
Genomics of Postprandial Lipidomics in the Genetics of Lipid-Lowering Drugs and Diet Network Study


 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. High-Fat Meal and Measurement of Postprandial Lipemia
2.3. Genomics
2.4. Lipidomics
2.5. Replication Population
3. Statistical Methods
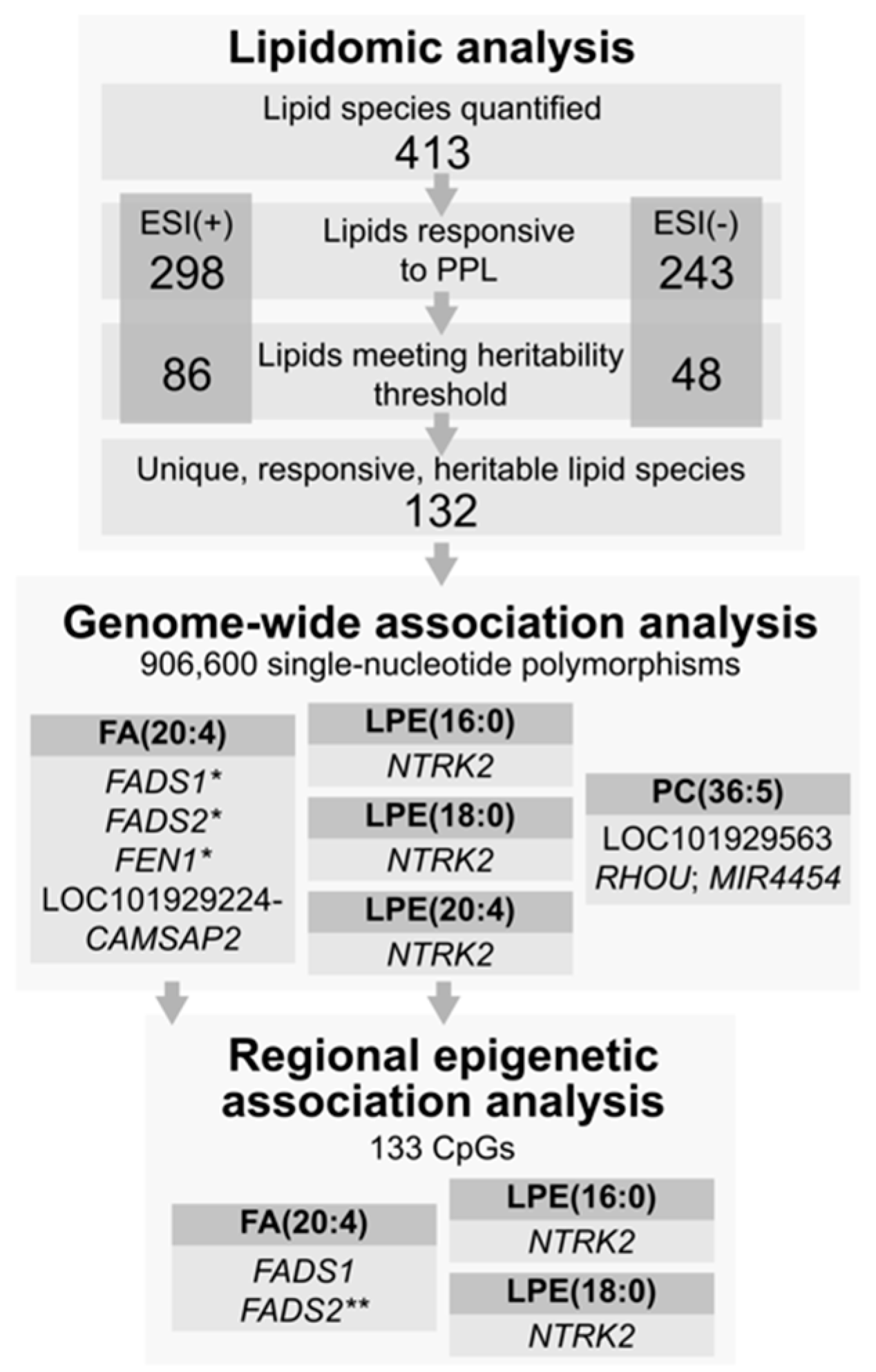
4. Results
5. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Teeman, C.S.; Kurti, S.P.; Cull, B.J.; Emerson, S.R.; Haub, M.D.; Rosenkranz, S.K. Postprandial lipemic and inflammatory responses to high-fat meals: A review of the roles of acute and chronic exercise. Nutr. Metab. 2016, 13, 80. [Google Scholar] [CrossRef]
- O’Keefe, J.H.; Bell, D.S. Postprandial hyperglycemia/hyperlipidemia (postprandial dysmetabolism) is a cardiovascular risk factor. Am. J. Cardiol. 2007, 100, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Sanders, T.A. Dietary fat and postprandial lipids. Curr. Atheroscler. Rep. 2003, 5, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Tholstrup, T.; Miller, G.J.; Bysted, A.; Sandstrom, B. Effect of individual dietary fatty acids on postprandial activation of blood coagulation factor VII and fibrinolysis in healthy young men. Am. J. Clin. Nutr. 2003, 77, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Duttaroy, A.K. Postprandial activation of hemostatic factors: Role of dietary fatty acids. Prostaglandins Leukot. Essent. Fatty Acids 2005, 72, 381–391. [Google Scholar] [CrossRef]
- Hodis, H.N. Triglyceride-rich lipoprotein remnant particles and risk of atherosclerosis. Circulation 1999, 99, 2852–2854. [Google Scholar] [CrossRef] [PubMed]
- Cohn, J.S.; McNamara, J.R.; Cohn, S.D.; Ordovas, J.M.; Schaefer, E.J. Postprandial plasma lipoprotein changes in human subjects of different ages. J. Lipid Res. 1988, 29, 469–479. [Google Scholar] [CrossRef]
- Steiner, G. Triglyceride-rich lipoproteins and atherosclerosis, from fast to feast. Ann. Med. 1993, 25, 431–435. [Google Scholar] [CrossRef]
- Sandesara, P.B.; Virani, S.S.; Fazio, S.; Shapiro, M.D. The Forgotten Lipids: Triglycerides, Remnant Cholesterol, and Atherosclerotic Cardiovascular Disease Risk. Endocr. Rev. 2019, 40, 537–557. [Google Scholar] [CrossRef]
- Patsch, J.R.; Miesenbock, G.; Hopferwieser, T.; Muhlberger, V.; Knapp, E.; Dunn, J.K.; Gotto, A.M., Jr.; Patsch, W. Relation of triglyceride metabolism and coronary artery disease. Studies in the postprandial state. Arterioscler. Thromb. 1992, 12, 1336–1345. [Google Scholar] [CrossRef]
- Meyer, E.; Westerveld, H.T.; de Ruyter-Meijstek, F.C.; van Greevenbroek, M.M.; Rienks, R.; van Rijn, H.J.; Erkelens, D.W.; de Bruin, T.W. Abnormal postprandial apolipoprotein B-48 and triglyceride responses in normolipidemic women with greater than 70% stenotic coronary artery disease: A case-control study. Atherosclerosis 1996, 124, 221–235. [Google Scholar] [CrossRef]
- Mero, N.; Malmstrom, R.; Steiner, G.; Taskinen, M.R.; Syvanne, M. Postprandial metabolism of apolipoprotein B-48- and B-100-containing particles in type 2 diabetes mellitus: Relations to angiographically verified severity of coronary artery disease. Atherosclerosis 2000, 150, 167–177. [Google Scholar] [CrossRef]
