
Aberrance of Zinc Metalloenzymes-Induced Human Diseases and Its Potential Mechanisms

Abstract
1. Introduction
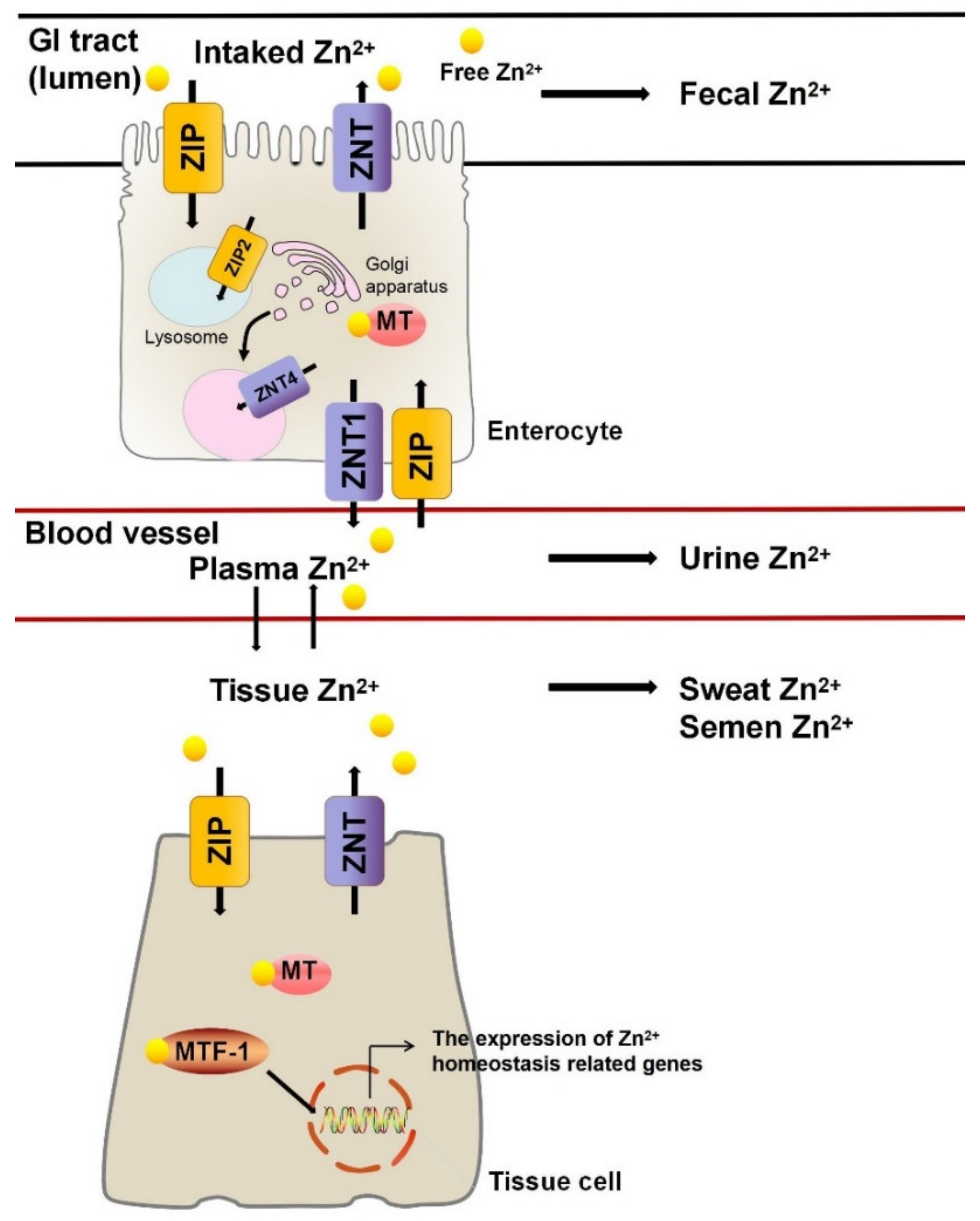
2. Zn2+ Homeostasis
3. Role of Zn2+ and Zn2+ Metalloenzymes in Physiological Processes
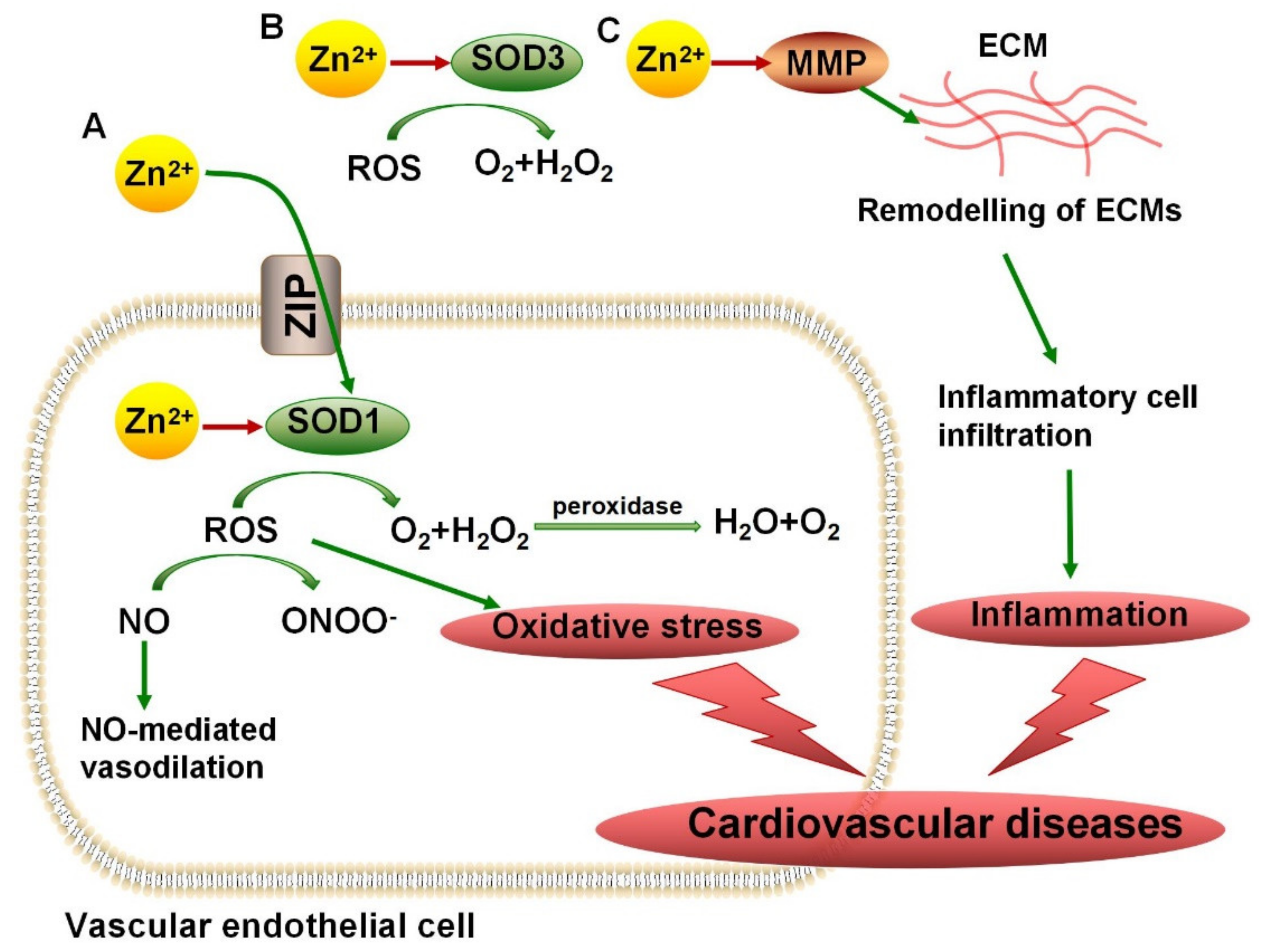
3.1. Zn2+ Metalloenzymes Regulate Antioxidant Activity
3.2. Zn2+ Metalloenzymes Regulate Inflammation
3.3. Zn2+ Metalloenzymes Regulate the Immune Response
3.4. Zn2+ Metalloenzymes Regulate Apoptosis
3.5. Zn2+ Metalloenzymes Regulate Other Physiological Processes
4. Zn2+ Metalloenzymes and Diseases
4.1. SODs in Cardiovascular Diseases, ALS, and AD
4.2. MMPs in Vascular Diseases
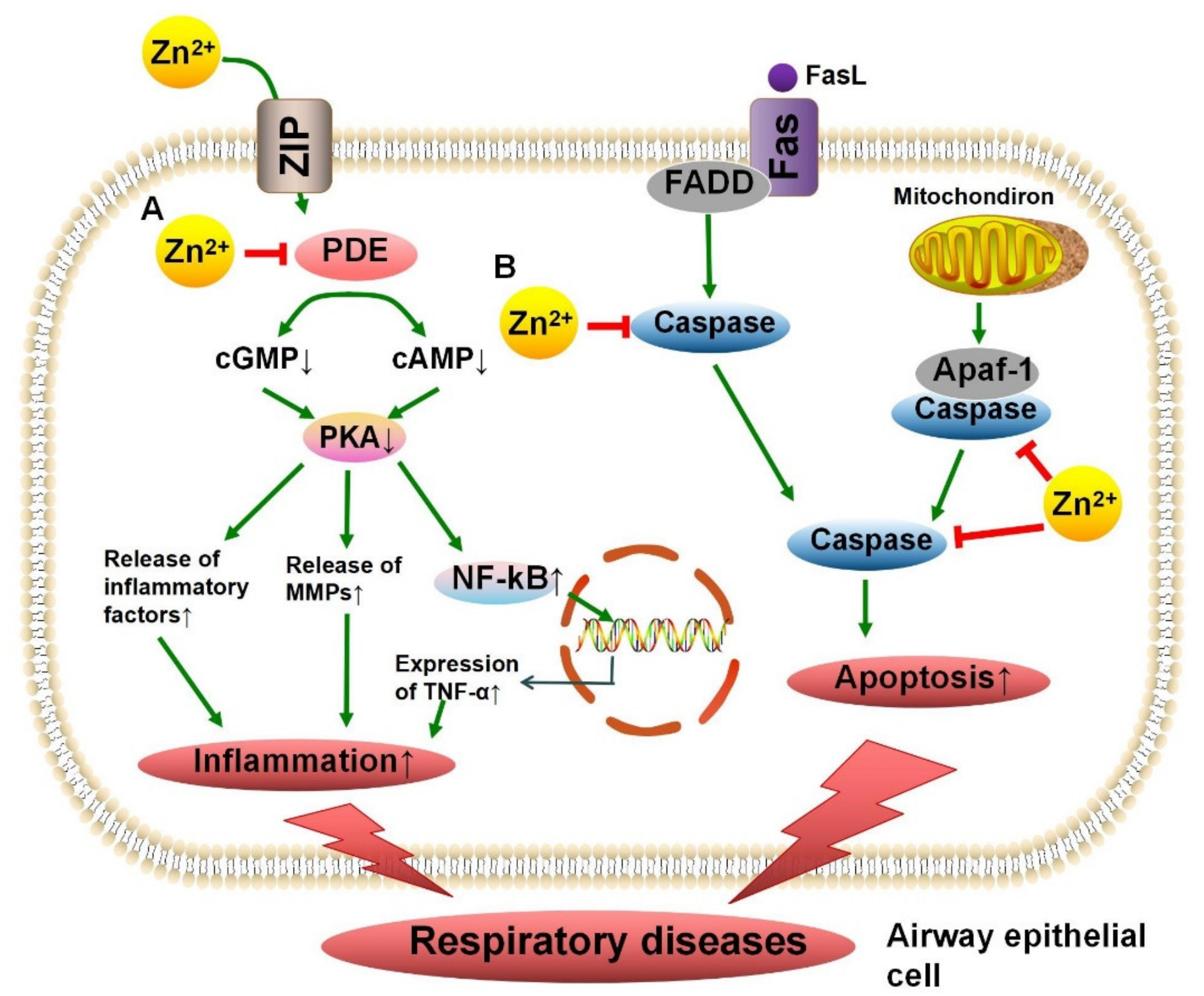
4.3. Phosphodiesterase in Chronic Obstructive Pulmonary Disease
4.4. Protein Kinase C in Immune Diseases
4.5. Caspase in Asthma
4.6. Carbonic Anhydrase in Hypogeusia
4.7. Alkaline Phosphatase in Bone Disorder
5. Zn2+ and Zn2+ Metalloenzymes in Cancer
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Prasad, A.S. Discovery of Human Zinc Deficiency: Its Impact on Human Health and Disease. Adv. Nutr. 2013, 4, 176–190. [Google Scholar] [CrossRef]
- Prasad, A.S.; Miale, A., Jr.; Farid, Z.; Sandstead, H.H.; Schulert, A.R. Zinc metabolism in patients with the syndrome of iron deficiency anemia, hepatosplenomegaly, dwarfism, and hypognadism. J. Lab. Clin. Med. 1963, 61, 537–549. [Google Scholar]
- Sanna, A.; Firinu, D.; Zavattari, P.; Valera, P. Zinc Status and Autoimmunity: A Systematic Review and Meta-Analysis. Nutrients 2018, 10, 68. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Kinoshita, M.; Shimada, S.; Kawamura, T. Zinc and Skin Disorders. Nutrients 2018, 10, 199. [Google Scholar] [CrossRef] [PubMed]
- Wastney, M.E.; Aamodt, R.L.; Rumble, W.F.; Henkin, R.I. Kinetic analysis of zinc metabolism and its regulation in normal humans. Am. J. Physiol. 1986, 251, R398–R408. [Google Scholar] [CrossRef]
- Mariani, E.; Mangialasche, F.; Feliziani, F.; Cecchetti, R.; Malavolta, M.; Bastiani, P.; Baglioni, M.; Dedoussis, G.; Fulop, T.; Herbein, G.; et al. Effects of zinc supplementation on antioxidant enzyme activities in healthy old subjects. Exp. Gerontol. 2008, 43, 445–451. [Google Scholar] [CrossRef]
- Von Bülow, V.; Rink, L.; Haase, H. Zinc-Mediated Inhibition of Cyclic Nucleotide Phosphodiesterase Activity and Expression Suppresses TNF-α and IL-1β Production in Monocytes by Elevation of Guanosine 3′,5′-Cyclic Monophosphate. J. Immunol. 2005, 175, 4697–4705. [Google Scholar] [CrossRef] [PubMed]
- Wessels, I.; Maywald, M.; Rink, L. Zinc as a Gatekeeper of Immune Function. Nutrients 2017, 9, 1286. [Google Scholar] [CrossRef]
- Shankar, A.H.; Prasad, A.S. Zinc and immune function: The biological basis of altered resistance to infection. Am. J. Clin. Nutr. 1998, 68, 447S–463S. [Google Scholar] [CrossRef]
- Maret, W. Zinc Biochemistry: From a Single Zinc Enzyme to a Key Element of Life. Adv. Nutr. 2013, 4, 82–91. [Google Scholar] [CrossRef]
- Prasad, A.S. Discovery of human zinc deficiency: 50 years later. J. Trace Elem. Med. Biol. 2012, 26, 66–69. [Google Scholar] [CrossRef]
- Himoto, T.; Masaki, T. Associations between Zinc Deficiency and Metabolic Abnormalities in Patients with Chronic Liver Disease. Nutrients 2018, 10, 88. [Google Scholar] [CrossRef]
- Lim, K.H.C.; Riddell, L.J.; Nowson, C.A.; Booth, A.O.; Szymlek-Gay, E.A. Iron and Zinc Nutrition in the Economically-Developed World: A Review. Nutrients 2013, 5, 3184–3211. [Google Scholar] [CrossRef] [PubMed]
