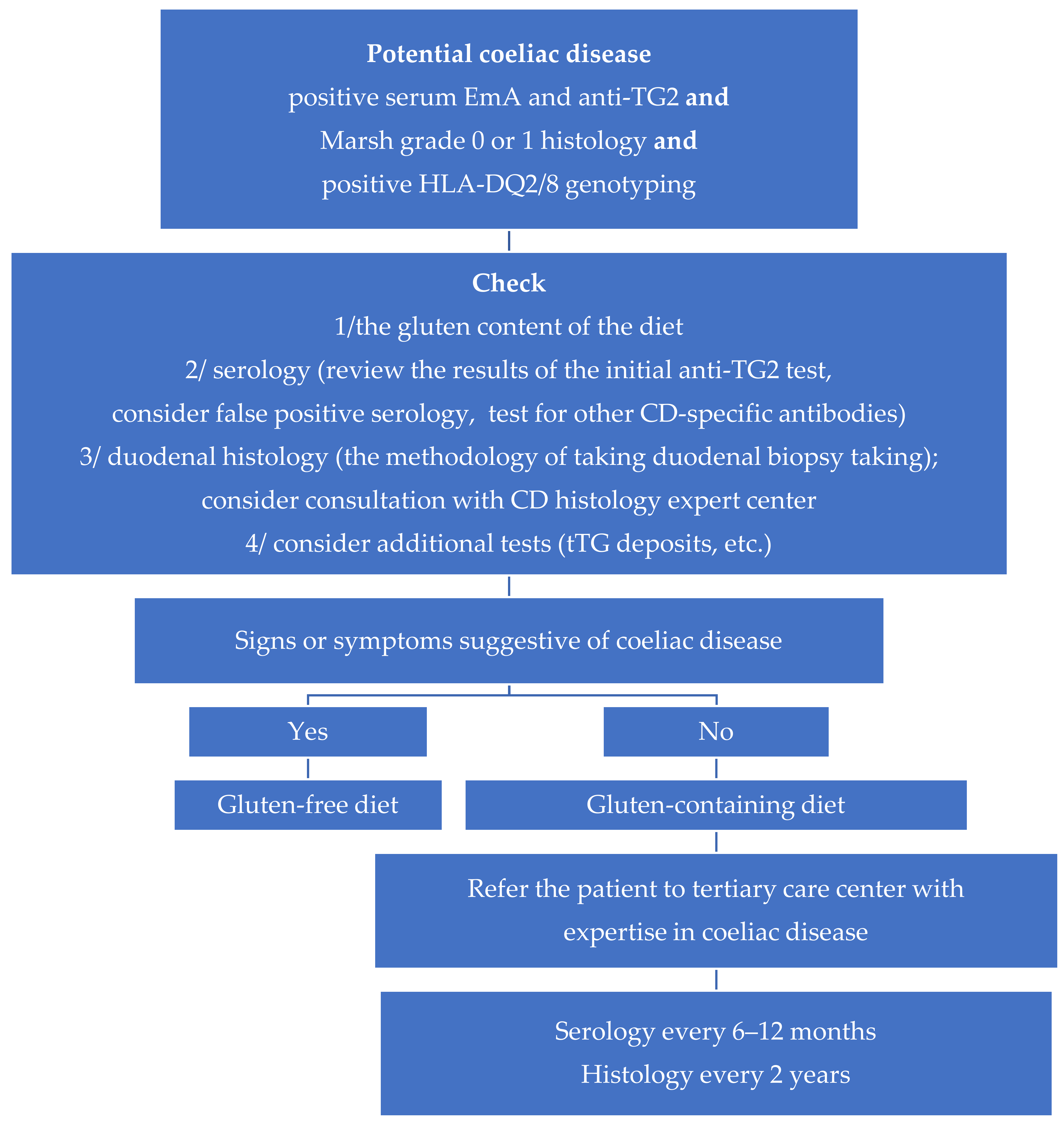
3. Verification of a Diagnosis of PCD
4. Histological Features
5. Clinical Picture
6. Prognostic Markers
7. Natural Course
8. Treatment Principles
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Ludvigsson, J.F.; Leffler, D.A.; Bai, J.C.; Biagi, F.; Fasano, A.; Green, P.H.R.; Hadjivassiliou, M.; Kaukinen, K.; Kelly, C.P.; Leonard, J.N.; et al. The Oslo definitions for coeliac disease and related terms. Gut 2012, 62, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Al-Toma, A.; Volta, U.; Auricchio, R.; Castillejo, G.; Sanders, D.S.; Cellier, C.; Mulder, C.J.; Lundin, K.E.A. European Society for the Study of Coeliac Disease (ESsCD) guideline for coeliac disease and other gluten-related disorders. United Eur. Gastroenterol. J. 2019, 7, 583–613. [Google Scholar] [CrossRef] [PubMed]
- Husby, S.; Koletzko, S.; Korponay-Szabó, I.; Kurppa, K.; Mearin, M.L.; Ribes-Koninckx, C.; Shamir, R.; Troncone, R.; Auricchio, R.; Castillejo, G.; et al. European Society Paediatric Gastroenterology, Hepatology and Nutrition Guidelines for Diagnosing Coeliac Disease 2020. J. Pediatr. Gastroenterol. Nutr. 2020, 70, 141–156. [Google Scholar] [CrossRef]
- Ferguson, A.; Arranz, E.; O’Mahony, S. Clinical and pathological spectrum of celiac disease: Active, silent, latent, potential. Gut 1993, 34, 150–151. [Google Scholar] [CrossRef] [PubMed]
- Itzlinger, A.; Branchi, F.; Elli, L.; Schumann, M. Gluten-Free Diet in Celiac Disease—Forever and for All? Nutrients 2018, 10, 1796. [Google Scholar] [CrossRef] [PubMed]
- Biagi, F.; Trotta, L.; Alfano, C.; Balduzzi, D.; Staffieri, V.; Bianchi, P.I.; Marchese, A.; Vattiato, C.; Zilli, A.; Luinetti, O.; et al. Prevalence and natural history of potential celiac disease in adult patients. Scand. J. Gastroenterol. 2013, 48, 537–542. [Google Scholar] [CrossRef]
- Lionetti, E.; Castellaneta, S.; Pulvirenti, A.; Tonutti, E.; Francavilla, R.; Fasano, A.; Catassi, C. Prevalence and Natural History of Potential Celiac Disease in At-Family-Risk Infants Prospectively Investigated from Birth. J. Pediatr. 2012, 161, 908–914.e2. [Google Scholar] [CrossRef]
- Volta, U.; Caio, G.; Giancola, F.; Rhoden, K.J.; Ruggeri, E.; Boschetti, E.; Stanghellini, V.; De Giorgio, R. Features and pro-gression of potential celiac disease in adults. Clin. Gastroenterol. Hepatol. 2016, 14, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Mandile, R.; Discepolo, V.; Scapaticci, S.; Del Vecchio, M.R.; Maglio, M.A.; Greco, L.; Troncone, R.; Auricchio, R. The Effect of Gluten-free Diet on Clinical Symptoms and the Intestinal Mucosa of Patients With Potential Celiac Disease. J. Pediatr. Gastroenterol. Nutr. 2018, 66, 654–656. [Google Scholar] [CrossRef]
