
Inhibition of Soluble Epoxide Hydrolase Is Protective against the Multiomic Effects of a High Glycemic Diet on Brain Microvascular Inflammation and Cognitive Dysfunction

 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
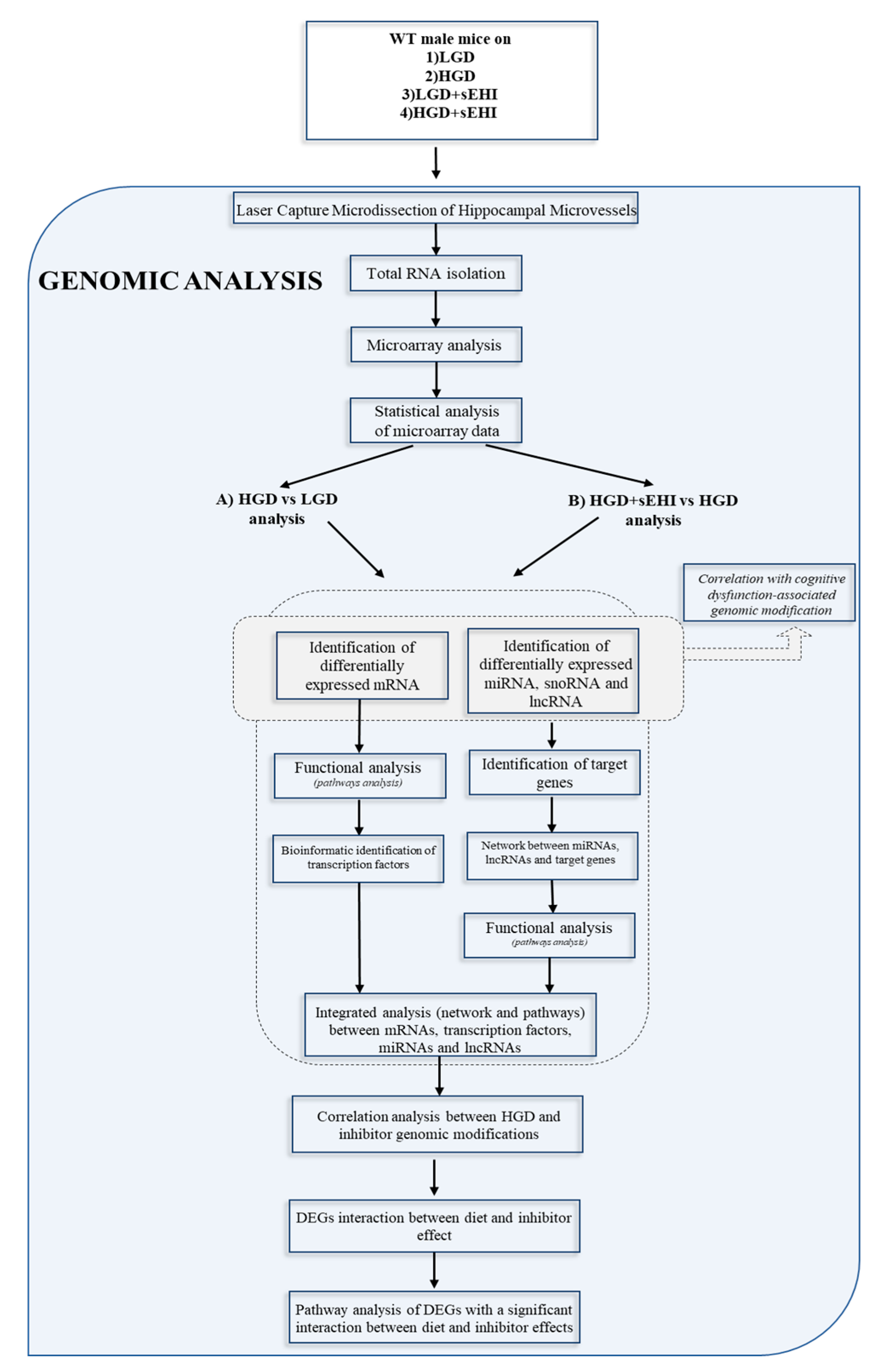
2. Materials and Methods
2.1. Experimental Animals and Soluble Epoxide Hydrolase Inhibitor (sEHI) Treatment
2.2. Serum Lipid, Glucose, and Insulin Assays
2.3. Isolation and Cryosection of Murine Brain Hippocampus
2.4. Laser Capture Microdissection (LCM) of Hippocampal Microvessels
2.5. RNA Extraction from Laser Captured Brain Microvessels
2.6. Microarray Hybridization and Transcriptome Analysis
2.7. Bioinformatic Analysis
2.8. Statistical Methods
3. Results
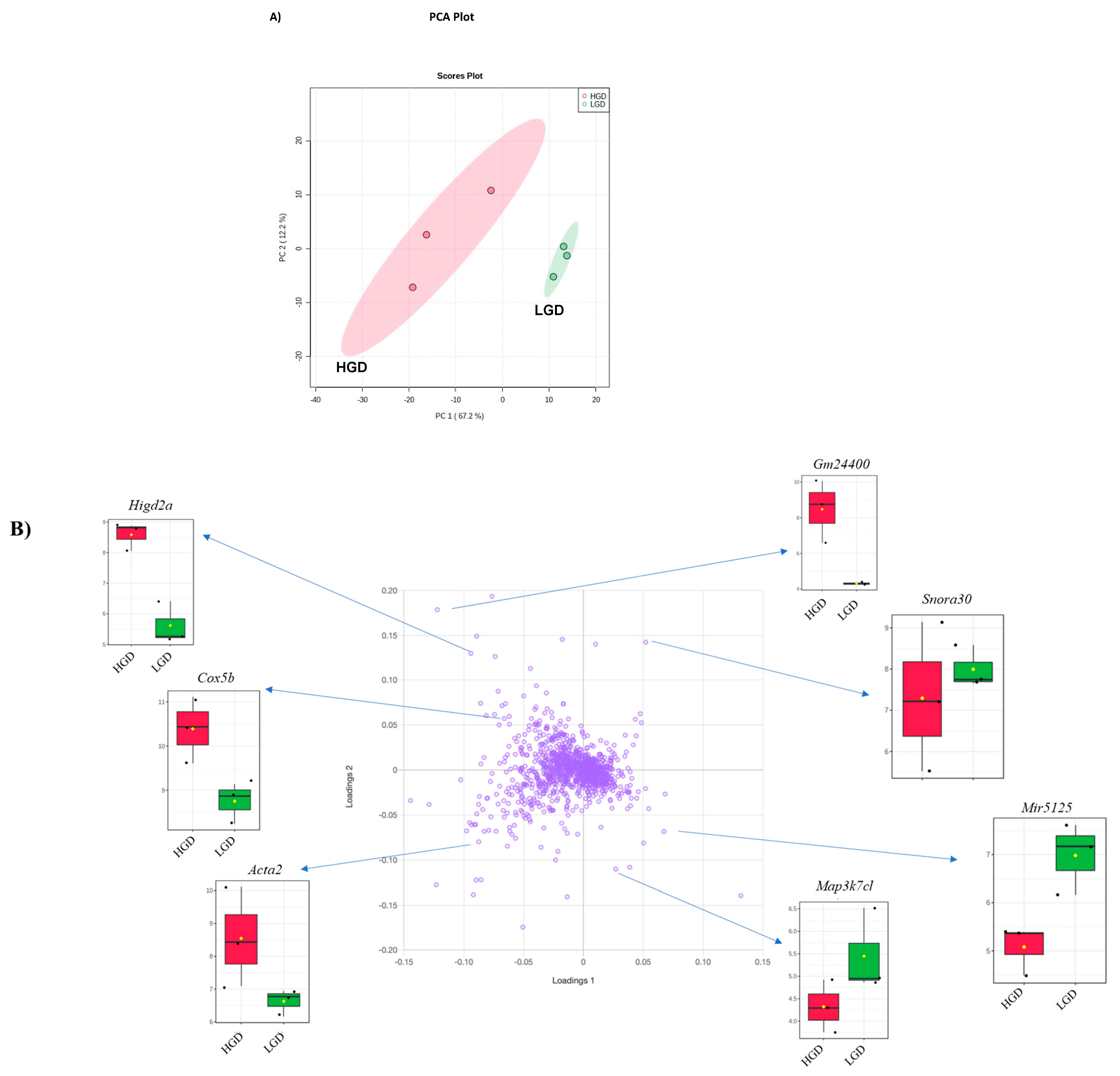
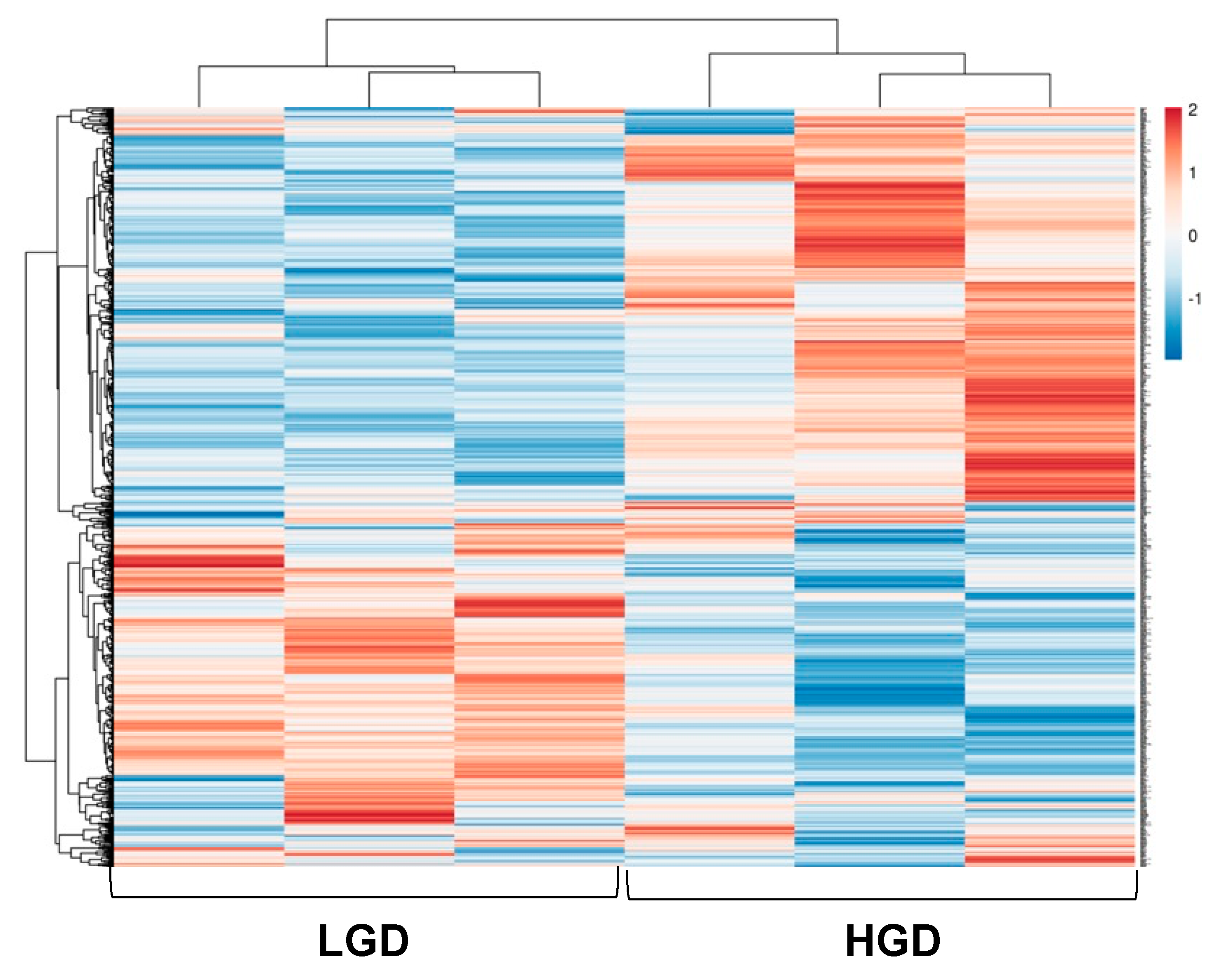
3.1. Effect of the High Glycemic Diet on the Hippocampal Microvascular Genome
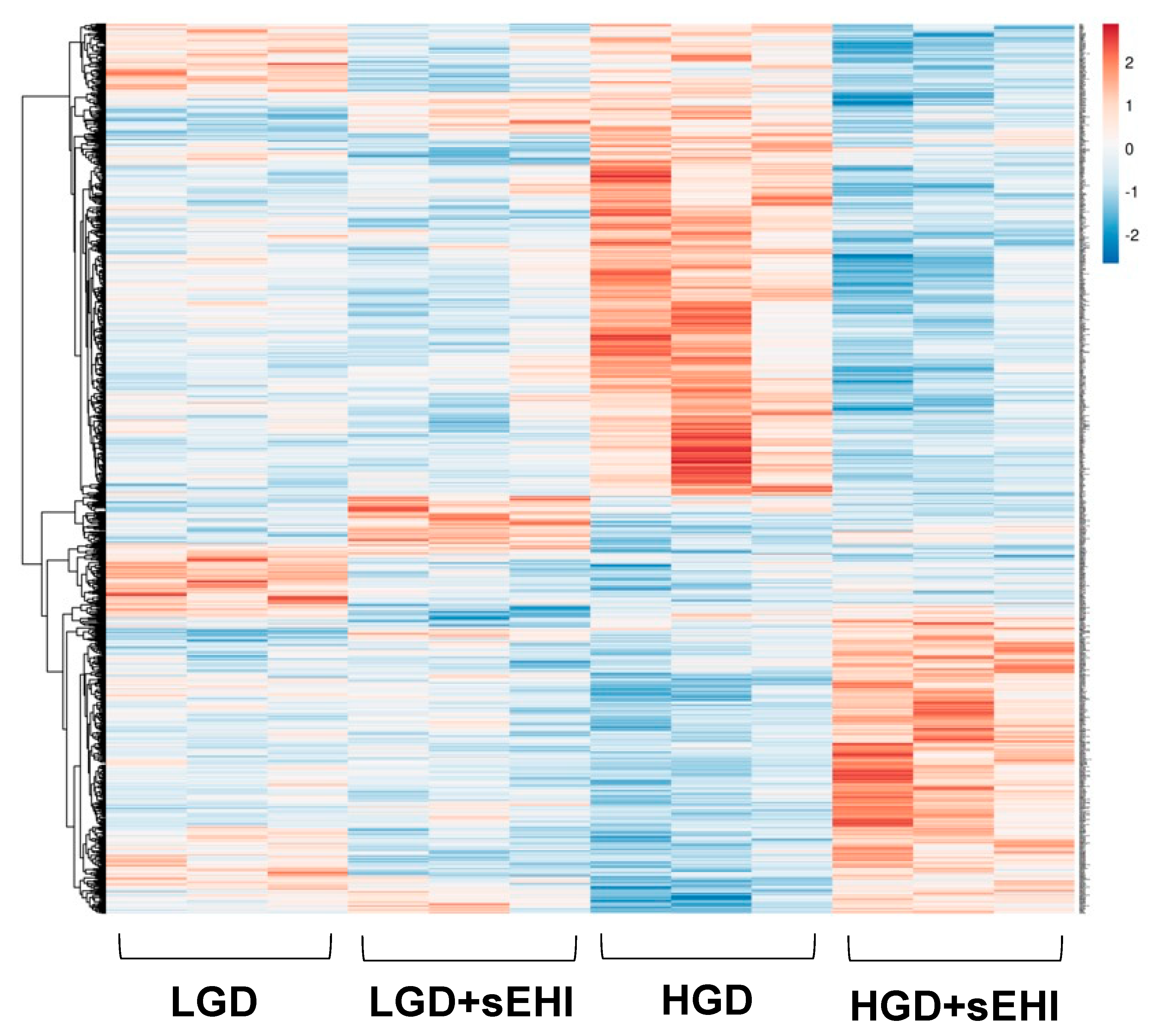
