Astaxanthin Prevents Atrophy in Slow Muscle Fibers by Inhibiting Mitochondrial Reactive Oxygen Species via a Mitochondria-Mediated Apoptosis Pathway

 ,
,
Abstract
1. Introduction
2. Materials and Methods
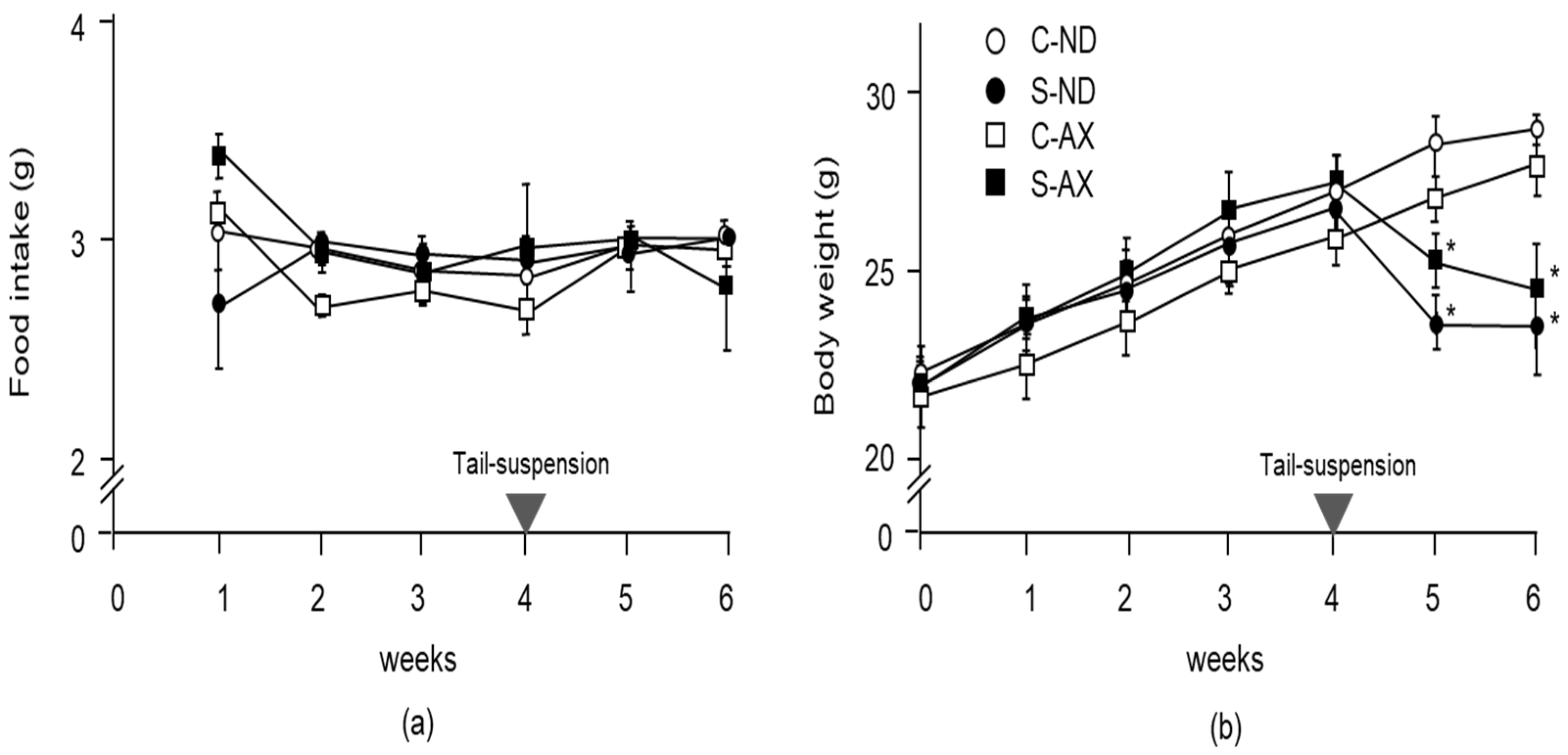
2.1. Animal Model
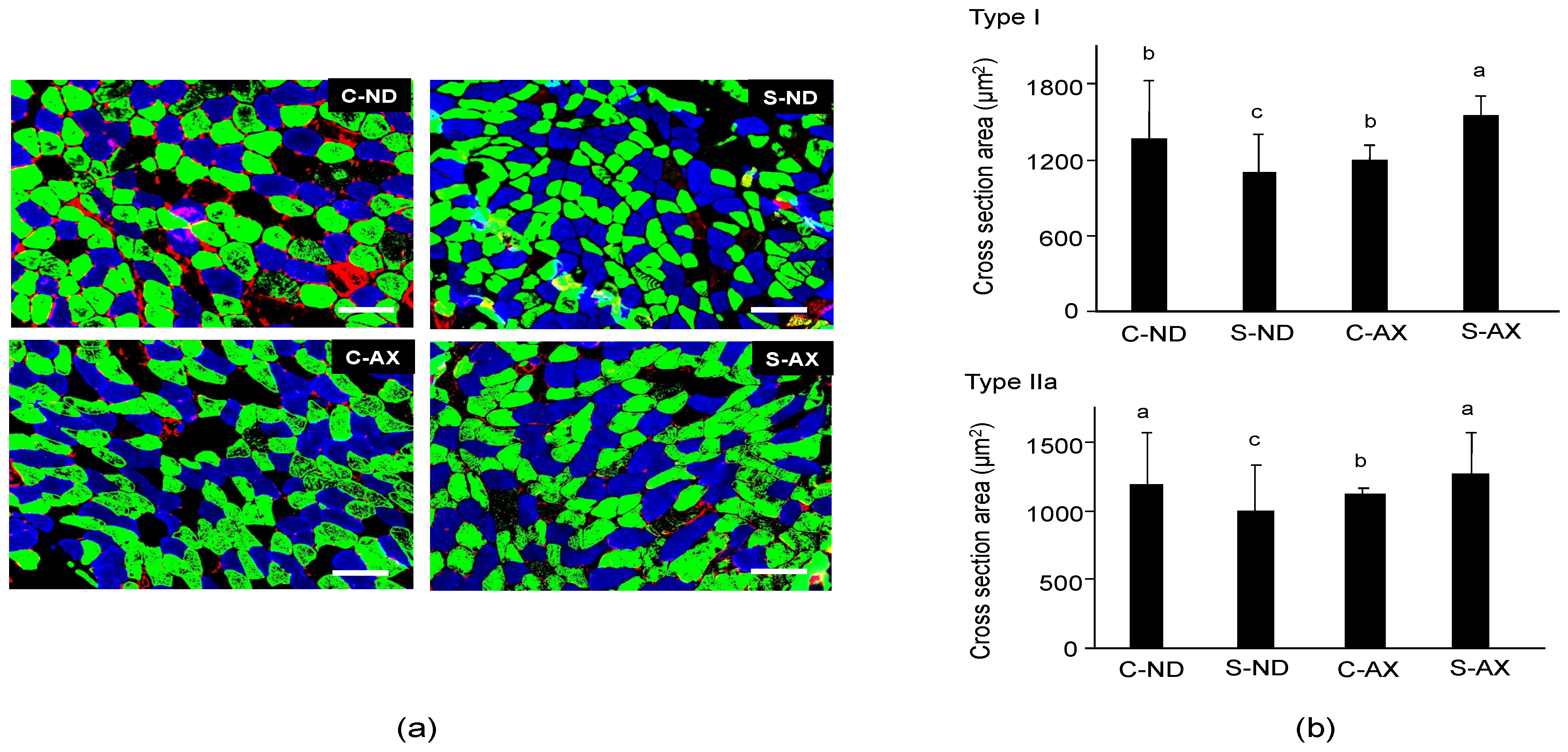
2.2. Multicolor Immunofluorescence Staining and Measurement of Cross-Sectional Area (CSA)
2.3. Cell Culture
2.4. Isolation of Mitochondria
2.5. Detection of AX in Mitochondrial and Cytosolic Fractions
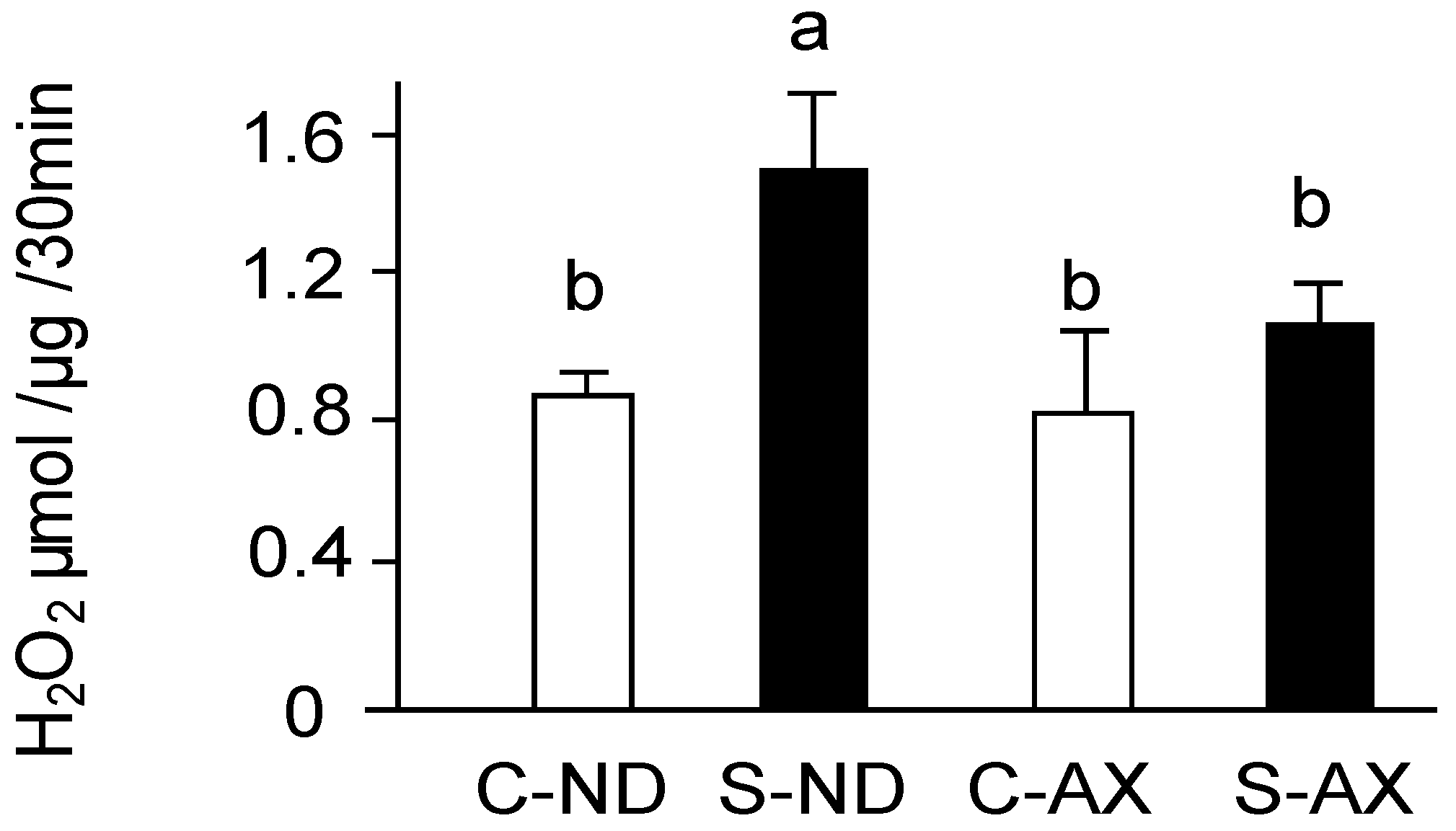
2.6. Detection of H2O2 Production
2.7. Measurement of Mitochondrial Superoxide Levels and Mitochondrial Membrane Potential (MMP)
2.8. Real-Time Reverse Transcription (RT)–Polymerase Chain Reaction (PCR)
2.9. Immunoblotting
2.10. Statistical Analyses
3. Results
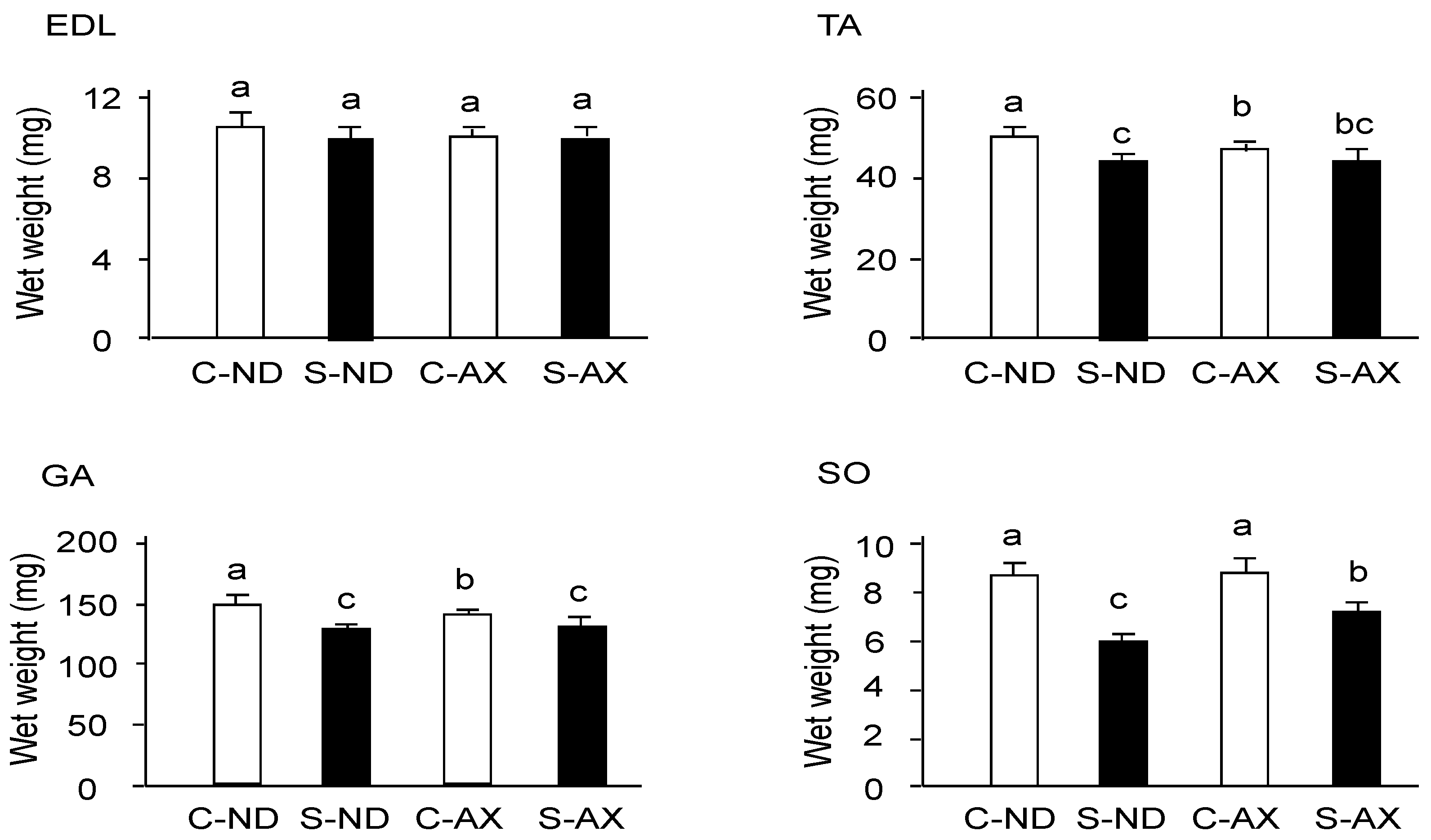
3.1. Effect of Dietary AX on Muscle Mass and Fiber Size in Tail-Suspension Mice
3.2. Effect of Dietary AX on H2O2 Production in the Muscle of Tail-Suspension Mice
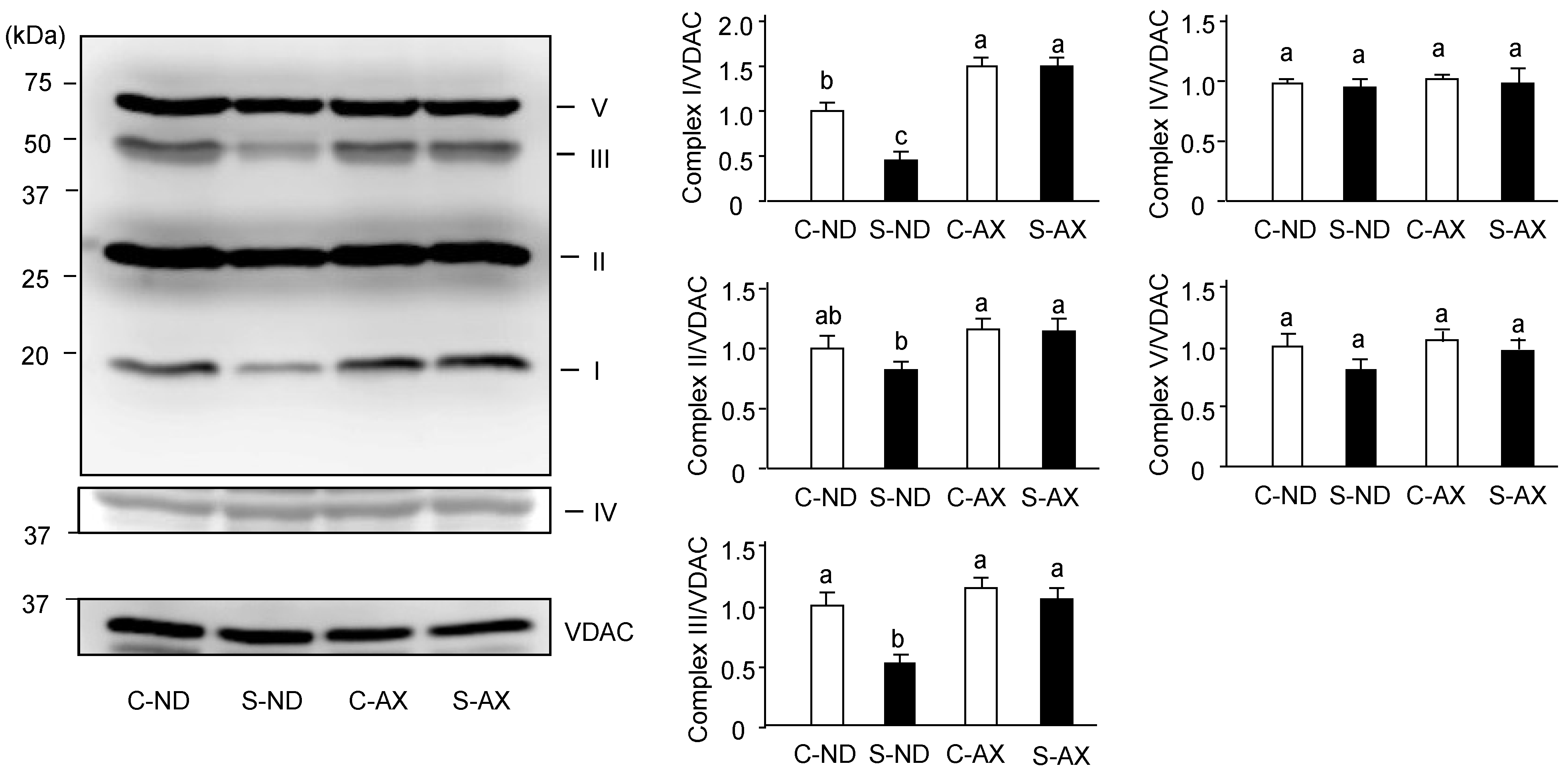
3.3. Effect of Dietary AX on Oxidative Phosphorylation Respiration in the Muscle of Tail-Suspension Mice
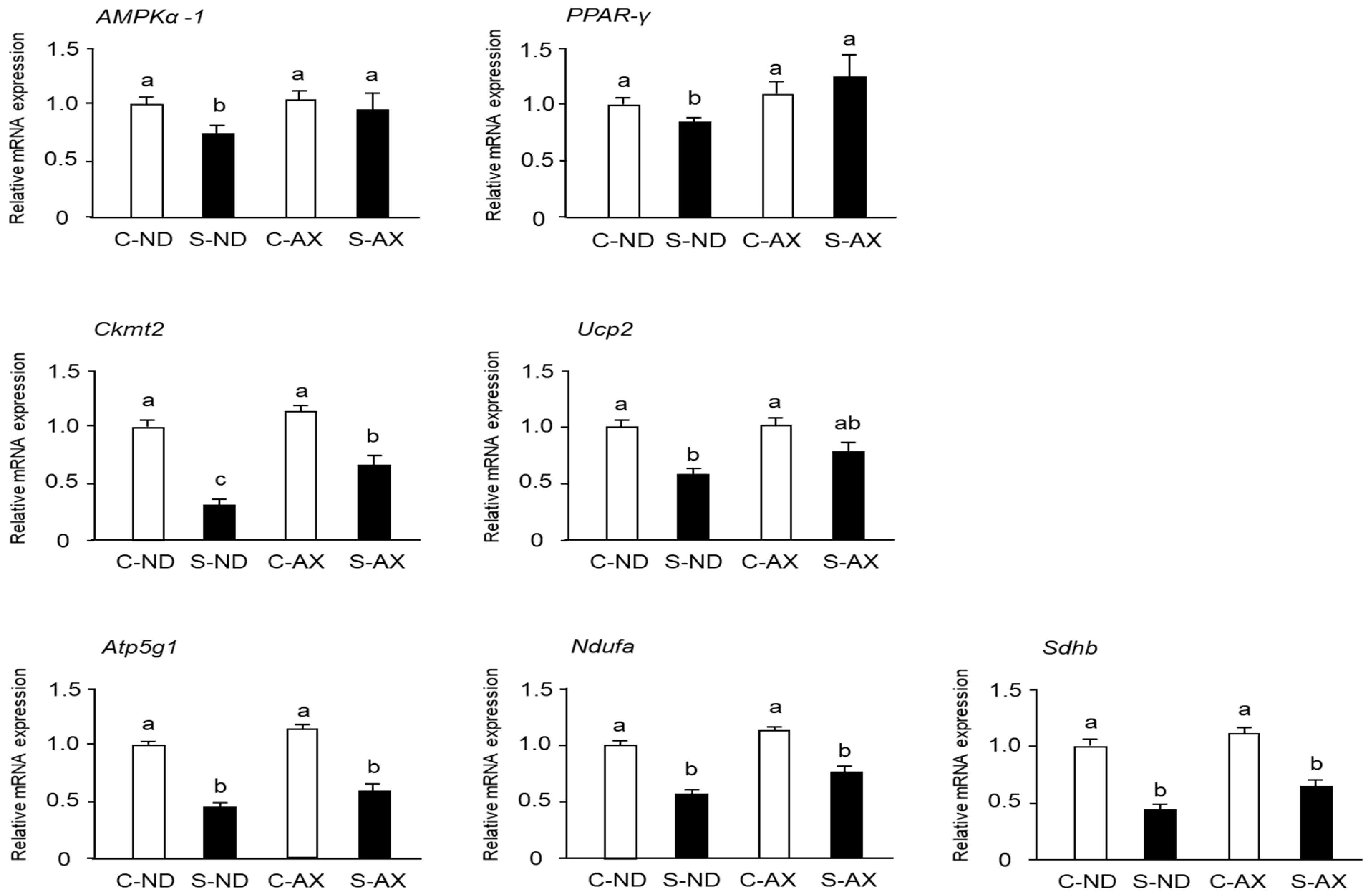
3.4. Effect of AX on Mitochondrial Biogenesis in the Muscle of Tail-Suspension Mice
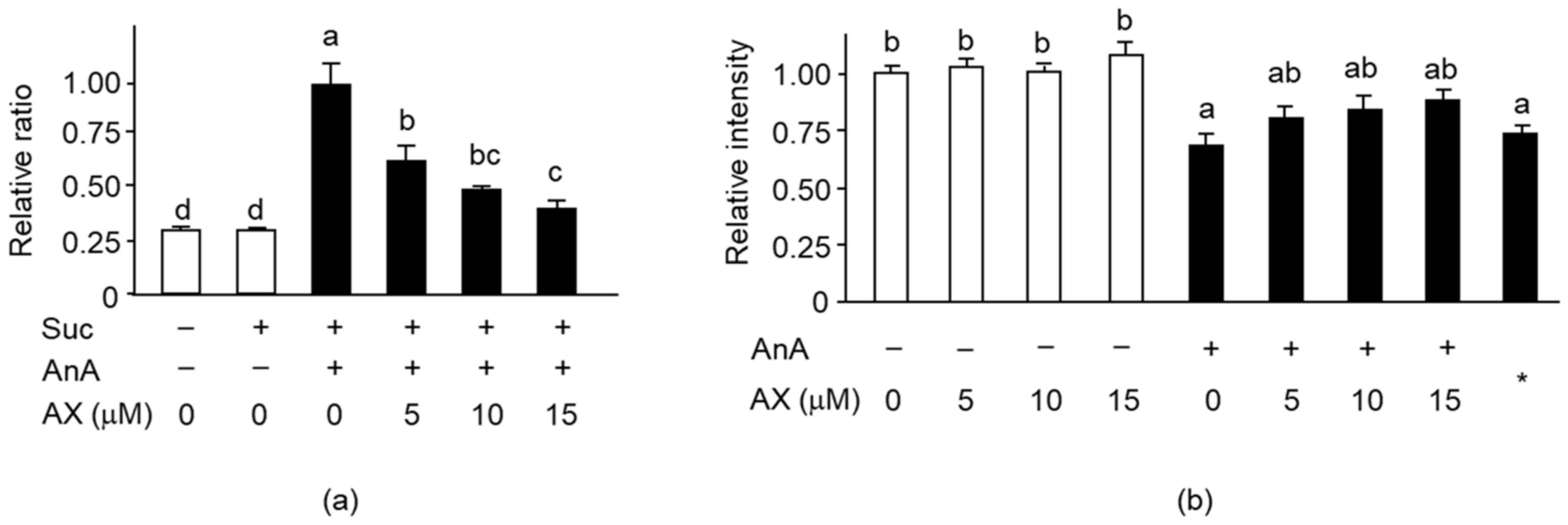
3.5. Effect of AX on Mitochondrial Function in Sol8 Myotubes
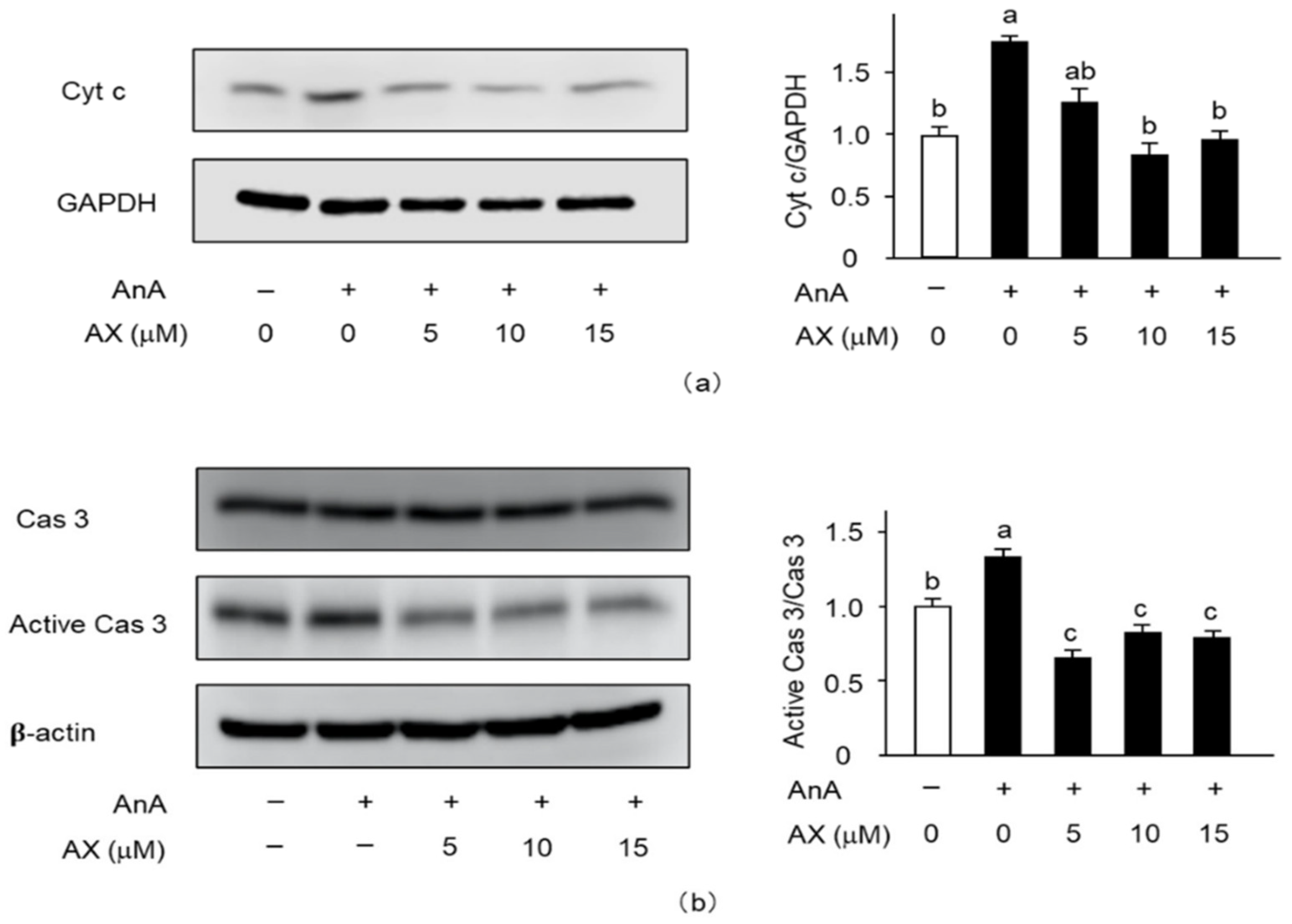
3.6. The Effect of AX on the Expression of Apoptosis-Related Proteins in AnA-Treated Sol8 Myotubes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| AMPK | adenosine 5′-monophosphate–activated protein kinase |
| AnA | antimycin A |
| ATP | adenosine triphosphate |
| AX | astaxanthin |
| BCA | bicinchoninic acid |
| CCCP | cytochrome cyanide m-chlorophenyl |
| CSA | cross-sectional area |
| DHE | dihydroethidium |
| DMEM | Dulbecco’s modified Eagle medium |
| DMSO | dimethyl sulfoxide |
| EDL | extensor digitorum longus |
| EDTA | ethylenediaminetetraacetic acid |
| FBS | fetal bovine serum |
| GA | gastrocnemius |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| HBSS | Hank’s balanced salt solutions |
| H2O2 | hydrogen peroxide |
| HPLC | High-performance liquid chromatography |
| HS | horse serum |
| MHC | myosin heavy chain |
| MMP | mitochondrial membrane potential |
| MOPS | 3-(N-morpholino) propanesulfonic acid |