- Schiepatti, A.; Savioli, J.; Vernero, M.; De Andreis, F.B.; Perfetti, L.; Meriggi, A.; Biagi, F. Pitfalls in the Diagnosis of Coeliac Disease and Gluten-Related Disorders. Nutrients 2020, 12, 1711. [Google Scholar] [CrossRef] [PubMed]
- Biagi, F.; Bianchi, P.I.; Vattiato, C.; Marchese, A.; Trotta, L.; Badulli, C.; De Silvestri, A.; Martinetti, M.; Corazza, G.R. Influence of HLA-DQ2 and DQ8 on Severity in Celiac Disease. J. Clin. Gastroenterol. 2012, 46, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Sperandeo, M.P.; Tosco, A.; Izzo, V.; Tucci, F.; Troncone, R.; Auricchio, R.; Romanos, J.; Trynka, G.; Auricchio, S.; Jabri, B.; et al. Potential Celiac Patients: A Model of Celiac Disease Pathogenesis. PLoS ONE 2011, 6, e21281. [Google Scholar] [CrossRef] [PubMed]
- Trovato, C.M.; Montuori, M.; Valitutti, F.; Leter, B.; Cucchiara, S.; Oliva, S. The Challenge of Treatment in Potential Celiac Disease. Gastroenterol. Res. Pr. 2019, 2019, 1–6. [Google Scholar] [CrossRef]
- Bizzaro, N.; Villalta, D.; Tonutti, E.; Doria, A.; Tampoia, M.; Bassetti, D.; Tozzoli, R. IgA and IgG Tissue Transglutaminase Antibody Prevalence and Clinical Significance in Connective Tissue Diseases, Inflammatory Bowel Disease, and Primary Biliary Cirrhosis. Dig. Dis. Sci. 2003, 48, 2360–2365. [Google Scholar] [CrossRef]
- Sárdy, M.; Csikós, M.; Geisen, C.; Preisz, K.; Kornseé, Z.; Tomsits, E.; Töx, U.; Hunzelmann, N.; Wieslander, J.; Kárpáti, S.; et al. Tissue transglutaminase ELISA positivity in autoimmune disease independent of gluten-sensitive disease. Clin. Chim. Acta 2007, 376, 126–135. [Google Scholar] [CrossRef]
- Parkkola, A.; Härkönen, T.; Ryhänen, S.J.; Uibo, R.; Ilonen, J.; Knip, M.; Register, A.T.F.P.D. Transglutaminase antibodies and celiac disease in children with type 1 diabetes and in their family members. Pediatr. Diabetes 2017, 19, 305–313. [Google Scholar] [CrossRef]
- Villalta, D.; Crovatto, M.; Stella, S.; Tonutti, E.; Tozzoli, R.; Bizzaro, N. False positive reactions for IgA and IgG anti-tissue transglutaminase antibodies in liver cirrhosis are common and method-dependent. Clin. Chim. Acta 2005, 356, 102–109. [Google Scholar] [CrossRef]
- Nisihara, R.M.; Kotze, L.M.S.; Utiyama, S.R.R.; Oliveira, N.P.; Fiedler, P.T.; Messias-Reason, I.T. Celiac disease in children and adolescents with Down syndrome. J. Pediatr. 2005, 81, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Stenberg, R.; Dahle, C.; Lindberg, E.; Schollin, J. Increased prevalence of anti-gliadin antibodies and anti-tissue transglu-taminase antibodies in children with cerebral palsy. J. Pediatr. Gastroenterol. Nutr. 2009, 49, 424–429. [Google Scholar] [CrossRef] [PubMed]