- Boren, J.; Matikainen, N.; Adiels, M.; Taskinen, M.R. Postprandial hypertriglyceridemia as a coronary risk factor. Clin. Chim. Acta 2014, 431, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Boquist, S.; Ruotolo, G.; Tang, R.; Bjorkegren, J.; Bond, M.G.; de Faire, U.; Karpe, F.; Hamsten, A. Alimentary lipemia, postprandial triglyceride-rich lipoproteins, and common carotid intima-media thickness in healthy, middle-aged men. Circulation 1999, 100, 723–728. [Google Scholar] [CrossRef]
- Samson, C.E.; Galia, A.L.; Llave, K.I.; Zacarias, M.B.; Mercado-Asis, L.B. Postprandial Peaking and Plateauing of Triglycerides and VLDL in Patients with Underlying Cardiovascular Diseases Despite Treatment. Int. J. Endocrinol. Metab. 2012, 10, 587–593. [Google Scholar] [CrossRef]
- Groot, P.H.; van Stiphout, W.A.; Krauss, X.H.; Jansen, H.; van Tol, A.; van Ramshorst, E.; Chin-On, S.; Hofman, A.; Cresswell, S.R.; Havekes, L. Postprandial lipoprotein metabolism in normolipidemic men with and without coronary artery disease. Arterioscler. Thromb. 1991, 11, 653–662. [Google Scholar] [CrossRef]
- Uiterwaal, C.S.; Grobbee, D.E.; Witteman, J.C.; van Stiphout, W.A.; Krauss, X.H.; Havekes, L.M.; de Bruijn, A.M.; van Tol, A.; Hofman, A. Postprandial triglyceride response in young adult men and familial risk for coronary atherosclerosis. Ann. Intern. Med. 1994, 121, 576–583. [Google Scholar] [CrossRef]
- Tiret, L.; Gerdes, C.; Murphy, M.J.; Dallongeville, J.; Nicaud, V.; O’Reilly, D.S.; Beisiegel, U.; De Backer, G. Postprandial response to a fat tolerance test in young adults with a paternal history of premature coronary heart disease—The EARS II study (European Atherosclerosis Research Study). Eur. J. Clin. Investig. 2000, 30, 578–585. [Google Scholar] [CrossRef]
- Wojczynski, M.K.; Parnell, L.D.; Pollin, T.I.; Lai, C.Q.; Feitosa, M.F.; O’Connell, J.R.; Frazier-Wood, A.C.; Gibson, Q.; Aslibekyan, S.; Ryan, K.A.; et al. Genome-wide association study of triglyceride response to a high-fat meal among participants of the NHLBI Genetics of Lipid Lowering Drugs and Diet Network (GOLDN). Metabolism 2015, 64, 1359–1371. [Google Scholar] [CrossRef]
- Lai, C.Q.; Parnell, L.D.; Smith, C.E.; Guo, T.; Sayols-Baixeras, S.; Aslibekyan, S.; Tiwari, H.K.; Irvin, M.R.; Bender, C.; Fei, D.; et al. Carbohydrate and fat intake associated with risk of metabolic diseases through epigenetics of CPT1A. Am. J. Clin. Nutr. 2020, 112, 1200–1211. [Google Scholar] [CrossRef]
- LaBarre, J.L.; Singer, K.; Burant, C.F. Advantages of Studying the Metabolome in Response to Mixed-Macronutrient Challenges and Suggestions for Future Research Designs. J. Nutr. 2021, 151, 2868–2881. [Google Scholar] [CrossRef] [PubMed]
- Wenk, M.R. The emerging field of lipidomics. Nat. Rev. Drug Discov. 2005, 4, 594–610. [Google Scholar] [CrossRef]
- Rhee, E.P.; Cheng, S.; Larson, M.G.; Walford, G.A.; Lewis, G.D.; McCabe, E.; Yang, E.; Farrell, L.; Fox, C.S.; O’Donnell, C.J.; et al. Lipid profiling identifies a triacylglycerol signature of insulin resistance and improves diabetes prediction in humans. J. Clin. Investig. 2011, 121, 1402–1411. [Google Scholar] [CrossRef] [PubMed]
- Meikle, P.J.; Wong, G.; Barlow, C.K.; Kingwell, B.A. Lipidomics: Potential role in risk prediction and therapeutic monitoring for diabetes and cardiovascular disease. Pharmacol. Ther. 2014, 143, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Bou Khalil, M.; Hou, W.; Zhou, H.; Elisma, F.; Swayne, L.A.; Blanchard, A.P.; Yao, Z.; Bennett, S.A.; Figeys, D. Lipidomics era: Accomplishments and challenges. Mass Spectrom. Rev. 2010, 29, 877–929. [Google Scholar] [CrossRef]