| Ndufa | NADH: ubiquinone oxidoreductase complex assembly factor |
| OXPHOS | oxidative phosphorylation |
| PPAR | peroxisome proliferator–activated receptor |
| PVDF | polyvinylidene difluoride |
| ROS | reactive oxygen species |
| Sdhb | succinate dehydrogenase complex, subunit B |
| SDS-PAGE | odium dodecyl sulfate—polyacrylamide gel electrophoresis |
| SO | soleus |
| TA | tibialis anterior |
| UCP | uncoupling protein |
References
- Goto, K.; Okuyama, R.; Honda, M.; Uchida, H.; Akema, T.; Ohira, Y.; Yoshioka, T. Profiles of Connectin (Titin) in Atrophied Soleus Muscle Induced by Unloading of Rats. J. Appl. Physiol. 2003, 94, 897–902. [Google Scholar] [CrossRef] [PubMed]
- Ohira, Y. Neuromuscular Adaptation to Microgravity Environment. Jpn. J. Physiol. 2000, 50, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Indo, H.P.; Davidson, M.; Yen, H.C.; Suenaga, S.; Tomita, K.; Nishii, T.; Higuchi, M.; Koga, Y.; Ozawa, T.; Majima, H.J. Evidence of ROS Generation by Mitochondria in Cells with Impaired Electron Transport Chain and Mitochondrial DNA Damage. Mitochondrion 2007, 7, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide Metabolism in Mammalian Organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef]
- Nikawa, T.; Ishidoh, K.; Hirasaka, K.; Ishihara, I.; Ikemoto, M.; Kano, M.; Kominami, E.; Nonaka, I.; Ogawa, T.; Adams, G.R.; et al. Skeletal Muscle Gene Expression in Space-Flown Rats. FASEB J. 2004, 18, 522–524. [Google Scholar] [CrossRef]
- Powers, S.K.; Smuder, A.J.; Criswell, D.S. Mechanistic Links between Oxidative Stress and Disuse Muscle Atrophy. Antioxid. Redox Signal. 2011, 15, 2519–2528. [Google Scholar] [CrossRef]
- Romanello, V.; Guadagnin, E.; Gomes, L.; Roder, I.; Sandri, C.; Petersen, Y.; Milan, G.; Masiero, E.; Del, P.P.; Foretz, M.; et al. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J. 2010, 29, 1774–1785. [Google Scholar] [CrossRef]
- Adhihetty, P.L.; Ljubicic, V.; Menzies, K.J.; Hood, D.A. Differential susceptibility of subsarcolemmal and intermyofibrillar mitochondria to apoptotic stimuli. Am. J. Physiol. Cell Physiol. 2005, 289, C994–C1001. [Google Scholar] [CrossRef]
- Kowaltowski, A.J.; Vercesi, A.E. Mitochondrial damage induced by conditions of oxidative stress. Free Radic. Biol. Med. 1999, 26, 463–471. [Google Scholar] [CrossRef]