- Gać, P.; Czerwińska, K.; Macek, P.; Jaremków, A.; Mazur, G.; Pawlas, K.; Poręba, R. The importance of selenium and zinc deficiency in cardiovascular disorders. Environ. Toxicol. Pharmacol. 2021, 82, 103553. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.; Samman, S. Vegetarian Diets across the Lifecycle: Impact on zinc intake and status. Adv. Food Nutr. Res. 2015, 74, 93–131. [Google Scholar] [CrossRef] [PubMed]
- Murgia, C.; Grosser, D.; Truong-Tran, A.Q.; Roscioli, E.; Michalczyk, A.; Ackland, M.L.; Stoltenberg, M.; Danscher, G.; Lang, C.; Knight, D.; et al. Apical Localization of Zinc Transporter ZnT4 in Human Airway Epithelial Cells and Its Loss in a Murine Model of Allergic Airway Inflammation. Nutrients 2011, 3, 910–928. [Google Scholar] [CrossRef]
- Lichten, L.A.; Cousins, R.J. Mammalian Zinc Transporters: Nutritional and Physiologic Regulation. Annu. Rev. Nutr. 2009, 29, 153–176. [Google Scholar] [CrossRef]
- Dufner-Beattie, J.; Wang, F.; Kuo, Y.-M.; Gitschier, J.; Eide, D.; Andrews, G.K. The Acrodermatitis Enteropathica Gene ZIP4 Encodes a Tissue-specific, Zinc-regulated Zinc Transporter in Mice. J. Biol. Chem. 2003, 278, 33474–33481. [Google Scholar] [CrossRef]
- Kimura, T.; Kambe, T. The Functions of Metallothionein and ZIP and ZnT Transporters: An Overview and Perspective. Int. J. Mol. Sci. 2016, 17, 336. [Google Scholar] [CrossRef]
- Nishito, Y.; Kambe, T. Zinc transporter 1 (ZNT1) expression on the cell surface is elaborately controlled by cellular zinc levels. J. Biol. Chem. 2019, 294, 15686–15697. [Google Scholar] [CrossRef]
- Liuzzi, J.P.; Blanchard, R.K.; Cousins, R.J. Differential Regulation of Zinc Transporter 1, 2, and 4 mRNA Expression by Dietary Zinc in Rats. J. Nutr. 2001, 131, 46–52. [Google Scholar] [CrossRef]
- Chowanadisai, W.; Lönnerdal, B.; Kelleher, S.L. Identification of a Mutation in SLC30A2 (ZnT-2) in Women with Low Milk Zinc Concentration That Results in Transient Neonatal Zinc Deficiency. J. Biol. Chem. 2006, 281, 39699–39707. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhou, B. Dietary zinc absorption: A play of Zips and ZnTs in the gut. IUBMB Life 2010, 62, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Maret, W.; Sandstead, H.H. Zinc requirements and the risks and benefits of zinc supplementation. J. Trace Elem. Med. Biol. 2006, 20, 3–18. [Google Scholar] [CrossRef]
- Andrews, G.K. Regulation of metallothionein gene expression by oxidative stress and metal ions. Biochem. Pharmacol. 2000, 59, 95–104. [Google Scholar] [CrossRef]
- Sivalingam, N.; Pichandi, S.; Chapla, A.; Dinakaran, A.; Jacob, M. Zinc protects against indomethacin-induced damage in the rat small intestine. Eur. J. Pharmacol. 2011, 654, 106–116. [Google Scholar] [CrossRef]
- Lu, J.; Stewart, A.J.; Sadler, P.J.; Pinheiro, T.J.; Blindauer, C.A. Albumin as a zinc carrier: Properties of its high-affinity zinc-binding site. Biochem. Soc. Trans. 2008, 36, 1317–1321. [Google Scholar] [CrossRef]
- Laity, J.H.; Andrews, G.K. Understanding the mechanisms of zinc-sensing by metal-response element binding transcription factor-1 (MTF-1). Arch. Biochem. Biophys. 2007, 463, 201–210. [Google Scholar] [CrossRef]
- Liuzzi, J.P.; Bobo, J.A.; Lichten, L.A.; Samuelson, D.A.; Cousins, R.J. Responsive transporter genes within the murine intestinal-pancreatic axis form a basis of zinc homeostasis. Proc. Natl. Acad. Sci. USA 2004, 101, 14355–14360. [Google Scholar] [CrossRef]
- Guo, L.; Lichten, L.A.; Ryu, M.-S.; Liuzzi, J.P.; Wang, F.; Cousins, R.J. STAT5-glucocorticoid receptor interaction and MTF-1 regulate the expression of ZnT2 (Slc30a2) in pancreatic acinar cells. Proc. Natl. Acad. Sci. USA 2010, 107, 2818–2823. [Google Scholar] [CrossRef]