- German, J.B.; Gillies, L.A.; Smilowitz, J.T.; Zivkovic, A.M.; Watkins, S.M. Lipidomics and lipid profiling in metabolomics. Curr. Opin. Lipidol. 2007, 18, 66–71. [Google Scholar] [CrossRef]
- Fernandis, A.Z.; Wenk, M.R. Membrane lipids as signaling molecules. Curr. Opin. Lipidol. 2007, 18, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Bellis, C.; Kulkarni, H.; Mamtani, M.; Kent, J.W., Jr.; Wong, G.; Weir, J.M.; Barlow, C.K.; Diego, V.; Almeida, M.; Dyer, T.D.; et al. Human plasma lipidome is pleiotropically associated with cardiovascular risk factors and death. Circ. Cardiovasc. Genet. 2014, 7, 854–863. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.H.; Hauser, E.R.; Bain, J.R.; Muehlbauer, M.J.; Haynes, C.; Stevens, R.D.; Wenner, B.R.; Dowdy, Z.E.; Granger, C.B.; Ginsburg, G.S.; et al. High heritability of metabolomic profiles in families burdened with premature cardiovascular disease. Mol. Syst. Biol. 2009, 5, 258. [Google Scholar] [CrossRef]
- Hicks, A.A.; Pramstaller, P.P.; Johansson, A.; Vitart, V.; Rudan, I.; Ugocsai, P.; Aulchenko, Y.; Franklin, C.S.; Liebisch, G.; Erdmann, J.; et al. Genetic determinants of circulating sphingolipid concentrations in European populations. PLoS Genet. 2009, 5, e1000672. [Google Scholar] [CrossRef] [PubMed]
- Illig, T.; Gieger, C.; Zhai, G.; Romisch-Margl, W.; Wang-Sattler, R.; Prehn, C.; Altmaier, E.; Kastenmuller, G.; Kato, B.S.; Mewes, H.W.; et al. A genome-wide perspective of genetic variation in human metabolism. Nat. Genet. 2010, 42, 137–141. [Google Scholar] [CrossRef]
- Mitchell, B.D.; McArdle, P.F.; Shen, H.; Rampersaud, E.; Pollin, T.I.; Bielak, L.F.; Jaquish, C.; Douglas, J.A.; Roy-Gagnon, M.H.; Sack, P.; et al. The genetic response to short-term interventions affecting cardiovascular function: Rationale and design of the Heredity and Phenotype Intervention (HAPI) Heart Study. Am. Heart J. 2008, 155, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Higgins, M.; Province, M.; Heiss, G.; Eckfeldt, J.; Ellison, R.C.; Folsom, A.R.; Rao, D.C.; Sprafka, J.M.; Williams, R. NHLBI Family Heart Study: Objectives and design. Am. J. Epidemiol. 1996, 143, 1219–1228. [Google Scholar] [CrossRef][Green Version]
- Irvin, M.R.; Kabagambe, E.K.; Tiwari, H.K.; Parnell, L.D.; Straka, R.J.; Tsai, M.; Ordovas, J.M.; Arnett, D.K. Apolipoprotein E polymorphisms and postprandial triglyceridemia before and after fenofibrate treatment in the Genetics of Lipid Lowering and Diet Network (GOLDN) Study. Circ. Cardiovasc. Genet. 2010, 3, 462–467. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bibikova, M.; Barnes, B.; Tsan, C.; Ho, V.; Klotzle, B.; Le, J.M.; Delano, D.; Zhang, L.; Schroth, G.P.; Gunderson, K.L.; et al. High density DNA methylation array with single CpG site resolution. Genomics 2011, 98, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Irvin, M.R.; Zhi, D.; Joehanes, R.; Mendelson, M.; Aslibekyan, S.; Claas, S.A.; Thibeault, K.S.; Patel, N.; Day, K.; Jones, L.W.; et al. Epigenome-wide association study of fasting blood lipids in the Genetics of Lipid-lowering Drugs and Diet Network study. Circulation 2014, 130, 565–572. [Google Scholar] [CrossRef]