- Davinelli, S.; Nielsen, M.E.; Scapagnini, G. Astaxanthin in Skin Health, Repair, and Disease: A Comprehensive Review. Nutrients 2018, 10, 522. [Google Scholar] [CrossRef]
- Yuan, J.P.; Peng, J.; Yin, K.; Wang, J.H. Potential Health-Promoting Effects of Astaxanthin: A High-Value Carotenoid Mostly from Microalgae. Mol. Nutr. Food Res. 2011, 55, 150–165. [Google Scholar] [CrossRef] [PubMed]
- Ohno, M.; Darwish, W.S.; Ikenaka, Y.; Miki, W.; Ishizuka, M. Astaxanthin Can Alter CYP1A-dependent Activities via Two Different Mechanisms: Induction of Protein Expression and Inhibition of NADPH P450 Reductase Dependent Electron Transfer. Food Chem. Toxicol. 2011, 49, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.M.; Asoh, S.; Hiranuma, H.; Ohsawa, I.; Iio, K.; Satou, A.; Ishikura, M.; Ohta, S. Astaxanthin Protects Mitochondrial Redox State and Functional Integrity Against Oxidative Stress. J. Nutr. Biochem. 2010, 21, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.W.; Xu, X.C.; Liu, T.; Yuan, S. Mitochondrion-Permeable Antioxidants to Treat ROS-Burst-Mediated Acute Diseases. Oxid. Med. Cell Longev. 2016, 1, 6859523. [Google Scholar] [CrossRef]
- Kuroki, T.; Ikeda, S.; Okada, T.; Maoka, T.; Kitamura, A.; Sugimoto, M.; Kume, S. Astaxanthin Ameliorates Heat Stress-Induced Impairment of Blastocyst Development in Vitro: Astaxanthin Colocalization with and Action on Mitochondria. J. Assist. Reprod. Genet. 2013, 30, 623–631. [Google Scholar] [CrossRef]
- Song, X.D.; Wang, B.S.; Lin, S.C.; Jing, L.L.; Mao, C.P.; Xu, P.; Lv, C.J.; Liu, W.; Zuo, J. Astaxanthin Inhibits Apoptosis in Alveolar Epithelial Cells Type II in Vivo and in Vitro Through the ROS-dependent Mitochondrial Signalling Pathway. J. Cell. Mol. Med. 2014, 18, 2198–2212. [Google Scholar] [CrossRef]
- Mukai, R.; Matsui, N.; Fujikura, Y.; Matsumoto, N.; Hou, D.X.; Kanzaki, N.; Shibata, H.; Horikawa, M.; Iwasa, K.; Hirasaka, K.; et al. Preventive Effect of Dietary Quercetin on Disuse Muscle Atrophy by Targeting Mitochondria in Denervated Mice. J. Nutr. Biochem. 2016, 31, 67–76. [Google Scholar] [CrossRef]
- Hiramoto, S.; Yahata, N.; Saitoh, K.; Yoshimura, T.; Wang, Y.; Taniyama, S.; Nikawa, T.; Tachibana, K.; Hirasaka, K. Dietary Supplementation with Alkylresorcinols Prevents Muscle Atrophy Through a Shift of Energy Supply. J. Nutr. Biochem. 2018, 61, 147–154. [Google Scholar] [CrossRef]