- De Leo, L.; Quaglia, S.; Ziberna, F.; Vatta, S.; Martelossi, S.; Maschio, M.; Not, T. Serum anti-tissue transglutaminase anti-bodies detected during febrile illness may not be produced by the intestinal mucosa. J. Pediatr. 2015, 166, 761–763. [Google Scholar] [CrossRef]
- Peracchi, M.; Trovato, C.; Longhi, M.; Gasparin, M.; Conte, D.; Tarantino, C.; Prati, D.; Bardella, M.T. Tissue transglutami-nase antibodies in patients with end-stage heart failure. Am. J. Gastroenterol. 2002, 97, 2850–2854. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.; Ramesh, A.; Matthias, T. Serologic Diagnosis of Celiac Disease: New Biomarkers. Gastroenterol. Clin. N. Am. 2019, 48, 307–317. [Google Scholar] [CrossRef]
- Popp, A.; Mäki, M. Gluten-Induced Extra-Intestinal Manifestations in Potential Celiac Disease—Celiac Trait. Nutrents 2019, 11, 320. [Google Scholar] [CrossRef] [PubMed]
- Castellaneta, S.; Piccinno, E.; Oliva, M.; Cristofori, F.; Vendemiale, M.; Ortolani, F.; Papadia, F.; Catassi, C.; Cavallo, L.; Francavilla, R. High Rate of Spontaneous Normalization of Celiac Serology in a Cohort of 446 Children With Type 1 Diabetes: A Prospective Study. Diabetes Care 2015, 38, 760–766. [Google Scholar] [CrossRef]
- Tosco, A.; Salvati, V.M.; Auricchio, R.; Maglio, M.; Borrelli, M.; Coruzzo, A.; Paparo, F.; Boffardi, M.; Esposito, A.; D’Adamo, G.; et al. Natural History of Potential Celiac Disease in Children. Clin. Gastroenterol. Hepatol. 2011, 9, 320–325. [Google Scholar] [CrossRef]
- Kurppa, K.; Ashorn, M.; Iltanen, S.; Koskinen, L.L.; Saavalainen, P.; Koskinen, O.; Mäki, M.; Kaukinen, K. Celiac Disease without Villous Atrophy in Children: A Prospective Study. J. Pediatr. 2010, 157, 373–380.e1. [Google Scholar] [CrossRef] [PubMed]
- Husnoo, N.; Ahmed, W.; Shiwani, M.H. Duodenal biopsies for the diagnosis of coeliac disease: Are we adhering to current guidance? BMJ Open Gastroenterol. 2017, 4, e000140. [Google Scholar] [CrossRef]
- Ravelli, A.; Villanacci, V. Tricks of the trade: How to avoid histological Pitfalls in celiac disease. Pathol.-Res. Pr. 2012, 208, 197–202. [Google Scholar] [CrossRef]
- Montén, C.; Bjelkenkrantz, K.; Gudjonsdottir, A.H.; Browaldh, L.; Arnell, H.; Naluai, Å.T.; Agardh, D. Validity of histology for the diagnosis of paediatric coeliac disease: A Swedish multicentre study. Scand. J. Gastroenterol. 2015, 51, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Silvester, J.A.; Kelly, C.P. The Potential for Treatment of Potential Celiac Disease. Clin. Gastroenterol. Hepatol. 2016, 14, 694–695. [Google Scholar] [CrossRef]
- Szaflarska-Popławska, A.; Parzęcka, M.; Kuczyńska, R. The Range of Lesions in the Small Intestine of Children with Celiac Disease Determined by Capsule Endoscopy. Adv. Clin. Exp. Med. 2014, 23, 785–790. [Google Scholar] [CrossRef]