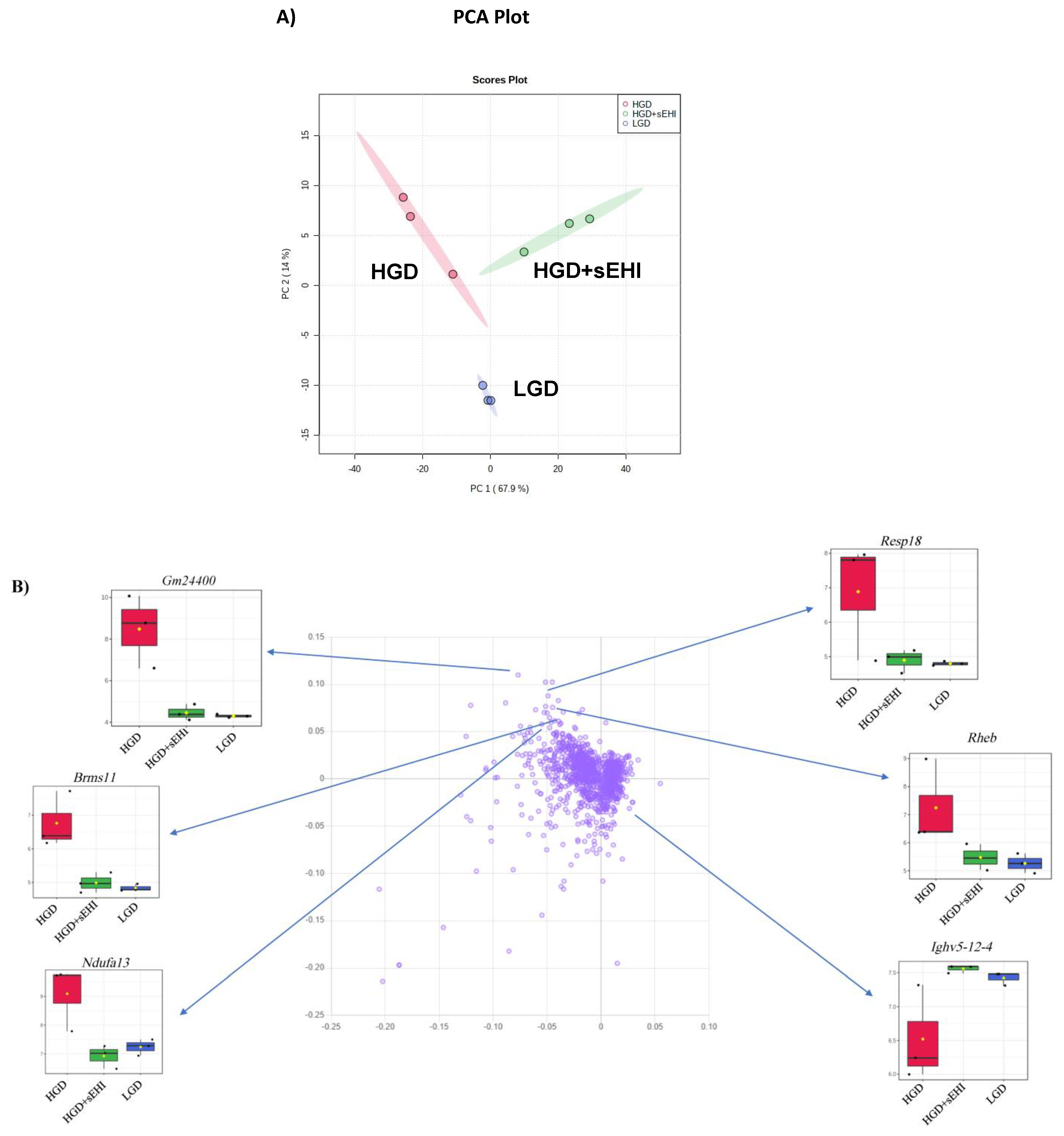
3.1.1. Global Gene Expression and Hierarchical Clustering
3.1.2. Differential Gene Expression
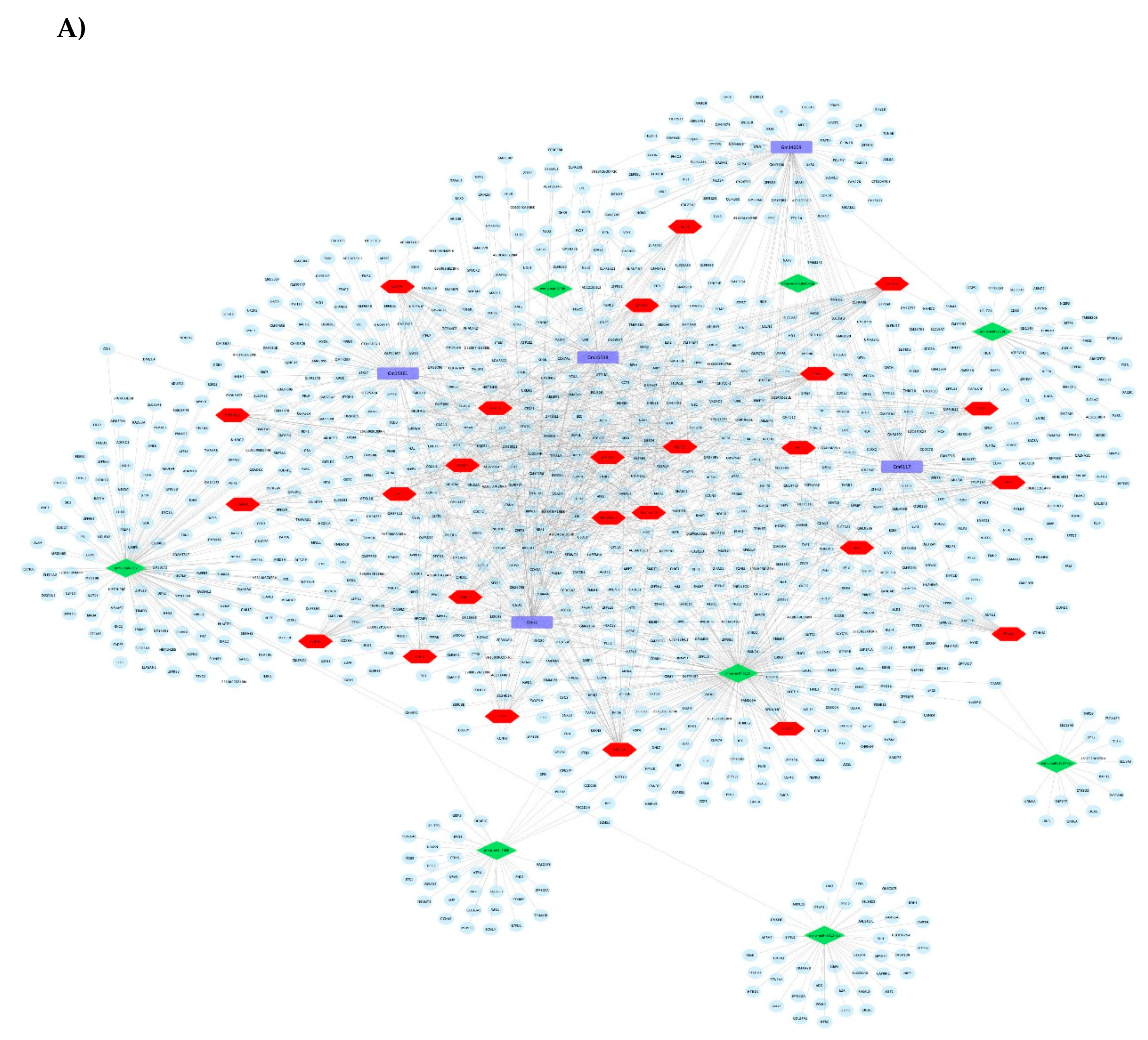
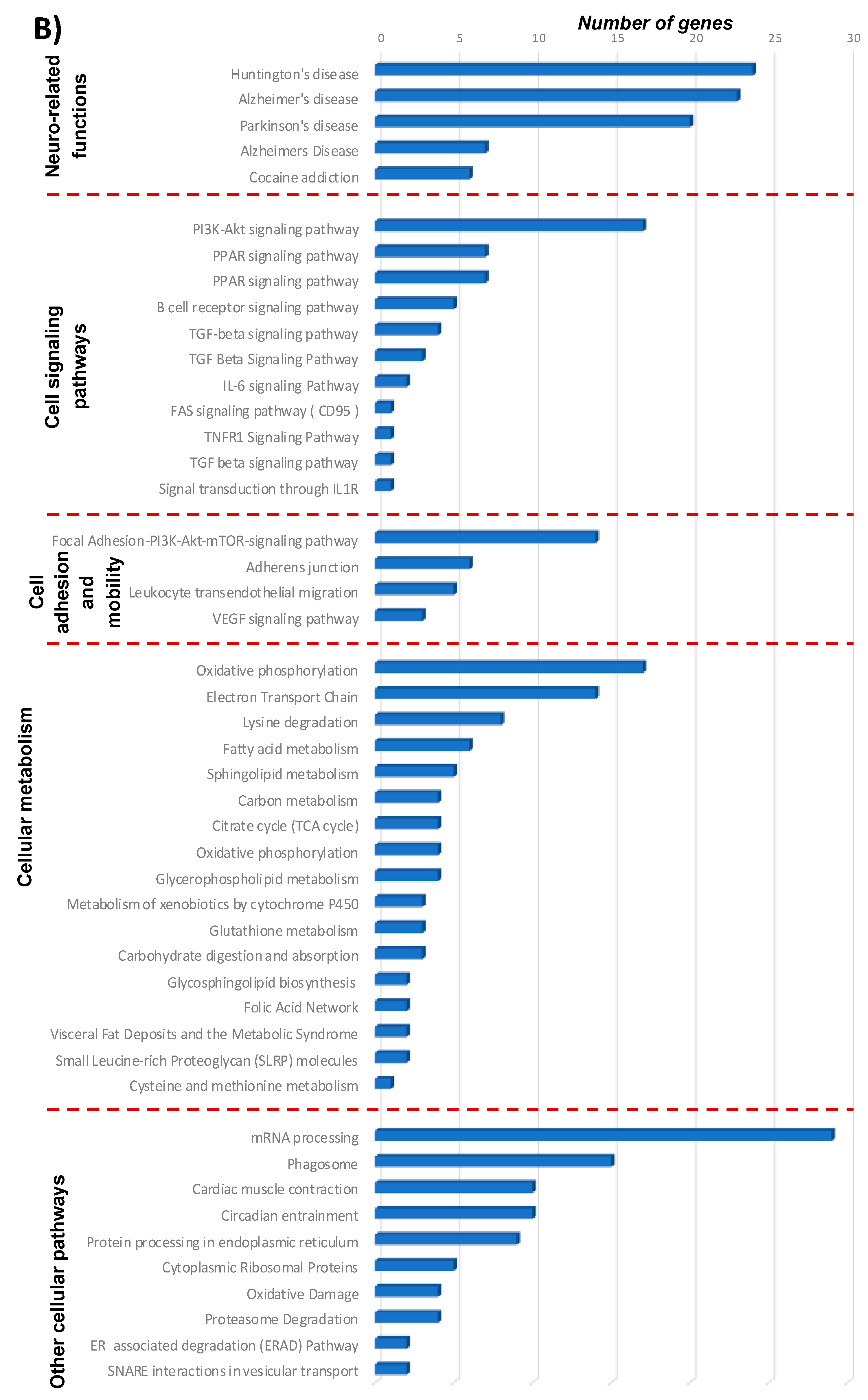
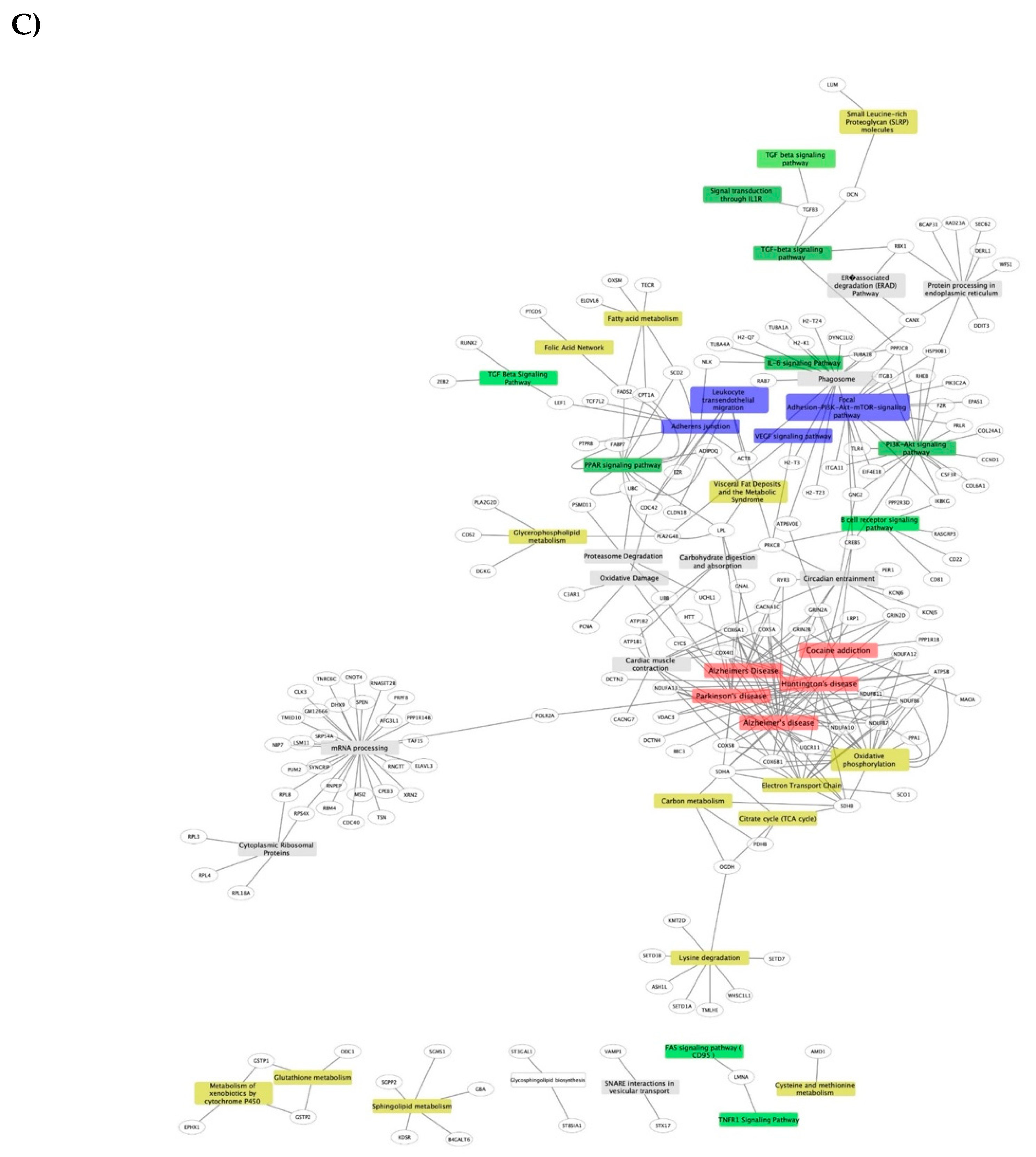
3.1.3. Pathways and Networks for Coding and Non-Coding Differentially Expressed Genes
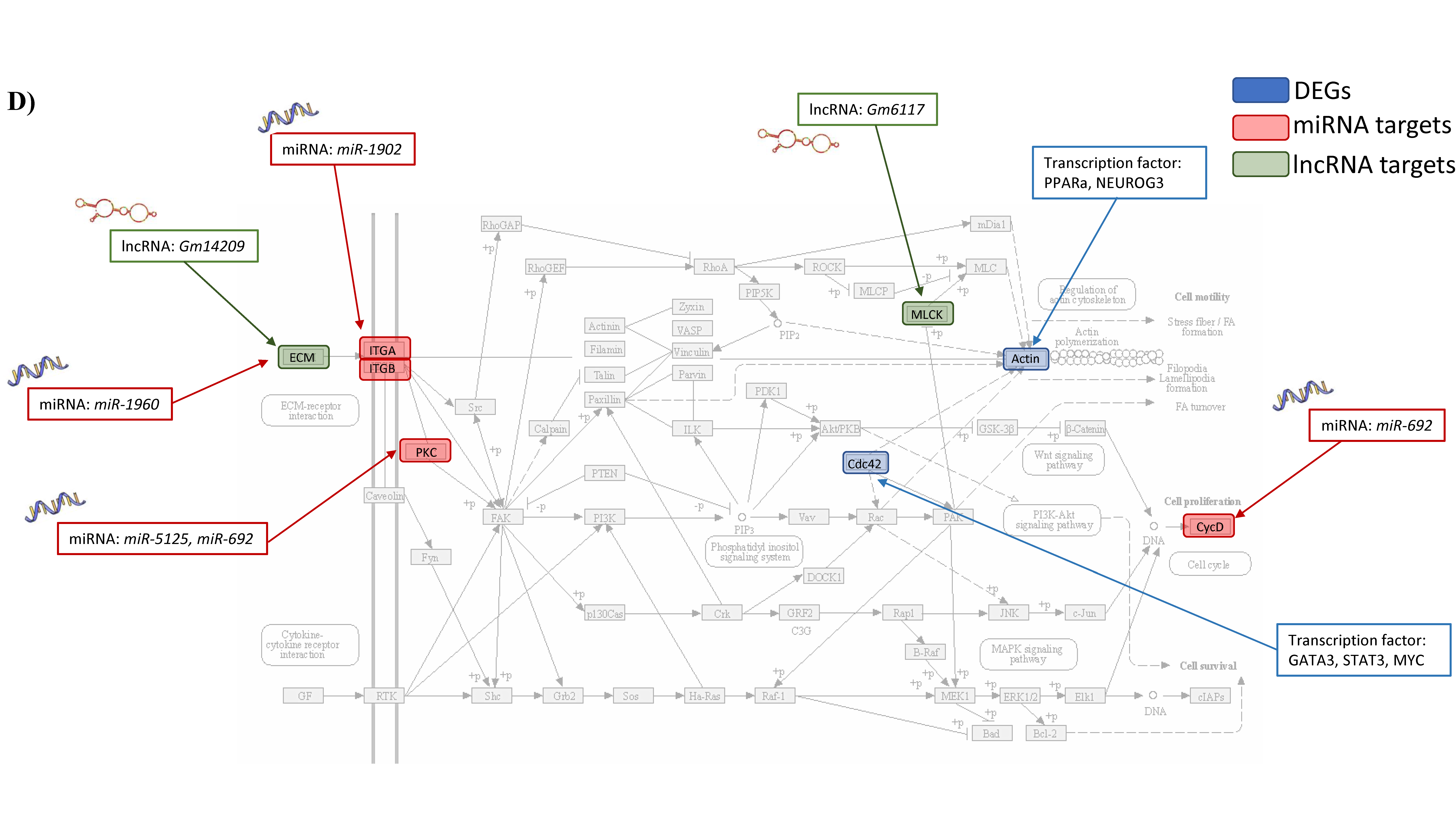
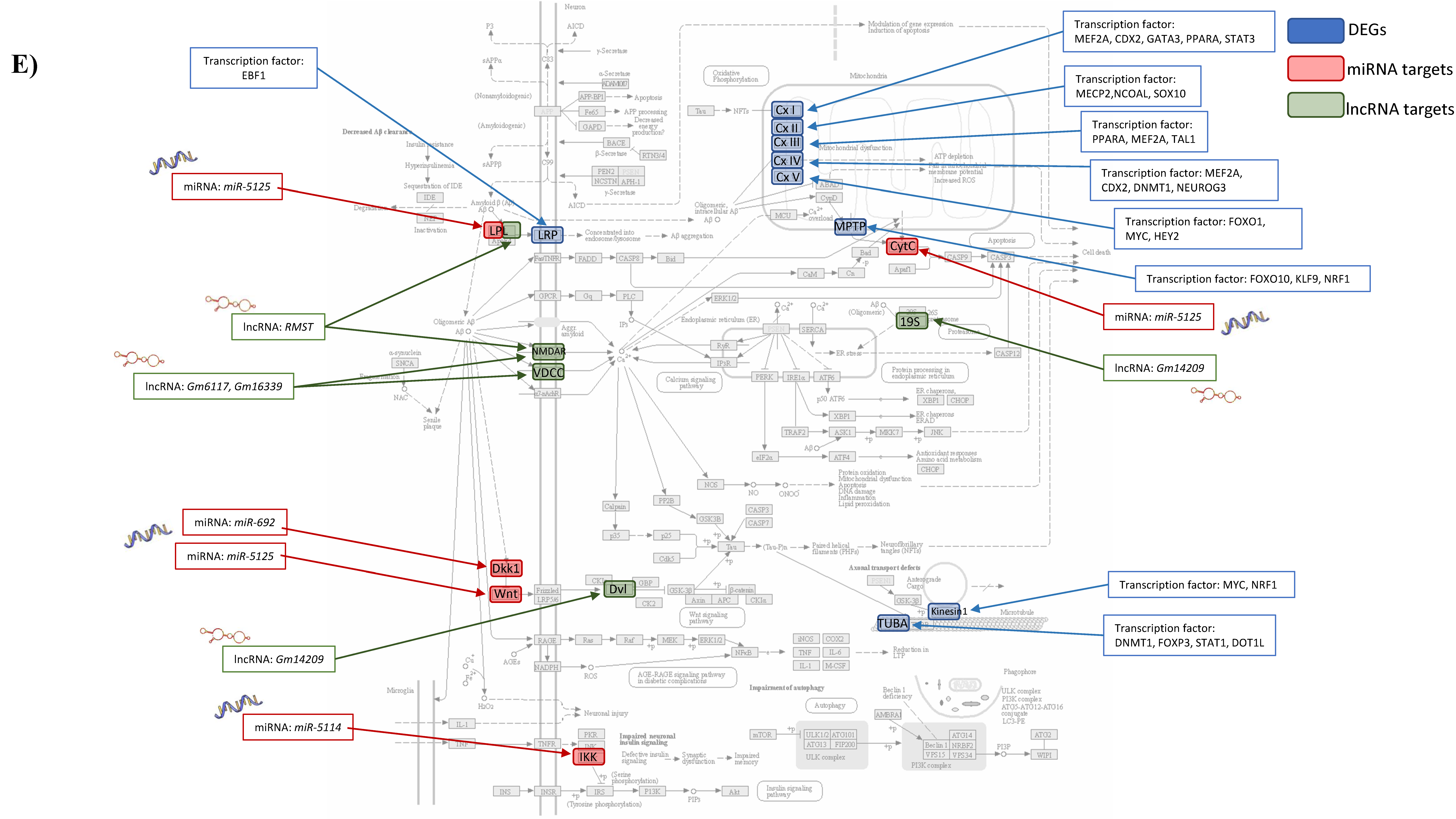
3.1.4. Integrated Analysis of Differentially Expressed Genes, Key Pathways and Networks
3.2. Effect of the Soluble Epoxide Hydrolase Inhibitor (sEHI) on the Hippocampal Microvascular Genome of Mice fed the High Glycemic Diet
3.3. Correlation of Genomics with Human Alzheimer’s Disease Data
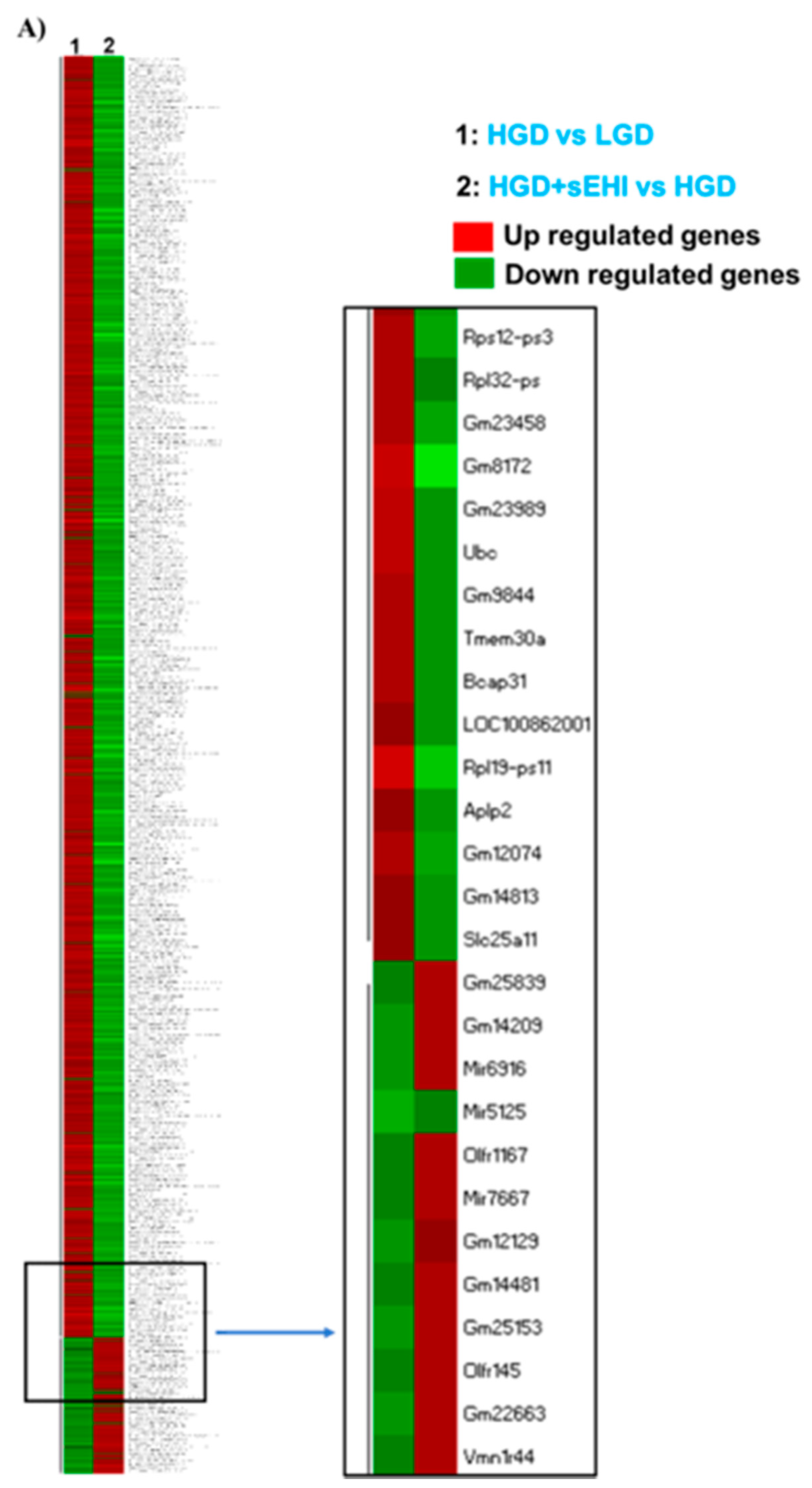
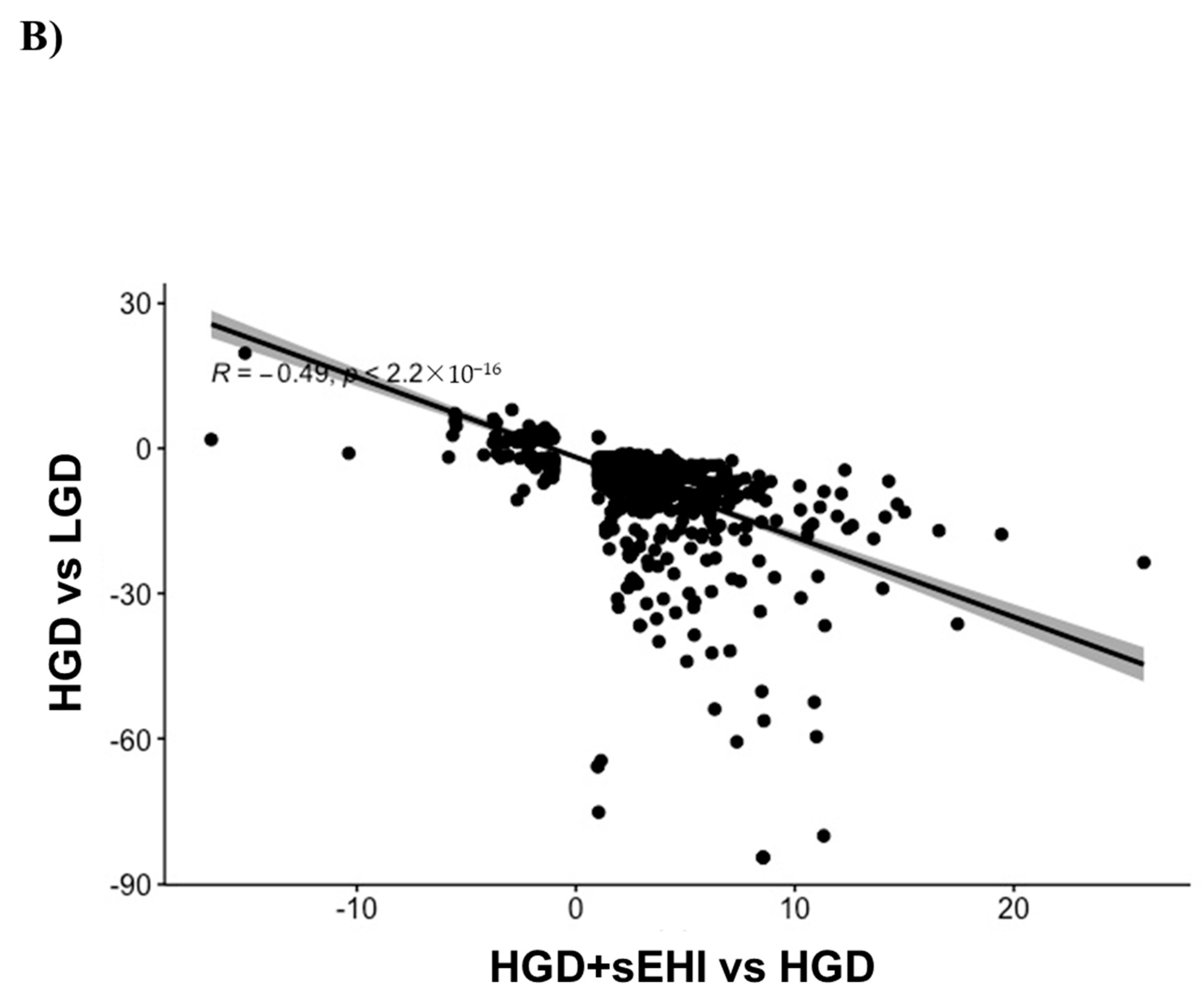
3.4. Interaction of Effects of the Glycemic Diet and the Soluble Epoxide Hydrolase Inhibitor
4. Discussion
5. Conclusions and Clinical Implications