- Cajka, T.; Fiehn, O. LC–MS-Based Lipidomics and Automated Identification of Lipids Using the LipidBlast In-Silico MS/MS Library. In Lipidomics: Methods and Protocols; Bhattacharya, S.K., Ed.; Springer: New York, NY, USA, 2017; pp. 149–170. [Google Scholar] [CrossRef]
- Showalter, M.R.; Nonnecke, E.B.; Linderholm, A.L.; Cajka, T.; Sa, M.R.; Lönnerdal, B.; Kenyon, N.J.; Fiehn, O. Obesogenic diets alter metabolism in mice. PLoS ONE 2018, 13, e0190632. [Google Scholar] [CrossRef]
- Fan, S.; Kind, T.; Cajka, T.; Hazen, S.L.; Tang, W.H.W.; Kaddurah-Daouk, R.; Irvin, M.R.; Arnett, D.K.; Barupal, D.K.; Fiehn, O. Systematic Error Removal Using Random Forest for Normalizing Large-Scale Untargeted Lipidomics Data. Anal. Chem. 2019, 91, 3590–3596. [Google Scholar] [CrossRef]
- Kind, T.; Liu, K.H.; Lee, D.Y.; DeFelice, B.; Meissen, J.K.; Fiehn, O. LipidBlast in silico tandem mass spectrometry database for lipid identification. Nat. Methods 2013, 10, 755–758. [Google Scholar] [CrossRef]
- Bagheri, M.; Tiwari, H.K.; Murillo, A.L.; Al-Tobasei, R.; Arnett, D.K.; Kind, T.; Barupal, D.K.; Fan, S.; Fiehn, O.; O’connell, J.; et al. A lipidome-wide association study of the lipoprotein insulin resistance index. Lipids Health Dis. 2020, 19, 153. [Google Scholar] [CrossRef]
- Montasser, M.E.; Aslibekyan, S.; Srinivasasainagendra, V.; Tiwari, H.K.; Patki, A.; Bagheri, M.; Kind, T.; Barupal, D.K.; Fan, S.; Perry, J.; et al. Leveraging a founder population to identify novel rare-population genetic determinants of lipidome. bioRxiv 2021. [Google Scholar] [CrossRef]
- Das, S.; Forer, L.; Schönherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef] [PubMed]
- Loh, P.R.; Palamara, P.F.; Price, A.L. Fast and accurate long-range phasing in a UK Biobank cohort. Nat. Genet. 2016, 48, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Fuchsberger, C.; Abecasis, G.R.; Hinds, D.A. minimac2: Faster genotype imputation. Bioinformatics 2015, 31, 782–784. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Benyamin, B.; McEvoy, B.P.; Gordon, S.; Henders, A.K.; Nyholt, D.R.; Madden, P.A.; Heath, A.C.; Martin, N.G.; Montgomery, G.W.; et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010, 42, 565–569. [Google Scholar] [CrossRef]
- Golan, D.; Rosset, S. Accurate estimation of heritability in genome wide studies using random effects models. Bioinformatics 2011, 27, i317–i323. [Google Scholar] [CrossRef]
- Lourenco, V.M.; Pires, A.M.; Kirst, M. Robust linear regression methods in association studies. Bioinformatics 2011, 27, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Zhi, D.; Aslibekyan, S.; Irvin, M.R.; Claas, S.A.; Borecki, I.B.; Ordovas, J.M.; Absher, D.M.; Arnett, D.K. SNPs located at CpG sites modulate genome-epigenome interaction. Epigenetics 2013, 8, 802–806. [Google Scholar] [CrossRef]
- Gromovsky, A.D.; Schugar, R.C.; Brown, A.L.; Helsley, R.N.; Burrows, A.C.; Ferguson, D.; Zhang, R.; Sansbury, B.E.; Lee, R.G.; Morton, R.E.; et al. Δ-5 Fatty Acid Desaturase. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Glaser, C.; Heinrich, J.; Koletzko, B. Role of FADS1 and FADS2 polymorphisms in polyunsaturated fatty acid metabolism. Metabolism 2010, 59, 993–999. [Google Scholar] [CrossRef]
- Chen, H.Y.; Cairns, B.J.; Small, A.M.; Burr, H.A.; Ambikkumar, A.; Martinsson, A.; Thériault, S.; Munter, H.M.; Steffen, B.; Zhang, R.; et al. Association of FADS1/2 Locus Variants and Polyunsaturated Fatty Acids With Aortic Stenosis. JAMA Cardiol. 2020, 5, 694–702. [Google Scholar] [CrossRef]