- Kowalski, K.; Mulak, A.; Jasińska, M.; Paradowski, L. Diagnostic challenges in celiac disease. Adv. Clin. Exp. Med. 2017, 26, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Zingone, F.; Marsilio, I.; Fassan, M.; Pilotto, V.; Maddalo, G.; Lorenzon, G.; Savarino, E.V.; Farinati, F. Duodenal Histological Findings and Risk of Coeliac Disease in Subjects with Autoimmune Atrophic Gastritis: A Retrospective Evaluation. Digestion 2020, 19, 1–7. [Google Scholar] [CrossRef]
- Paparo, F.; Petrone, E.; Tosco, A.; Maglio, M.; Borrelli, M.; Salvati, V.M.; Miele, E.; Greco, L.; Auricchio, S.; Troncone, R. Clinical, HLA, and Small Bowel Immunohistochemical Features of Children with Positive Serum Antiendomysium Antibodies and Architecturally Normal Small Intestinal Mucosa. Am. J. Gastroenterol. 2005, 100, 2294–2298. [Google Scholar] [CrossRef] [PubMed]
- Nijeboer, P.; van Gils, T.; Reijm, M.; Ooijevaar, R.; Lissenberg-Witte, B.I.; Bontkes, H.J.; Mulder, C.J.J.; Bouma, G.; Gam-ma-Delta, T. Lymphocytes in the Diagnostic Approach of Coeliac Disease. J. Clin. Gastroenterol. 2019, 53, e208–e213. [Google Scholar] [CrossRef]
- Tosco, A.; Aitoro, R.; Auricchio, R.; Ponticelli, D.; Miele, E.; Paparo, F.; Greco, L.; Troncone, R.; Maglio, M. Intestinal an-ti-tissue transglutaminase antibodies in potential coeliac disease. Clin. Exp. Med. 2012, 171, 69–75. [Google Scholar]
- De Leo, L.; Bramuzzo, M.; Ziberna, F.; Villanacci, V.; Martelossi, S.; Di Leo, G.; Zanchi, C.; Giudici, F.; Pandullo, M.; Riznik, P.; et al. Diagnostic accuracy and applicability of intestinal auto-antibodies in the wide clinical spectrum of coeliac disease. EBioMedicine 2020, 51, 102567. [Google Scholar] [CrossRef] [PubMed]
- Bernini, P.; Bertini, I.; Calabrò, A.; La Marca, G.; Lami, G.; Luchinat, C.; Renzi, D.; Tenori, L. Are Patients with Potential Celiac Disease Really Potential? The Answer of Metabonomics. J. Proteome Res. 2011, 10, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Auricchio, R.; Mandile, R.; Del Vecchio, M.R.; Scapaticci, S.; Galatola, M.; Maglio, M.; Discepolo, V.; Miele, E.; Cielo, D.; Troncone, R.; et al. Progression of celiac disease in children with antibodies against tissue transglutaminase and normal duodenal architecture. Gastroenterology 2019, 157, 413–420. [Google Scholar] [CrossRef]
- Lionetti, E.; Castellaneta, S.; Francavilla, R.; Pulvirenti, A.; Catassi, G.N.; Catassi, C. The SIGENP Working Group of Weaning and CD Risk Long-Term Outcome of Potential Celiac Disease in Genetically at-Risk Children: The Prospective CELIPREV Cohort Study. J. Clin. Med. 2019, 8, 186. [Google Scholar] [CrossRef]