- Kondaiah, P.; Yaduvanshi, P.S.; Sharp, P.A.; Pullakhandam, R. Iron and Zinc Homeostasis and Interactions: Does Enteric Zinc Excretion Cross-Talk with Intestinal Iron Absorption? Nutrients 2019, 11, 1885. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef]
- Prasad, A.S. Zinc: Role in immunity, oxidative stress and chronic inflammation. Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-Y.; Steffen, J.; Eide, D.J. Cytosolic Superoxide Dismutase (SOD1) Is Critical for Tolerating the Oxidative Stress of Zinc Deficiency in Yeast. PLoS ONE 2009, 4, e7061. [Google Scholar] [CrossRef] [PubMed]
- Lewandowski, Ł.; Kepinska, M.; Milnerowicz, H. The copper-zinc superoxide dismutase activity in selected diseases. Eur. J. Clin. Investig. 2019, 49, e13036. [Google Scholar] [CrossRef]
- Lewandowski, Ł.; Kepinska, M.; Milnerowicz, H. Inhibition of copper-zinc superoxide dismutase activity by selected environmental xenobiotics. Environ. Toxicol. Pharmacol. 2018, 58, 105–113. [Google Scholar] [CrossRef]
- Perry, J.; Shin, D.; Getzoff, E.; Tainer, J. The structural biochemistry of the superoxide dismutases. Biochim. Biophys. Acta BBA—Proteins Proteom. 2010, 1804, 245–262. [Google Scholar] [CrossRef]
- Antonyuk, S.V.; Strange, R.W.; Marklund, S.L.; Hasnain, S.S. The Structure of Human Extracellular Copper–Zinc Superoxide Dismutase at 1.7 Å Resolution: Insights into Heparin and Collagen Binding. J. Mol. Biol. 2009, 388, 310–326. [Google Scholar] [CrossRef]
- Homma, K.; Fujisawa, T.; Tsuburaya, N.; Yamaguchi, N.; Kadowaki, H.; Takeda, K.; Nishitoh, H.; Matsuzawa, A.; Naguro, I.; Ichijo, H. SOD1 as a Molecular Switch for Initiating the Homeostatic ER Stress Response under Zinc Deficiency. Mol. Cell 2013, 52, 75–86. [Google Scholar] [CrossRef]
- Kara, E.; Gunay, M.; Cicioglu, I.; Ozal, M.; Kilic, M.; Mogulkoc, R.; Baltaci, A.K. Effect of Zinc Supplementation on Antioxidant Activity in Young Wrestlers. Biol. Trace Elem. Res. 2010, 134, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Jarosz, M.; Olbert, M.; Wyszogrodzka, G.; Mlyniec, K.; Librowski, T. Antioxidant and anti-inflammatory effects of zinc. Zinc-dependent NF-κB signaling. Inflammopharmacology 2017, 25, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Ibs, K.-H.; Rink, L. Zinc-Altered Immune function. J. Nutr. 2003, 133, 1452S–1456S. [Google Scholar] [CrossRef] [PubMed]
- Yasui, K.; Baba, A. Therapeutic potential of superoxide dismutase (SOD) for resolution of inflammation. Inflamm. Res. 2006, 55, 359–363. [Google Scholar] [CrossRef]
- Niwa, Y. Lipid Peroxides and Superoxide Dismutase (SOD) Induction in Skin Inflammatory Diseases, and Treatment with SOD Preparations. Dermatologica 1989, 179, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Laronha, H.; Caldeira, J. Structure and Function of Human Matrix Metalloproteinases. Cells 2020, 9, 1076. [Google Scholar] [CrossRef] [PubMed]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef]
- Krstić, J.; Santibañez, J.F. Transforming Growth Factor-Beta and Matrix Metalloproteinases: Functional Interactions in Tumor Stroma-Infiltrating Myeloid Cells. Sci. World J. 2014, 2014, 521754. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, H.-X.; Chen, Y.; Wang, Y.; Yang, M.; Guo, M. Zinc Deficiency Induces Oxidative Damage and Causes Spleen Fibrosis. Biol. Trace Elem. Res. 2020, 194, 203–209. [Google Scholar] [CrossRef]
- Xu, C.; Huang, Z.; Liu, L.; Luo, C.; Lu, G.; Li, Q.; Gao, X. Zinc Regulates Lipid Metabolism and MMPs Expression in Lipid Disturbance Rabbits. Biol. Trace Elem. Res. 2015, 168, 411–420. [Google Scholar] [CrossRef]