- Tallima, H.; El Ridi, R. Arachidonic acid: Physiological roles and potential health benefits—A review. J. Adv. Res. 2018, 11, 33–41. [Google Scholar] [CrossRef]
- Margioris, A.N. Fatty acids and postprandial inflammation. Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, L.M.; Dutta, R.; Seeds, M.C.; Lake, K.N.; Hallmark, B.; Mathias, R.A.; Howard, T.D.; Chilton, F.H. FADS genetic and metabolomic analyses identify the ∆5 desaturase (FADS1) step as a critical control point in the formation of biologically important lipids. Sci. Rep. 2020, 10, 15873. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, Y.; Ji, Y.; Xu, W.; Ullah, N.; Yu, H.; Wu, Y.; Xie, L. Association between. Br. J. Nutr. 2021, 126, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Dorajoo, R.; Sun, Y.; Han, Y.; Ke, T.; Burger, A.; Chang, X.; Low, H.Q.; Guan, W.; Lemaitre, R.N.; Khor, C.C.; et al. A genome-wide association study of n-3 and n-6 plasma fatty acids in a Singaporean Chinese population. Genes Nutr. 2015, 10, 53. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.; Steffen, B.T.; Lemaitre, R.N.; Wu, J.H.Y.; Tanaka, T.; Manichaikul, A.; Foy, M.; Rich, S.S.; Wang, L.; Nettleton, J.A.; et al. Genome-wide association study of plasma N6 polyunsaturated fatty acids within the cohorts for heart and aging research in genomic epidemiology consortium. Circ. Cardiovasc. Genet. 2014, 7, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, H.; Lu, L.; Manichaikul, A.; Zhu, J.; Chen, Y.D.; Sun, L.; Liang, S.; Siscovick, D.S.; Steffen, L.M.; et al. Genome-wide meta-analyses identify novel loci associated with n-3 and n-6 polyunsaturated fatty acid levels in Chinese and European-ancestry populations. Hum. Mol. Genet. 2016, 25, 1215–1224. [Google Scholar] [CrossRef]
- Larsson, S.C.; Carter, P.; Vithayathil, M.; Mason, A.M.; Michaëlsson, K.; Baron, J.A.; Burgess, S. Genetically predicted plasma phospholipid arachidonic acid concentrations and 10 site-specific cancers in UK biobank and genetic consortia participants: A mendelian randomization study. Clin. Nutr. 2021, 40, 3332–3337. [Google Scholar] [CrossRef]
- Takkunen, M.J.; de Mello, V.D.; Schwab, U.S.; Kuusisto, J.; Vaittinen, M.; Ågren, J.J.; Laakso, M.; Pihlajamäki, J.; Uusitupa, M.I. Gene-diet interaction of a common FADS1 variant with marine polyunsaturated fatty acids for fatty acid composition in plasma and erythrocytes among men. Mol. Nutr. Food Res. 2016, 60, 381–389. [Google Scholar] [CrossRef]
- Merino, D.M.; Johnston, H.; Clarke, S.; Roke, K.; Nielsen, D.; Badawi, A.; El-Sohemy, A.; Ma, D.W.; Mutch, D.M. Polymorphisms in FADS1 and FADS2 alter desaturase activity in young Caucasian and Asian adults. Mol. Genet. Metab. 2011, 103, 171–178. [Google Scholar] [CrossRef]
- de la Garza Puentes, A.; Montes Goyanes, R.; Chisaguano Tonato, A.M.; Torres-Espínola, F.J.; Arias García, M.; de Almeida, L.; Bonilla Aguirre, M.; Guerendiain, M.; Castellote Bargalló, A.I.; Segura Moreno, M.; et al. Association of maternal weight with FADS and ELOVL genetic variants and fatty acid levels- The PREOBE follow-up. PLoS ONE 2017, 12, e0179135. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.C.; Chang, W.T.; Chang, H.Y.; Chung, H.F.; Chen, F.P.; Huang, Y.F.; Hsu, C.C.; Hwang, S.J. FADS Gene Polymorphisms, Fatty Acid Desaturase Activities, and HDL-C in Type 2 Diabetes. Int. J. Environ. Res. Public Health 2017, 14, 572. [Google Scholar] [CrossRef] [PubMed]