- Salmi, T.T.; Collin, P.; Järvinen, O.; Haimila, K.; Partanen, J.; Laurila, K.; Korponay-Szabo, I.R.; Huhtala, H.; Reunala, T.; Maki, M.; et al. Immunoglobulin A autoantibodies against transglutaminase 2 in the small intestinal mucosa predict forthcoming coeliac disease. Aliment. Pharmacol. Ther. 2006, 24, 541–552. [Google Scholar] [CrossRef]
- Auricchio, R.; Tosco, A.; Piccolo, E.; Galatola, M.; Izzo, V.; Maglio, M.; Paparo, F.; Troncone, R.; Greco, L. Potential Celiac Children: 9-Year Follow-Up on a Gluten-Containing Diet. Am. J. Gastroenterol. 2014, 109, 913–921. [Google Scholar] [CrossRef]
- Kurppa, K.; Collin, P.; Viljamaa, M.; Haimila, K.; Saavalainen, P.; Partanen, J.; Laurila, K.; Huhtala, H.; Paasikivi, K.; Mäki, M.; et al. Diagnosing Mild Enteropathy Celiac Disease: A Randomized, Controlled Clinical Study. Gastroenterol. 2009, 136, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Kondala, R.; Puri, A.S.; Banka, A.K.; Sachdeva, S.; Sakhuja, P. Short-term prognosis of potential celiac disease in Indian patients. United Eur. Gastroenterol. J. 2016, 4, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Zong, G.; Lebwohl, B.; Hu, F.B.; Sampson, L.; Dougherty, L.W.; Willett, W.C.; Chan, A.T.; Sun, Q. Gluten intake and risk of type 2 diabetes in three large prospective cohort studies of US men and women. Diabetologia 2018, 61, 2164–2173. [Google Scholar] [CrossRef]
- Lebwohl, B.; Cao, Y.; Zong, G.; Hu, F.B.; Green, P.H.R.; Neugut, A.I.; Rimm, E.B.; Sampson, L.; Dougherty, L.W.; Giovannucci, E.; et al. Long term gluten consumption in adults without celiac disease and risk of coronary heart disease: Prospective cohort study. BMJ 2017, 357, j1892. [Google Scholar] [CrossRef] [PubMed]
- Blackett, J.W.; Shamsunder, M.; Reilly, N.R.; Green, P.H.; Lebwohl, B. Characteristics and comorbidities of inpatients without celiac disease on a gluten-free diet. Eur. J. Gastroenterol. Hepatol. 2018, 30, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Repo, M.; Lindfors, K.; Mäki, M.; Huhtala, H.; Laurila, K.; Lähdeaho, M.-L.; Saavalainen, P.; Kaukinen, K.; Kurppa, K. Anemia and Iron Deficiency in Children With Potential Celiac Disease. J. Pediatr. Gastroenterol. Nutr. 2017, 64, 56–62. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| First Author and Publication Date | Study Population | Results | Limitations |
|---|---|---|---|
| Pediatric Studies | |||
| Paparo F. 2005 [34] | 18/24 children with symptoms suggestive of CD or belonging to “at-risk” groups on a gluten-containing diet | antibody negativization in 3/18, villous atrophy development in 2/6 who underwent a second biopsy | small sample size |
| 6/24 children with symptoms suggestive of CD or belonging to “at-risk” groups on GFD | all demonstrated positive clinical and serological response | ||