- Olechnowicz, J.; Tinkov, A.; Skalny, A.; Suliburska, J. Zinc status is associated with inflammation, oxidative stress, lipid, and glucose metabolism. J. Physiol. Sci. 2018, 68, 19–31. [Google Scholar] [CrossRef]
- Grommes, J.; Binnebösel, M.; Klink, C.D.; Von Trotha, K.T.; Rosch, R.; Oettinger, A.P.; Lindlar, I.; Krones, C.J. Balancing zinc deficiency leads to an improved healing of colon anastomosis in rats. Int. J. Color. Dis. 2010, 26, 295–301. [Google Scholar] [CrossRef]
- Ke, H. Implications of PDE4 structure on inhibitor selectivity across PDE families. Int. J. Impot. Res. 2004, 16, S24–S27. [Google Scholar] [CrossRef][Green Version]
- Von Bülow, V.; Dubben, S.; Engelhardt, G.; Hebel, S.; Plümäkers, B.; Heine, H.; Rink, L.; Haase, H. Zinc-dependent suppression of TNF-alpha production is mediated by protein kinase A-induced inhibition of Raf-1, I kappa B kinase beta, and NF-kappa B. J. Immunol. 2007, 179, 4180–4186. [Google Scholar] [CrossRef]
- Ranju, V. Scope of adjuvant therapy using roflumilast, a PDE-4 inhibitor against COVID-19. Pulm. Pharmacol. Ther. 2021, 66, 101978. [Google Scholar] [CrossRef]
- Gopalakrishna, R.; Jaken, S. Protein kinase C signaling and oxidative stress. Free Radic. Biol. Med. 2000, 28, 1349–1361. [Google Scholar] [CrossRef]
- Baier, G.; Wagner, J. PKC inhibitors: Potential in T cell-dependent immune diseases. Curr. Opin. Cell Biol. 2009, 21, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Haase, H.; Hebel, S.; Engelhardt, G.; Rink, L. Flow cytometric measurement of labile zinc in peripheral blood mononuclear cells. Anal. Biochem. 2006, 352, 222–230. [Google Scholar] [CrossRef]
- Hayashi, K.; Ishizuka, S.; Yokoyama, C.; Hatae, T. Attenuation of interferon-γ mRNA expression in activated Jurkat T cells by exogenous zinc via down-regulation of the calcium-independent PKC–AP-1 signaling pathway. Life Sci. 2008, 83, 6–11. [Google Scholar] [CrossRef]
- Shalini, S.; Dorstyn, L.; Dawar, S.; Kumar, S. Old, new and emerging functions of caspases. Cell Death Differ. 2015, 22, 526–539. [Google Scholar] [CrossRef]
- Eron, S.J.; MacPherson, D.J.; Dagbay, K.B.; Hardy, J.A. Multiple Mechanisms of Zinc-Mediated Inhibition for the Apoptotic Caspases-3, -6, -7, and -8. ACS Chem. Biol. 2018, 13, 1279–1290. [Google Scholar] [CrossRef] [PubMed]
- Velázquez-Delgado, E.M.; Hardy, J.A. Zinc-mediated Allosteric Inhibition of Caspase-6. J. Biol. Chem. 2012, 287, 36000–36011. [Google Scholar] [CrossRef]
- Truong-Tran, A.Q.; Grosser, D.; Ruffin, R.E.; Murgia, C.; Zalewski, P.D. Apoptosis in the normal and inflamed airway epithelium: Role of zinc in epithelial protection and procaspase-3 regulation. Biochem. Pharmacol. 2003, 66, 1459–1468. [Google Scholar] [CrossRef]
- Sunderman, F.W. The influence of zinc on apoptosis. Ann. Clin. Lab. Sci. 1995, 25, 134–142. [Google Scholar] [PubMed]
- Supuran, C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.-L.; Supuran, C.; Scozzafava, A.; Winum, J.-Y. Design of Zinc Binding Functions for Carbonic Anhydrase Inhibitors. Curr. Pharm. Des. 2008, 14, 615–621. [Google Scholar] [CrossRef]
- Goto, T.; Komai, M.; Bryant, B.P.; Furukawa, Y. Reduction in Carbonic Anhydrase Activity in the Tongue Epithelium and Submandibular Gland in Zinc-Deficient Rats. Int. J. Vitam. Nutr. Res. 2000, 70, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Lukaski, H.C. Low dietary zinc decreases erythrocyte carbonic anhydrase activities and impairs cardiorespiratory function in men during exercise. Am. J. Clin. Nutr. 2005, 81, 1045–1051. [Google Scholar] [CrossRef]