- Howard, T.D.; Mathias, R.A.; Seeds, M.C.; Herrington, D.M.; Hixson, J.E.; Shimmin, L.C.; Hawkins, G.A.; Sellers, M.; Ainsworth, H.C.; Sergeant, S.; et al. DNA methylation in an enhancer region of the FADS cluster is associated with FADS activity in human liver. PLoS ONE 2014, 9, e97510. [Google Scholar] [CrossRef] [PubMed]
- Rahbar, E.; Ainsworth, H.C.; Howard, T.D.; Hawkins, G.A.; Ruczinski, I.; Mathias, R.; Seeds, M.C.; Sergeant, S.; Hixson, J.E.; Herrington, D.M.; et al. Uncovering the DNA methylation landscape in key regulatory regions within the FADS cluster. PLoS ONE 2017, 12, e0180903. [Google Scholar] [CrossRef] [PubMed]
- Rahbar, E.; Waits, C.M.K.; Kirby, E.H.; Miller, L.R.; Ainsworth, H.C.; Cui, T.; Sergeant, S.; Howard, T.D.; Langefeld, C.D.; Chilton, F.H. Allele-specific methylation in the FADS genomic region in DNA from human saliva, CD4+ cells, and total leukocytes. Clin. Epigenetics 2018, 10, 46. [Google Scholar] [CrossRef]
- He, Z.; Zhang, R.; Jiang, F.; Zhang, H.; Zhao, A.; Xu, B.; Jin, L.; Wang, T.; Jia, W.; Jia, W.; et al. FADS1-FADS2 genetic polymorphisms are associated with fatty acid metabolism through changes in DNA methylation and gene expression. Clin. Epigenetics 2018, 10, 113. [Google Scholar] [CrossRef] [PubMed]
- Anto, L.; Warykas, S.W.; Torres-Gonzalez, M.; Blesso, C.N. Milk Polar Lipids: Underappreciated Lipids with Emerging Health Benefits. Nutrients 2020, 12, 1001. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Goulding, E.H.; Zang, K.; Cepoi, D.; Cone, R.D.; Jones, K.R.; Tecott, L.H.; Reichardt, L.F. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat. Neurosci. 2003, 6, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.; Yeo, G.; Hung, C.; Keogh, J.; Clayton, P.; Banerjee, K.; McAulay, A.; O’Rahilly, S.; Farooqi, I.S. Functional characterization of human NTRK2 mutations identified in patients with severe early-onset obesity. Int. J. Obes. 2007, 31, 359–364. [Google Scholar] [CrossRef]


{kind=link}
| GOLDN | HAPI Heart | |
|---|---|---|
| N | 668 | 639 |
| Sex, Male | 50% | 56% |
| Age (years) | 49 (16) | 43 (13) |
| BMI (kg/m2) | 28 (5) | 26 (4) |
| Cholesterol (mg/dL) | 193 (40) | 207 (46) |
| Fasting LDL (mg/dL) | 124 (32) | 138 (42) |
| Fasting HDL (mg/dL) | 47 (13) | 55 (14) |
| Fasting Triglycerides (mg/dL) | 141 (100) | 67 (40) |
| 6 h Triglycerides (mg/dL) | 241 (186) | 173 (127) |
| Data recorded as Mean (SD) or % | ||
| Lipid | Variant Information | GWAS Discovery Results in GOLDN | Replication Results in HAPI Heart | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CHR/SNP | BP Build38 | Gene | Functional Annotation | EA/RA/EAF | SNP Beta (SE) | p-Value * | EA/RA/EAF | SNP Beta (SE) | p-Value | |
| FA (20:4) | 11/rs174547 | 61,803,311 | FADS1 | intronic | C/T/0.29 | −0.26 (0.05) | 1.68 × 10−7 | C/T/0.23 | −1.20 × 10−1 (0.04) | 1.52 × 10−3 |
| FA (20:4) | 11/rs174577 | 61,837,342 | FADS2 | intronic | A/C/0.28 | −0.26 (0.05) | 1.39 × 10−7 | A/C/0.25 | −1.08 × 10-1 (0.04) | 3.27 × 10−3 |
| FA (20:4) | 11/rs174583 | 61,842,278 | FADS2 | intronic | T/C/0.28 | −0.24 (0.04) | 7.03 × 10−7 | T/C/0.25 | −1.08 × 10−1 (0.04) | 3.27 × 10−3 |
| FA (20:4) | 11/rs4246215 | 61,796,827 | FEN1 | UTR3 | T/G/0.29 | −0.25 (0.04) | 1.22 × 10−7 | T/G/0.26 | −8.77 × 10−2 (0.04) | 1.84 × 10−2 |