| Kurppa K. 2010 [26] | 8/13 children with signs/conditions suggestive of CD on a GFD | villous atrophy in 5/8 after the first year and in next 2/8 after the second year | only four distal duodenal biopsies taken, short follow-up period |
| 5/13 children with signs/conditions suggestive of CD on a GFD | positive clinical and serological response in all after a year | ||
| Tosco A. 2011 [25] | 86 asymptomatic children on a gluten-containing diet | persistent positive serology in 52.9%, completely or persistently negative serology in 14.6%, fluctuation of antibody titers in 32.6%; villous atrophy in 12/39 (30.8%) who underwent a repeat biopsy within 3 years | only four distal duodenal biopsies taken |
| 20 children with persistent symptoms/conditions suggestive of CD put on a GFD | no clinical response in 9/20 | ||
| Lionetti E. 2012 [7] | 21 asymptomatic children left on a gluten-containing diet for two years | negative serology in 18/21 (86%), fluctuating antibody level in 2/21 (9%), histologically confirmed CD in 1/21 (5%) | |
| Auricchio R. 2014 [42] | 175 asymptomatic children on a gluten-containing diet | persistently elevated anti-TG2 level in 43%, negative anti-tTG in 20% and fluctuant anti-TG2 with transiently negative values in 37% during follow-up, normal duodenal architecture at 3, 6 and 9 years of follow-up in 86%, 73% and 67% patients, respectively | |
| Mandile R. 2018 [9] | 35 symptomatic children placed on GFD | positive clinical response in 19/35 (54%), partial clinical response in 2/35 (6%), no clinical response in 14/35 (40%), no significant differences in terms of Marsh grade, lamina propria CD25+ cells, CD3+, γδ+ intraepithelial lymphocytes density and intestinal anti-TG2 deposits after at least 1 year on GFD | |
| Lionetti E. 2019 [40] | 23 asymptomatic children on gluten-containing diet | negative serology up to 10 years of follow-up from the first biopsy in 19/23 (83%), fluctuating antibody values and persistently negative biopsy in 1/23 (4%), overt CD development in 3/23 (13%) | |
| Auricchio R. 2019 [39] | 280 children with symptoms, familiar risk or autoimmune comorbidity followed on a gluten-containing diet over a median follow-up of 60 months | a GFD introduction (without biopsy) for symptoms developed during the follow-up in 39/280 (13.9%); a flat mucosa development in 42/280 (15%); negativization of anti-TG2 or EMA in 89/280 (32%), 166/280 (59.2%) remained potential at 12 years of follow-up | |
| Adult Studies | |||
| Kurppa K. 2009 [43] | 10/23 adults with signs suggestive of CD on a gluten-containing diet | villous height/crypt depth ratio decreased, intraepithelial lymphocytosis and serum endomysial antibody titers remained increased in all; the symptoms persisted in all | Marsh II included in study population |