- Zaher, D.M.; El-Gamal, M.I.; Omar, H.A.; Aljareh, S.N.; Al-Shamma, S.A.; Ali, A.J.; Zaib, S.; Iqbal, J. Recent advances with alkaline phosphatase isoenzymes and their inhibitors. Arch. Pharm. 2020, 353, e2000011. [Google Scholar] [CrossRef]
- Coleman, J.E. Structure and mechanism of alkaline phosphatase. Annu. Rev. Biophys. Biomol. Struct. 1992, 21, 441–483. [Google Scholar] [CrossRef] [PubMed]
- Rashidi, A.A.; Salehi, M.; Piroozmand, A.; Sagheb, M.M. Effects of Zinc Supplementation on Serum Zinc and C-Reactive Protein Concentrations in Hemodialysis Patients. J. Ren. Nutr. 2009, 19, 475–478. [Google Scholar] [CrossRef]
- Naber, T.H.; Baadenhuysen, H.; Jansen, J.B.; Hamer, C.J.V.D.; Broek, W.V.D. Serum alkaline phosphatase activity during zinc deficiency and long-term inflammatory stress. Clin. Chim. Acta 1996, 249, 109–127. [Google Scholar] [CrossRef]
- Okegbile, E.O.; Odunuga, O.; Oyewo, A. Effect of dietary zinc deficiency on alkaline phosphatase and nucleic acids in rats. Afr. J. Med. Med. Sci. 1999, 27, 189–192. [Google Scholar]
- Kodama, H.; Tanaka, M.; Naito, Y.; Katayama, K.; Moriyama, M. Japan’s Practical Guidelines for Zinc Deficiency with a Particular Focus on Taste Disorders, Inflammatory Bowel Disease, and Liver Cirrhosis. Int. J. Mol. Sci. 2020, 21, 2941. [Google Scholar] [CrossRef] [PubMed]
- Didion, S.P.; Ryan, M.J.; Didion, L.A.; Fegan, P.E.; Sigmund, C.D.; Faraci, F.M. Increased superoxide and vascular dysfunction in CuZnSOD-deficient mice. Circ. Res. 2002, 91, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.J.; McKeown, S.R.; Rashid, S. Mutant SOD1 mediated pathogenesis of Amyotrophic Lateral Sclerosis. Gene 2016, 577, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Murata, N.; Noda, Y.; Tahara, S.; Kaneko, T.; Kinoshita, N.; Hatsuta, H.; Murayama, S.; Barnham, K.J.; Irie, K.; et al. SOD1 (Copper/Zinc Superoxide Dismutase) Deficiency Drives Amyloid β Protein Oligomerization and Memory Loss in Mouse Model of Alzheimer Disease. J. Biol. Chem. 2011, 286, 44557–44568. [Google Scholar] [CrossRef]
- Nazıroğlu, M.; Muhamad, S.; Pecze, L. Nanoparticles as potential clinical therapeutic agents in Alzheimer’s disease: Focus on selenium nanoparticles. Expert Rev. Clin. Pharmacol. 2017, 10, 773–782. [Google Scholar] [CrossRef]
- Wang, X.; Khalil, R.A. Matrix Metalloproteinases, Vascular Remodeling, and Vascular Disease. Adv. Pharmacol. 2018, 81, 241–330. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.M. Treating COPD with PDE 4 inhibitors. Int. J. Chronic Obstr. Pulm. Dis. 2007, 2, 517–533. [Google Scholar]
- Altman, A.; Kong, K.-F. Protein kinase C inhibitors for immune disorders. Drug Discov. Today 2014, 19, 1217–1221. [Google Scholar] [CrossRef] [PubMed]
- Paulazo, M.A.; Klecha, A.J.; Sterle, H.; Valli, E.; Torti, H.; Cayrol, F.; Arcos, M.L.B.; Cremaschi, G.A. Hypothyroidism-related zinc deficiency leads to suppression of T lymphocyte activity. Endocrine 2019, 66, 266–277. [Google Scholar] [CrossRef]
- Truong-Tran, A.Q.; Ruffin, R.E.; Zalewski, P.D. Visualization of labile zinc and its role in apoptosis of primary airway epithelial cells and cell lines. Am. J. Physiol. Cell. Mol. Physiol. 2000, 279, L1172–L1183. [Google Scholar] [CrossRef]
- Brown, D.; Garcia-Segura, L.; Orci, L. Carbonic anhydrase is associated with taste buds in rat tongue. Brain Res. 1984, 324, 346–348. [Google Scholar] [CrossRef]