- Kanazashi, M.; Tanaka, M.; Nakanishi, Y.; Maeshige, N.; Fujino, H. Effects of Astaxanthin Supplementation and Electrical Stimulation on Muscle Atrophy and Decreased Oxidative Capacity in Soleus Muscle During Hindlimb Unloading in Rats. J. Physiol. Sci. 2019, 69, 757–767. [Google Scholar] [CrossRef]
- Shibaguchi, T.; Yamaguchi, Y.; Miyaji, N.; Yoshihara, T.; Naito, H.; Goto, K.; Ohmori, D.; Yoshioka, T.; Sugiura, T. Astaxanthin Intake Attenuates Muscle Atrophy Caused by Immobilization in Rats. Physiol. Rep. 2016, 4, e12885. [Google Scholar] [CrossRef]
- Bloemberg, D.; Quadrilatero, J. Rapid Determination of Myosin Heavy Chain Expression in Rat, Mouse, and Human Skeletal Muscle Using Multicolor Immunofluorescence Analysis. PLoS ONE 2012, 7, e35273. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Hwang, S.H.; Lim, J.A.; Froehner, S.C.; Adams, M.E.; Kim, H.S. alpha-syntrophin modulates myogenin expression in differentiating myoblasts. PLoS ONE 2010, 5, e15355. [Google Scholar] [CrossRef] [PubMed]
- Sprague, J.E.; Yang, X.M.; Sommers, J.; Gilman, T.L.; Mills, E.M. Roles of Norepinephrine, Free Fatty Acids, Thyroid Status, and Skeletal Muscle Uncoupling Protein 3 Expression in Sympathomimetic-Induced Thermogenesis. J. Pharmacol. Exp. Ther. 2007, 320, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Hirasaka, K.; Lago, C.U.; Kenaston, M.A.; Fathe, K.; Nowinski, S.M.; Nikawa, T.; Mills, E.M. Identification of a Redox-Modulatory Interaction Between Uncoupling Protein 3 and Thioredoxin 2 in the Mitochondrial Intermembrane Space. Antioxid. Redox Signal. 2011, 15, 2645–2661. [Google Scholar] [CrossRef]
- Hirasaka, K.; Saito, S.; Yamaguchi, S.; Miyazaki, R.; Wang, Y.; Haruna, M.; Taniyama, S.; Higashitani, A.; Terao, J.; Nikawa, T.; et al. Dietary supplementation with Isoflavones Prevents Muscle Wasting in Tumor-Bearing Mice. J. Nutr. Sci. Vitaminol. 2016, 62, 178–184. [Google Scholar] [CrossRef]
- Ohira, T.; Ohira, T.; Kawano, F.; Shibaguchi, T.; Okabe, H.; Goto, K.; Ogita, F.; Sudoh, M.; Roy, R.R.; Edgerton, V.R.; et al. Effects of gravitational loading levels on protein expression related to metabolic and/or morphologic properties of mouse neck muscles. Physiol. Rep. 2014, 2, e00183. [Google Scholar] [CrossRef]
- Kitaoka, Y.; Takeda, K.; Tamura, Y.; Fujimaki, S.; Takemasa, T.; Hatta, H. Nrf2 Deficiency Does Not Affect Denervation-Induced Alterations in Mitochondrial Fission and Fusion Proteins in Skeletal Muscle. Physiol. Rep. 2016, 4, e13064. [Google Scholar] [CrossRef]