| 13/23 adults with signs suggestive of CD on a GFD | villous height/crypt depth ratio increased, intraepithelial lymphocytosis decreased, serum endomysial antibody titers normalized, the symptoms alleviated in all | ||
| Biagi F. 2013 [6] | 24 adult patients’ symptoms of malabsorption, associated diseases or familiarity for CD maintaining a GFD | flat mucosa development in 5/14 within 12 ± 8 months; preserved mucosal architecture in 9/14 within 30 ± 29 months, spontaneous clinical remission in 3/10 without subsequent biopsy | retrospective study |
| 23 adult patients with symptoms of malabsorption or associated diseases put on a GFD | clinical improvement in all (19/23) with gastrointestinal symptoms or dermatitis herpetiformis | ||
| Volta U. 2016 [8] | 16 asymptomatic adult patients left on a gluten-containing diet over a median follow-up of 3 years | diarrhea/anemia and subtotal villous atrophy development in 1/16 (6%), EmA/anti-TG2 disappearance in 4/16 (25%), antibody fluctuation in 1/16 (6.3%), antibody persistence in 10/16 (62.5%), no histologic changes in 10 patients with persistent or fluctuating antibody positivity | small sample size |
| 61 adult symptomatic patients put on a GFD over a median follow-up of 3 years | all demonstrated significant clinical improvement and negativization of antibodies | ||
| Mixed-Age Studies | |||
| Kondala R. 2016 [44] | 57 patients (children and adults) with IBS-like symptoms, iron-deficiency anemia or familiarity for CD on gluten-containing diet followed up for 12 months | serological negativization in 12/57 (21.1%), non-progressive duodenal histology in 46/57 (80%), histological worsening from Marsh-0-II to Marsh III in 4/57 (7%) | Marsh 2 included in the study population, short-term follow-up |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szaflarska-Popławska, A. Wait-and-See Approach or Gluten-Free Diet Administration—The Rational Management of Potential Coeliac Disease. Nutrients 2021, 13, 947. https://doi.org/10.3390/nu13030947
Szaflarska-Popławska A. Wait-and-See Approach or Gluten-Free Diet Administration—The Rational Management of Potential Coeliac Disease. Nutrients. 2021; 13(3):947. https://doi.org/10.3390/nu13030947
Chicago/Turabian StyleSzaflarska-Popławska, Anna. 2021. "Wait-and-See Approach or Gluten-Free Diet Administration—The Rational Management of Potential Coeliac Disease" Nutrients 13, no. 3: 947. https://doi.org/10.3390/nu13030947
APA StyleSzaflarska-Popławska, A. (2021). Wait-and-See Approach or Gluten-Free Diet Administration—The Rational Management of Potential Coeliac Disease. Nutrients, 13(3), 947. https://doi.org/10.3390/nu13030947