| FA (20:4) | 1/rs12042035 | 200,710,954 | LOC101929224-CAMSAP2 | intergenic | G/T/0.01 | 0.76 (0.12) | 6.31 × 10−9 | - | - | - |
| LPE (16:0) | 9/rs12552641 | 84,838,137 | NTRK2 | intronic | G/C/0.01 | 1.83 (0.31) | 4.38 × 10−8 | - | - | - |
| LPE (16:0) | 9/rs12555204 | 84,838,239 | NTRK2 | intronic | G/A/0.01 | 1.83 (0.31) | 4.43 × 10−8 | - | - | - |
| LPE (18:0) | 9/rs12552641 | 84,838,137 | NTRK2 | intronic | G/C/0.01 | 1.67 (0.31) | 3.78 × 10−7 | - | - | - |
| LPE (18:0) | 9/rs12555204 | 84,838,239 | NTRK2 | intronic | G/A/0.01 | 1.67 (0.31) | 3.88 × 10−7 | - | - | - |
| LPE (22:6) | 9/rs12555204 | 84,838,239 | NTRK2 | intronic | G/A/0.01 | 1.68 (0.32) | 9.49 × 10−7 | - | - | - |
| LPE (22:6) | 9/rs12552641 | 84,838,137 | NTRK2 | intronic | G/C/0.01 | 1.68 (0.32) | 9.50 × 10−7 | - | - | - |
| PC (36:5) A | 9/rs10965980 | 23,625,667 | LOC101929563 | ncRNA_intronic | T/C/0.01 | 1.53 (0.23) | 2.60 × 10−9 | T/C/0.02 | 1.21 × 10−1 (0.18) | 5.14 × 10−1 |
| PC (36:5) A | 9/rs10965998 | 23,634,930 | LOC101929563 | ncRNA_intronic | G/A/0.01 | 1.53 (0.23) | 2.90 × 10−9 | G/A/0.02 | 1.21 × 10−1 (0.18) | 5.14 × 10−1 |
| PC (36:5) A | 9/rs10965994 | 23,634,530 | LOC101929563 | ncRNA_intronic | A/T/0.01 | 1.40 (0.25) | 8.09 × 10−7 | A/T/0.02 | 1.21 × 10−1 (0.18) | 5.14 × 10−1 |
| PC (36:5) A | 9/rs17836336 | 23,619,256 | LOC101929563 | ncRNA_intronic | G/C/0.01 | 1.55 (0.26) | 1.34 × 10−7 | - | - | - |
| PC (36:5) A | 1/rs431675 | 229,169,403 | RHOU;MIR4454 | intergenic | C/T/0.38 | 0.25 (0.04) | 3.51 × 10−9 | C/T/0.36 | −3.05 × 10−2 (0.18) | 5.62 × 10−1 |
| Lipid | Gene | CpG | CHR:BP (hg38) | CpG Beta (SE) | CpG p-Value | SNP | Distance from CpG | meQTL Beta (se) | Z | meQTL p-Value |
|---|---|---|---|---|---|---|---|---|---|---|
| LPE (16:0) | NTRK2 | cg13504245 | 9:84667694 | −2.41 (0.90) | 7.44 × 10−3 | rs12552641 | +170,443 | −1.50 × 10−2 (1.28 × 10−2) | −1.17 | 2.42 × 10−1 |
| LPE (18:0) | NTRK2 | cg14447193 | 9:84818948 | −2.45 (0.85) | 5.47 × 10−3 | rs12552641 | +19,189 | −3.32 × 10−2 (1.35 × 10−2) | −2.45 | 1.46 × 10−2 |
| FA (20:4) | FADS1 | cg25448062 | 11:61812645 | −3.46 (1.32) | 8.83 × 10−3 | rs174547 | +9334 | −6.40 × 10−3 (6.59 × 10−4) | −9.70 | 6.79 × 10−21 |
| FA (20:4) | FADS1 | cg07689907 | 11:61815101 | 1.50 (0.45) | 9.05 × 10−4 | rs174547 | −11,790 | −4.09 × 10-2(3.49 × 10−3) | −11.71 | 6.87 × 10-29 |
| FA (20:4) | FADS1 | cg14725641 | 11:61815290 | 7.20 (2.63) | 6.22 × 10−3 | rs174547 | −11,979 | −4.58 × 10−3 (5.44 × 10−4) | −8.43 | 2.22 × 10−16 |
| FA (20:4) | FADS1 | cg25326896 | 11:61815312 | 8.59 (2.81) | 2.26 × 10−3 | rs174547 | −12,001 | −3.42 × 10−3 (5.48 × 10−4) | -6.24 | 7.97 × 10−10 |
| FA (20:4) | FADS1 | cg12517394 | 11:61815322 | 5.60 (2.14) | 9.07 × 10−3 | rs174547 | −12,011 | −4.21 × 10−3 (6.51 × 10−4) | −6.47 | 1.97 × 10−10 |
| FA (20:4) | FADS1 | cg15598662 | 11:61815417 | 7.19 (2.13) | 7.28 × 10−4 | rs174547 | −12,106 | 3.42 × 10−3 (1.12 × 10-3) | 3.04 | 2.44 × 10−3 |
| FA (20:4) | FADS1 | cg23992449 | 11:61816483 | 14.82 (5.08) | 3.57 × 10−3 | rs174547 | −13,172 | −7.17 × 10−4 (2.60 × 10−4) | −2.75 | 6.06 × 10−3 |
| FA (20:4) | FADS1 | cg27173322 | 11:61816639 | 17.47 (4.99) | 4.68 × 10−4 | rs174547 | −13,328 | −6.15 × 10−4 (2.72 × 10−4) | −2.26 | 2.42 × 10−2 |
| FA (20:4) | FADS2 | cg27386326 | 11:61820507 | −2.03 (0.41) | 5.18 × 10−7 | rs174577 | +16,835 | 4.57 × 10−2 (4.49 × 10−3) | 10.18 | 1.01 × 10−22 |