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duplantier, S.C.; Gardner, C.D. A Critical Review of the Study of Neuroprotective Diets to Reduce Cognitive Decline. Nutrients 2021, 13, 2264. [Google Scholar] [CrossRef]
- Wiȩckowska-Gacek, A.; Mietelska-Porowska, A.; Chutorański, D.; Wydrych, M.; Długosz, J.; Wojda, U. Western Diet Induces Impairment of Liver-Brain Axis Accelerating Neuroinflammation and Amyloid Pathology in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 654509. [Google Scholar] [CrossRef]
- Leigh, S.J.; Morris, M.J. Diet, Inflammation and the gut microbiome: Mechanisms for obesity-associated cognitive impairment. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165767. [Google Scholar] [CrossRef]
- Buie, J.J.; Watson, L.S.; Smith, C.J.; Sims-Robinson, C. Obesity-related cognitive impairment: The role of endothelial dysfunction. Neurobiol. Dis. 2019, 132, 104580. [Google Scholar] [CrossRef]
- Rutkowsky, J.M.; Lee, L.L.; Puchowicz, M.; Golub, M.S.; Befroy, D.E.; Wilson, D.W.; Anderson, S.; Cline, G.; Bini, J.; Borkowski, K.; et al. Reduced cognitive function, increased blood-brain-barrier transport and inflammatory responses, and altered brain metabolites in LDLr -/- and C57BL/6 mice fed a western diet. PLoS ONE 2018, 13, e0191909. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, J.C.; Killcross, A.S.; Jenkins, T.A. Obesity and cognitive decline: Role of inflammation and vascular changes. Front. Neurosci. 2014, 8, 375. [Google Scholar] [CrossRef] [PubMed]
- Gorelick, P.B.; Counts, S.E.; Nyenhuis, D. Vascular cognitive impairment and dementia. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2016, 1862, 860–868. [Google Scholar] [CrossRef]
- Aung, H.H.; Altman, R.; Nyunt, T.; Kim, J.; Nuthikattu, S.; Budamagunta, M.; Voss, J.C.; Wilson, D.; Rutledge, J.C.; Villablanca, A.C. Lipotoxic brain microvascular injury