- Nizet, A.; Cavalier, E.; Stenvinkel, P.; Haarhaus, M.; Magnusson, P. Bone alkaline phosphatase: An important biomarker in chronic kidney disease—Mineral and bone disorder. Clin. Chim. Acta 2020, 501, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Linglart, A.; Duplan, M.B. Hypophosphatasia. Curr. Osteoporos. Rep. 2016, 14, 95–105. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Superoxide Dismutases: Role in Redox Signaling, Vascular Function, and Diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef]
- Laursen, J.B.; Rajagopalan, S.; Galis, Z.; Tarpey, M.; Freeman, B.A.; Harrison, D.G. Role of Superoxide in Angiotensin II–Induced but Not Catecholamine-Induced Hypertension. Circulation 1997, 95, 588–593. [Google Scholar] [CrossRef]
- Skene, K.; Walsh, S.K.; Okafor, O.; Godsman, N.; Barrows, C.; Meier, P.; Gordon, M.J.; Beattie, J.H.; Wainwright, C.L. Acute dietary zinc deficiency in rats exacerbates myocardial ischaemia–reperfusion injury through depletion of glutathione. Br. J. Nutr. 2019, 121, 961–973. [Google Scholar] [CrossRef]
- Majewski, M.; Ognik, K.; Thoene, M.; Rawicka, A.; Juskiewicz, J. Resveratrol modulates the blood plasma levels of Cu and Zn, the antioxidant status and the vascular response of thoracic arteries in copper deficient Wistar rats. Toxicol. Appl. Pharmacol. 2020, 390, 114877. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Hardiman, O. The epidemiology of ALS: A conspiracy of genes, environment and time. Nat. Rev. Neurol. 2013, 9, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Ermilova, I.P.; Ermilov, V.B.; Levy, M.; Ho, E.; Pereira, C.; Beckman, J.S. Protection by dietary zinc in ALS mutant G93A SOD transgenic mice. Neurosci. Lett. 2005, 379, 42–46. [Google Scholar] [CrossRef]
- Estévez, A.G.; Crow, J.P.; Sampson, J.B.; Reiter, C.; Zhuang, Y.; Richardson, G.J.; Tarpey, M.M.; Barbeito, L.; Beckman, J.S. Induction of Nitric Oxide-Dependent Apoptosis in Motor Neurons by Zinc-Deficient Superoxide Dismutase. Science 1999, 286, 2498–2500. [Google Scholar] [CrossRef]
- McAllum, E.J.; Roberts, B.R.; Hickey, J.L.; Dang, T.N.; Grubman, A.; Donnelly, P.S.; Liddell, J.R.; White, A.R.; Crouch, P.J. ZnII(atsm) is protective in amyotrophic lateral sclerosis model mice via a copper delivery mechanism. Neurobiol. Dis. 2015, 81, 20–24. [Google Scholar] [CrossRef]
- Greenough, M.A.; Volitakis, I.; Li, Q.-X.; Laughton, K.; Evin, G.; Ho, M.; Dalziel, A.H.; Camakaris, J.; Bush, A.I. Presenilins Promote the Cellular Uptake of Copper and Zinc and Maintain Copper Chaperone of SOD1-dependent Copper/Zinc Superoxide Dismutase Activity. J. Biol. Chem. 2011, 286, 9776–9786. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, P.M.; Shimokawa, H.; Feletou, M.; Tang, E.H.C. Endothelial dysfunction and vascular disease—A 30th anniversary update. Acta Physiol. 2017, 219, 22–96. [Google Scholar] [CrossRef]
- Teng, L.; Yu, M.; Li, J.-M.; Tang, H.; Yu, J.; Mo, L.-H.; Jin, J.; Liu, X.-Z. Matrix metalloproteinase-9 as new biomarkers of severity in multiple organ dysfunction syndrome caused by trauma and infection. Mol. Cell. Biochem. 2011, 360, 271–277. [Google Scholar] [CrossRef]
- Mittal, R.; Patel, A.P.; Debs, L.H.; Nguyen, D.; Patel, K.; Grati, M.; Mittal, J.; Yan, D.; Chapagain, P.; Liu, X.Z. Intricate Functions of Matrix Metalloproteinases in Physiological and Pathological Conditions. J. Cell. Physiol. 2016, 231, 2599–2621. [Google Scholar] [CrossRef]
- Onal, I.K.; Altun, B.; Onal, E.D.; Kırkpantur, A.; Oz, S.G.; Turgan, C. Serum levels of MMP-9 and TIMP-1 in primary hypertension and effect of antihypertensive treatment. Eur. J. Intern. Med. 2009, 20, 369–372. [Google Scholar] [CrossRef]
- Huang, S.-T.; Yang, R.-C.; Wu, H.-T.; Wang, C.-N.; Pang, J.-H. Zinc-Chelation Contributes to the Anti-Angiogenic Effect of Ellagic Acid on Inhibiting MMP-2 Activity, Cell Migration and Tube Formation. PLoS ONE 2011, 6, e18986. [Google Scholar] [CrossRef]