- Yeo, D.; Ji, L.L. Mitochondrial dysfunction and muscle disuse atrophy. F1000Research 2019, 8, F1000 Faculty Rev-1621. [Google Scholar]
- Nishida, Y.; Nawaz, A.; Kado, T.; Takikawa, A.; Igarashi, Y.; Onogi, Y.; Wada, T.; Sasaoka, T.; Yamamoto, S.; Sasahara, M.; et al. Astaxanthin stimulates mitochondrial biogenesis in insulin resistant muscle via activation of AMPK pathway. J. Cachexia Sarcopenia Muscle 2020, 11, 241–258. [Google Scholar] [CrossRef]
- Manabe, E.; Handa, O.; Naito, Y.; Mizushima, K.; Akagiri, S.; Adachi, S.; Takagi, T.; Kokura, S.; Maoka, T.; Yoshikawa, T. Astaxanthin Protects Mesangial Cells from Hyperglycemia-Induced Oxidative Signaling. J. Cell. Biochem. 2008, 103, 1925–1937. [Google Scholar] [CrossRef]
- Musacchia, X.J.; Steffen, J.M.; Deavers, D.R. Rat Hindlimb Muscle Responses to Suspension Hypokinesia/Hypodynamia. Aviat. Space Environ. Med. 1983, 54, 1015–1020. [Google Scholar] [PubMed]
- Desplanches, D.; Mayet, M.H.; Sempore, B.; Flandrois, R. Structural and Functional Responses to Prolonged Hindlimb Suspension in Rat Muscle. J. Appl. Physiol. 1987, 63, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Stein, T.P.; Schluter, M.D.; Galante, A.T.; Soteropoulos, P.; Ramirez, M.; Bigbee, A.; Grindeland, R.E.; Wade, C.E. Effect of hind limb muscle unloading on liver metabolism of rats. J. Nutr. Biochem. 2005, 16, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Marzetti, E.; Hwang, J.C.Y.; Lees, H.A.; Wohlgemuth, S.E.; Dupont-Versteegden, E.E.; Carter, C.S.; Bernabei, R.; Leeuwenburgh, C. Mitochondrial Death Effectors: Relevance to Sarcopenia and Disuse Muscle Atrophy. Biochim. Biophys. Acta 2010, 1800, 235–244. [Google Scholar] [CrossRef]
- Romanello, V.; Sandri, M. Mitochondrial Biogenesis and Fragmentation as Regulators of Protein Degradation in Striated Muscles. J. Mol. Cell. Cardiol. 2013, 55, 64–72. [Google Scholar] [CrossRef]
- Brand, M.D. The Sites and Topology of Mitochondrial Superoxide Production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef]
- Choi, M.H.; Ow, J.R.; Yang, N.D.; Taneja, R. Oxidative Stress-Mediated Skeletal Muscle Degeneration: Molecules, Mechanisms, and Therapies. Oxid. Med. Cell. Longev. 2016, 2016, 6842568. [Google Scholar] [CrossRef]