is mediated by activating transcription factor 3-dependent inflammatory and oxidative stress pathways. J. Lipid Res. 2016, 57, 955–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuthikattu, S.; Milenkovic, D.; Rutledge, J.; Villablanca, A. The Western Diet Regulates Hippocampal Microvascular Gene Expression: An Integrated Genomic Analyses in Female Mice. Sci Rep. 2019, 9, 19058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuthikattu, S.; Milenkovic, D.; Rutledge, J.C.; Villablanca, A.C. Lipotoxic Injury Differentially Regulates Brain Microvascular Gene Expression in Male Mice. Nutrients 2020, 12, 1771. [Google Scholar] [CrossRef]
- Nuthikattu, S.; Milenkovic, D.; Rutledge, J.C.; Villablanca, A.C. Sex-Dependent Molecular Mechanisms of Lipotoxic Injury in Brain Microvasculature: Implications for Dementia. Int. J. Mol. Sci. 2020, 21, 8146. [Google Scholar] [CrossRef] [PubMed]
- Seetharaman, S.; Andel, R.; McEvoy, C.; Dahl Aslan, A.K.; Finkel, D.; Pedersen, N.L. Blood glucose, Diet-based glycemic load and cognitive aging among dementia-free older adults. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 471–479. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.K.; Sullivan, D.K.; Swerdlow, R.H.; Vidoni, E.D.; Morris, J.K.; Mahnken, J.D.; Burns, J.M. A high-glycemic diet is associated with cerebral amyloid burden in cognitively normal older adults. Am. J. Clin. Nutr. 2017, 106, 1463–1470. [Google Scholar] [CrossRef]
- Gentreau, M.; Raymond, M.; Chuy, V.; Samieri, C.; Féart, C.; Berticat, C.; Artero, S. High Glycemic Load Is Associated with Cognitive Decline in Apolipoprotein E ε4 Allele Carriers. Nutrients 2020, 12, 3619. [Google Scholar] [CrossRef]
- Saikrishna, K.; Kumari, R.; Chaitanya, K.; Biswas, S.; Nayak, P.G.; Mudgal, J.; Kishore, A.; Nandakumar, K. Combined Administration of Monosodium Glutamate and High Sucrose Diet Accelerates the Induction of Type 2 Diabetes, Vascular Dysfunction, and Memory Impairment in Rats. J. Environ. Pathol. Toxicol. Oncol. 