| FA (20:4) | FADS2 | cg06781209 | 11:61827524 | 1.16 (0.41) | 4.69 × 10−3 | rs174577 | +9818 | −2.92 × 10−2 (4.44 × 10−3) | −6.58 | 9.61 × 10−11 |
| FA (20:4) | FADS2 | cg16576620 | 11:61828596 | 9.80 (3.45) | 4.52 × 10−3 | rs174577 | +8746 | −9.60 × 10−4 (4.01 × 10−4) | −2.40 | 1.69 × 10−2 |
| FA (20:4) | FADS2 | cg19610905 | 11:61828860 | 7.06 (1.59) | 8.68 × 10−6 | rs174577 | +8482 | −1.56 × 10−2 (6.45 × 10−4) | −24.20 | 4.07 × 10−93 |
| FA (20:4) | FADS2 | cg00603274 | 11:61829153 | 1.68 (0.55) | 2.41 × 10−3 | rs174577 | +8189 | −2.15 × 10−2 (2.91 × 10−3) | −7.38 | 4.87 × 10−13 |
| FA (20:4) | FADS2 | cg14911132 | 11:61829282 | 2.86 (1.02) | 4.97 × 10−3 | rs174577 | +8060 | −7.46 × 10−3 (1.48 × 10−3) | −5.05 | 5.86 × 10−7 |
| Outcome | Treatment | Mediator | Effect | Estimate | SE | 95% CI | Z | p-Value | |
|---|---|---|---|---|---|---|---|---|---|
| Lower | Upper | ||||||||
| FA (20:4) (6hr diff) | rs174577 | cg27386326 | Total Effect | −0.24 | 0.04 | −0.32 | −0.15 | −5.39 | <0.0001 |
| Natural Direct Effect (Treatment) | −0.18 | 0.06 | −0.29 | −0.06 | −2.98 | 0.0029 | |||
| Natural Indirect Effect (Mediator) | −0.06 | 0.04 | −0.14 | 0.02 | −1.57 | 0.1173 | |||
| Percentage Mediated | 25.95 | 17.24 | −7.84 | 59.74 | 1.51 | 0.1322 | |||
| cg19610905 | Total Effect | −0.24 | 0.04 | −0.32 | −0.15 | −5.39 | <0.0001 | ||
| Natural Direct Effect (Treatment) | −0.18 | 0.05 | −0.27 | −0.09 | −3.83 | 0.0001 | |||
| Natural Indirect Effect (Mediator) | −0.06 | 0.02 | −0.09 | −0.02 | −3.18 | 0.0015 | |||
| Percentage Mediated | 24.17 | 8.69 | 7.15 | 41.20 | 2.78 | 0.0054 | |||
| rs174547 | cg07689907 | Total Effect | −0.24 | 0.04 | −0.32 | −0.15 | −5.36 | <0.0001 | |
| Natural Direct Effect (Treatment) | −0.22 | 0.05 | −0.31 | −0.12 | −4.50 | <0.0001 | |||
| Natural Indirect Effect (Mediator) | −0.02 | 0.02 | −0.06 | 0.02 | −0.96 | 0.3394 | |||
| Percentage Mediated | 7.99 | 8.49 | −8.65 | 24.62 | 0.94 | 0.3468 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Irvin, M.R.; Montasser, M.E.; Kind, T.; Fan, S.; Barupal, D.K.; Patki, A.; Tanner, R.M.; Armstrong, N.D.; Ryan, K.A.; Claas, S.A.; et al. Genomics of Postprandial Lipidomics in the Genetics of Lipid-Lowering Drugs and Diet Network Study. Nutrients 2021, 13, 4000. https://doi.org/10.3390/nu13114000
Irvin MR, Montasser ME, Kind T, Fan S, Barupal DK, Patki A, Tanner RM, Armstrong ND, Ryan KA, Claas SA, et al. Genomics of Postprandial Lipidomics in the Genetics of Lipid-Lowering Drugs and Diet Network Study. Nutrients. 2021; 13(11):4000. https://doi.org/10.3390/nu13114000
Chicago/Turabian StyleIrvin, Marguerite R., May E. Montasser, Tobias Kind, Sili Fan, Dinesh K. Barupal, Amit Patki, Rikki M. Tanner, Nicole D. Armstrong, Kathleen A. Ryan, Steven A. Claas, and et al. 2021. "Genomics of Postprandial Lipidomics in the Genetics of Lipid-Lowering Drugs and Diet Network Study" Nutrients 13, no. 11: 4000. https://doi.org/10.3390/nu13114000
APA StyleIrvin, M. R., Montasser, M. E., Kind, T., Fan, S., Barupal, D. K., Patki, A., Tanner, R. M., Armstrong, N. D., Ryan, K. A., Claas, S. A., O’Connell, J. R., Tiwari, H. K., & Arnett, D. K. (2021). Genomics of Postprandial Lipidomics in the Genetics of Lipid-Lowering Drugs and Diet Network Study. Nutrients, 13(11), 4000. https://doi.org/10.3390/nu13114000