- Bhuvaneswari, S.; Anuradha, C.V. Astaxanthin Prevents Loss of Insulin Signaling and Improves Glucose Metabolism in Liver of Insulin Resistant Mice. Can. J. Physiol. Pharmacol. 2012, 90, 1544–1552. [Google Scholar] [CrossRef]
- Ni, Y.; Nagashimada, M.; Zhuge, F.; Zhan, L.L.; Nagata, N.; Tsutsui, A.; Nakanuma, Y.; Kaneko, S.; Ota, T. Astaxanthin Prevents and Reverses Diet-Induced Insulin Resistance and Steatohepatitis in Mice: A Comparison with Vitamin E. Sci. Rep. 2015, 5, 17192. [Google Scholar] [CrossRef]
- Kim, S.H.; Lim, J.W.; Kim, H. Astaxanthin Inhibits Mitochondrial Dysfunction and Interleukin-8 Expression in Helicobacter pylori-Infected Gastric Epithelial Cells. Nutrients 2018, 10, 1320. [Google Scholar] [CrossRef]
- Fan, C.D.; Sun, J.Y.; Fu, X.T.; Hou, Y.J.; Li, Y.; Yang, M.F.; Fu, X.Y.; Sun, B.Y. Astaxanthin Attenuates Homocysteine-Induced Cardiotoxicity in Vitro and in Vivo by Inhibiting Mitochondrial Dysfunction and Oxidative Damage. Front. Physiol. 2017, 8, 1041. [Google Scholar] [CrossRef]
- Krestinina, O.; Baburina, Y.; Krestinin, R.; Odinokova, I.; Fadeeva, I.; Sotnikova, L. Astaxanthin Prevents Mitochondrial Impairment Induced by Isoproterenol in Isolated Rat Heart Mitochondria. Antioxidants 2020, 9, 262. [Google Scholar] [CrossRef] [PubMed]
- Higuera-Ciapara, I.; Félix-Valenzuela, L.; Goycoolea, F.M. Astaxanthin: A Review of Its Chemistry and Applications. Crit. Rev. Food Sci. Nutr. 2006, 46, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Ambati, R.R.; Phang, S.M.; Ravi, S.; Aswathanarayana, R.G. Astaxanthin: Sources, Extraction, Stability, Biological Activities and Its Commercial Applications—A Review. Mar. Drugs 2014, 12, 128–152. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F.; Alexandre, A.; Lehninger, A.L. Ubisemiquinone Is the Electron Donor for Superoxide Formation by Complex III of Heart Mitochondria. Arch. Biochem. Biophys. 1985, 237, 408–414. [Google Scholar] [CrossRef]
- Kitakaze, T.; Harada, N.; Imagita, H.; Yamaji, R. β-Carotene Increases Muscle Mass and Hypertrophy in the Soleus Muscle in Mice. J. Nutr. Sci. Vitaminol. 2015, 61, 481–487. [Google Scholar] [CrossRef]
- Ogawa, M.; Kariya, Y.; Kitakaze, T.; Yamaji, R.; Harada, N.; Sakamoto, T.; Hosotani, K.; Nakano, Y.; Inui, H. The Preventive Effect of β-Carotene on Denervation-Induced Soleus Muscle Atrophy in Mice. Br. J. Nutr. 2013, 109, 1349–1358. [Google Scholar] [CrossRef]