- Raffetto, J.D.; Khalil, R.A. Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem. Pharmacol. 2008, 75, 346–359. [Google Scholar] [CrossRef] [PubMed]
- MacNee, W. Pathogenesis of chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2005, 2, 58–266. [Google Scholar] [CrossRef] [PubMed]
- Karadag, F.; Cildag, O.; Altinisik, M.; Kozaci, L.D.; Kiter, G.; Altun, C. Trace elements as a component of oxidative stress in COPD. Respirology 2004, 9, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.J.; Hansen, M.; Roscioli, E.; Jones, J.; Murgia, C.; Ackland, M.L.; Zalewski, P.; Anderson, G.; Ruffin, R. Dietary zinc mediates inflammation and protects against wasting and metabolic derangement caused by sustained cigarette smoke exposure in mice. Biometals 2010, 24, 23–39. [Google Scholar] [CrossRef]
- Wellinghausen, N.; Martin, M.; Rink, L. Zinc inhibits interleukin-1-dependent T cell stimulation. Eur. J. Immunol. 1997, 27, 2529–2535. [Google Scholar] [CrossRef]
- Zhong, C.; Wu, Y.; Chang, H.; Liu, C.; Zhou, L.; Zou, J.; Qi, Z. Effect of PKC inhibitor on experimental autoimmune myocarditis in Lewis rats. Oncotarget 2017, 8, 54187–54198. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.; Zheng, J.; Xiao, W.; Chang, S.; Wei, Q.; Wu, H.; Tao, Y.; Yang, G.; Xie, B.; Lan, X.; et al. PKC inhibition of sotrastaurin has antitumor activity in diffuse large B-cell lymphoma via regulating the expression of MCT-1. Acta Biochim. Biophys. Sin. 2018, 50, 399–407. [Google Scholar] [CrossRef]
- Bucchieri, F.; Puddicombe, S.M.; Lordan, J.L.; Richter, A.; Buchanan, D.; Wilson, S.J.; Ward, J.; Zummo, G.; Howarth, P.H.; Djukanović, R.; et al. Asthmatic Bronchial Epithelium Is More Susceptible to Oxidant-Induced Apoptosis. Am. J. Respir. Cell Mol. Biol. 2002, 27, 179–185. [Google Scholar] [CrossRef]
- Roscioli, E.; Hamon, R.; Lester, S.; Murgia, C.; Grant, J.; Zalewski, P. Zinc-rich inhibitor of apoptosis proteins (IAPs) as regulatory factors in the epithelium of normal and inflamed airways. Biometals 2013, 26, 205–227. [Google Scholar] [CrossRef]
- Riccioni, G.; D’Orazio, N. The role of selenium, zinc and antioxidant vitamin supplementation in the treatment of bronchial asthma: Adjuvant therapy or not? Expert Opin. Investig. Drugs 2005, 14, 1145–1155. [Google Scholar] [CrossRef]
- Yagi, T.; Asakawa, A.; Ueda, H.; Ikeda, S.; Miyawaki, S.; Inui, A. The Role of Zinc in the Treatment of Taste Disorders. Recent Pat. Food Nutr. Agric. 2013, 5, 44–51. [Google Scholar] [CrossRef]
- Komai, M.; Goto, T.; Suzuki, H.; Takeda, T.; Furukawa, Y. Zinc deficiency and taste dysfunction; Contribution of carbonic anhydrase, a zinc-metalloenzyme, to normal taste sensation. BioFactors 2000, 12, 65–70. [Google Scholar] [CrossRef]
- Henkin, R.I.; Martin, B.M.; Agarwal, R.P. Efficacy of exogenous oral zinc in treatment of patients with carbonic anhydrase VI deficiency. Am. J. Med. Sci. 1999, 318, 392–405. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.-J.; Cho, Y.-E.; Kim, T.; Shin, H.-I.; Kwun, I.-S. Zinc may increase bone formation through stimulating cell proliferation, alkaline phosphatase activity and collagen synthesis in osteoblastic MC3T3-E1 cells. Nutr. Res. Pract. 2010, 4, 356–361. [Google Scholar] [CrossRef]
- Sadighi, A.; Mahdavi-Roshan, M.; Moradi, A.; Ostadrahimi, A. The effects of zinc supplementation on serum zinc, alkaline phosphatase activity and fracture healing of bones. Saudi Med. J. 2008, 29, 1276–1279. [Google Scholar] [PubMed]
- Peretz, A.; Papadopoulos, T.; Willems, D.; Hotimsky, A.; Michiels, N.; Siderova, V.; Bergmann, P.; Nève, J. Zinc supplementation increases bone alkaline phosphatase in healthy men. J. Trace Elem. Med. Biol. 2001, 15, 175–178. [Google Scholar] [CrossRef]
- Skrajnowska, D.; Bobrowska-Korczak, B. Role of Zinc in Immune System and Anti-Cancer Defense Mechanisms. Nutrients 2019, 11, 2273. [Google Scholar] [CrossRef]
- Zhang, Y.; Tian, Y.; Zhang, H.; Xu, B.; Chen, H. Potential pathways of zinc deficiency-promoted tumorigenesis. Biomed. Pharmacother. 2021, 133, 110983. [Google Scholar] [CrossRef] [PubMed]
- Eide, D.J. The oxidative stress of zinc deficiency. Metallomics 2011, 3, 1124–1129. [Google Scholar] [CrossRef] [PubMed]
- Kocdor, H.; Ates, H.; Aydin, S.; Cehreli, R.; Soyarat, F.; Kemanli, P.; Harmanci, D.; Cengiz, H.; Kocdor, M. Zinc supplementation induces apoptosis and enhances antitumor efficacy of docetaxel in non-small-cell lung cancer. Drug Des. Dev. Ther. 2015, 9, 3899–3909. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Taccioli, C.; Jiang, Y.; Chen, H.; Smalley, K.J.; Huang, K.; Liu, X.; Farber, J.L.; Croce, C.M.; Fong, L.Y.Y. Zinc deficiency activates S100A8 inflammation in the absence of COX-2 and promotes murine oral-esophageal tumor progression. Int. J. Cancer 2011, 129, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Taccioli, C.; Chen, H.; Jiang, Y.; Liu, X.P.; Huang, K.; Smalley, K.J.; Farber, J.L.; Croce, C.M.; Fong, L. Dietary zinc deficiency fuels esophageal cancer development by inducing a distinct inflammatory signature. Oncogene 2011, 31, 4550–4558. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Zn2+ Metalloenzyme | Zn2+ Binding Sites | The Effect of Zn2+ on Zn2+ Metalloenzyme | Related Diseases | Ref |
|---|---|---|---|---|
| Copper/Zinc superoxide dismutase | Three histidine residues and one aspartic acid | Activator | CVDs, ALS and AD | [36,74,75,76,77] |
| Matrix metalloproteinase | Three histidine residues of the catalytic domain | Activator | Vascular diseases | [45,46,78] |
| Phosphodiesterase | Two histidine and two aspartic acid residues | Inhibitor | COPD | [52,79] |
| Protein kinase C | Cysteine residues in the regulatory domain | Activator | Immune diseases | [55,80,81] |
| Caspase | Caspase-6: one lysine, one glutamic acid and one histidine residue out of the active site; Caspase-8: one cysteine in the active site and the second binding site is unknown | Inhibitor | Asthma | [60,61,82] |
| Carbonic anhydrase | α-, γ-, and δ-CAs: three histidine residues and a hydroxide ion Type I β-CAs: two cysteine residues, one histidine residues, and a hydroxide ion Type II β-CAs: two cysteine residues, one histidine, and one aspartate residues | Activator | Hypogeusia | [64,83] |
| Alkaline phosphatase | Three metal binding sites in active center | Activator | Bone disorder | [69,84,85] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, Y.; Chen, H. Aberrance of Zinc Metalloenzymes-Induced Human Diseases and Its Potential Mechanisms. Nutrients 2021, 13, 4456. https://doi.org/10.3390/nu13124456
Cheng Y, Chen H. Aberrance of Zinc Metalloenzymes-Induced Human Diseases and Its Potential Mechanisms. Nutrients. 2021; 13(12):4456. https://doi.org/10.3390/nu13124456
Chicago/Turabian StyleCheng, Yunqi, and Hongping Chen. 2021. "Aberrance of Zinc Metalloenzymes-Induced Human Diseases and Its Potential Mechanisms" Nutrients 13, no. 12: 4456. https://doi.org/10.3390/nu13124456
APA StyleCheng, Y., & Chen, H. (2021). Aberrance of Zinc Metalloenzymes-Induced Human Diseases and Its Potential Mechanisms. Nutrients, 13(12), 4456. https://doi.org/10.3390/nu13124456