- Yoshihara, T.; Yamamoto, Y.; Shibaguchi, T.; Miyaji, N.; Kakigi, R.; Naito, H.; Goto, K.; Ohmori, D.; Yoshioka, T.; Sugiura, T. Dietary Astaxanthin Supplementation Attenuates Disuse-Induced Muscle Atrophy and Myonuclear Apoptosis in the Rat Soleus Muscle. J. Physiol. Sci. 2017, 67, 181–190. [Google Scholar] [CrossRef]
- Li, J.; Yu, W.; Li, X.T.; Qi, S.H.; Li, B. The Effects of Propofol on Mitochondrial Dysfunction Following Focal Cerebral Ischemia-Reperfusion in Rats. Neuropharmacology 2014, 77, 358–368. [Google Scholar] [CrossRef]
- He, G.D.; Feng, C.; Vinothkumar, R.; Chen, W.Q.; Dai, X.X.; Chen, X.; Ye, Q.Q.; Qiu, C.Y.; Zhou, H.P.; Wang, Y.; et al. Curcumin Analog EF24 Induces Apoptosis via ROS-dependent Mitochondrial Dysfunction in Human Colorectal Cancer Cells. Cancer Chemother. Pharmacol. 2016, 78, 1151–1161. [Google Scholar] [CrossRef]
- Shanmugapriya, K.; Kim, H.; Kang, H.W. In Vitro Antitumor Potential of Astaxanthin Nanoemulsion Against Cancer Cells via Mitochondrial Mediated Apoptosis. Int. J. Pharm. 2019, 560, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Pajaniradje, S.; Mohankumar, K.; Pamidimukkala, R.; Subramanian, S.; Rajagopalan, R. Antiproliferative and Apoptotic Effects of Sesbania Grandiflora Leaves in Human Cancer Cells. BioMed Res. Int. 2014, 474953. [Google Scholar]
- Meghani, N.; Patel, P.; Kansara, K.; Ranjan, S.; Dasgupta, N.; Ramalingam, C.; Kumar, A. Formulation of Vitamin D Encapsulated Cinnamon Oil Nanoemulsion: Its Potential Anti-Cancerous Activity in Human Alveolar Carcinoma Cells. Colloids Surf. B Biointerfaces 2018, 166, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Wang, X.N.; Miereles, C.; Bailey, J.L.; Debigare, R.; Zheng, B.; Price, S.R.; Mitch, W.E. Activation of caspase-3 Is an Initial Step Triggering Accelerated Muscle Proteolysis in Catabolic Conditions. J. Clin. Investig. 2004, 113, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Kavazis, A.N.; McClung, J.M. Oxidative Stress and Disuse Muscle Atrophy. J. Appl. Physiol. 2007, 102, 2389–2397. [Google Scholar] [CrossRef]
- Wu, Q.; Tang, Z.H.; Peng, J.; Liao, L.; Pan, L.H.; Wu, C.Y.; Jiang, Z.S.; Wang, G.X.; Liu, L.S. The Dual Behavior of PCSK9 in the Regulation of Apoptosis Is Crucial in Alzheimer’s Disease Progression (Review). Biomed. Rep. 2014, 2, 167–171. [Google Scholar] [CrossRef]
- Yang, Q.S.; Guo, S.F.; Wang, S.; Qian, Y.W.; Tai, H.P.; Chen, Z.X. Advanced Glycation End Products-Induced Chondrocyte Apoptosis Through Mitochondrial Dysfunction in Cultured Rabbit Chondrocyte. Fundam. Clin. Pharmacol. 2015, 29, 54–61. [Google Scholar] [CrossRef]
- Plant, P.J.; Bain, J.R.; Correa, J.E.; Woo, M.; Batt, J. Absence of caspase-3 Protects Against Denervation-Induced Skeletal Muscle Atrophy. J. Appl. Physiol. 2009, 107, 224–234. [Google Scholar] [CrossRef]
- Liu, X.B.; Shibata, T.; Hisaka, S.; Osawa, T. Astaxanthin Inhibits Reactive Oxygen Species-Mediated Cellular Toxicity in Dopaminergic SH-SY5Y Cells via Mitochondria-Targeted Protective Mechanism. Brain Res. 2009, 1254, 18–27. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AX Treatment | AX Content (nmol) | |
|---|---|---|
| Cytosol | Mitochondria | |
| 0 nmol | N.D. | N.D. |
| 100 nmol | 0.09 ± 0.01 | 1.07 ± 0.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, L.; Miyaji, N.; Yang, M.; Mills, E.M.; Taniyama, S.; Uchida, T.; Nikawa, T.; Li, J.; Shi, J.; Tachibana, K.; et al. Astaxanthin Prevents Atrophy in Slow Muscle Fibers by Inhibiting Mitochondrial Reactive Oxygen Species via a Mitochondria-Mediated Apoptosis Pathway. Nutrients 2021, 13, 379. https://doi.org/10.3390/nu13020379
Sun L, Miyaji N, Yang M, Mills EM, Taniyama S, Uchida T, Nikawa T, Li J, Shi J, Tachibana K, et al. Astaxanthin Prevents Atrophy in Slow Muscle Fibers by Inhibiting Mitochondrial Reactive Oxygen Species via a Mitochondria-Mediated Apoptosis Pathway. Nutrients. 2021; 13(2):379. https://doi.org/10.3390/nu13020379
Chicago/Turabian StyleSun, Luchuanyang, Nobuyuki Miyaji, Min Yang, Edward M. Mills, Shigeto Taniyama, Takayuki Uchida, Takeshi Nikawa, Jifeng Li, Jie Shi, Katsuyasu Tachibana, and et al. 2021. "Astaxanthin Prevents Atrophy in Slow Muscle Fibers by Inhibiting Mitochondrial Reactive Oxygen Species via a Mitochondria-Mediated Apoptosis Pathway" Nutrients 13, no. 2: 379. https://doi.org/10.3390/nu13020379
APA StyleSun, L., Miyaji, N., Yang, M., Mills, E. M., Taniyama, S., Uchida, T., Nikawa, T., Li, J., Shi, J., Tachibana, K., & Hirasaka, K. (2021). Astaxanthin Prevents Atrophy in Slow Muscle Fibers by Inhibiting Mitochondrial Reactive Oxygen Species via a Mitochondria-Mediated Apoptosis Pathway. Nutrients, 13(2), 379. https://doi.org/10.3390/nu13020379