2018, 37, 63–80. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.; Dogra, V.R.; Reichelt, A.C. High-sucrose diets in male rats disrupt aspects of decision making tasks, motivation and spatial memory, but not impulsivity measured by operant delay-discounting. Behav. Brain Res. 2017, 327, 144–154. [Google Scholar] [CrossRef]
- Żebrowska, E.; Chabowski, A.; Zalewska, A.; Maciejczyk, M. High-Sugar Diet Disrupts Hypothalamic but Not Cerebral Cortex Redox Homeostasis. Nutrients 2020, 12, 3181. [Google Scholar] [CrossRef]
- Pinto, B.A.; Melo, T.M.; Flister, K.F.; França, L.M.; Kajihara, D.; Tanaka, L.Y.; Laurindo, F.R.; Paes, A.M. Early and sustained exposure to high-sucrose diet triggers hippocampal ER stress in young rats. Metab. Brain Dis. 2016, 31, 917–927. [Google Scholar] [CrossRef]
- Yeh, S.H.; Shie, F.S.; Liu, H.K.; Yao, H.H.; Kao, P.C.; Lee, Y.H.; Chen, L.M.; Hsu, S.M.; Chao, L.J.; Wu, K.W.; et al. A high-sucrose diet aggravates Alzheimer’s disease pathology, attenuates hypothalamic leptin signaling, and impairs food-anticipatory activity in APPswe/PS1dE9 mice. Neurobiol. Aging 2020, 90, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Taha, A.Y.; Gao, F.; Ramadan, E.; Cheon, Y.; Rapoport, S.I.; Kim, H.W. Upregulated expression of brain enzymatic markers of arachidonic and docosahexaenoic acid metabolism in a rat model of the metabolic syndrome. BMC Neurosci. 2012, 13, 131. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Luo, G.; Zhang, L.F.; Geng, H.X. Neuroprotective effects of epoxyeicosatrienoic acids. Prostaglandins Other Lipid Mediat. 2018, 138, 9–14. [Google Scholar] [CrossRef]
- Zarriello, S.; Tuazon, J.P.; Corey, S.; Schimmel, S.; Rajani, M.; Gorsky, A.; Incontri, D.; Hammock, B.D.; Borlongan, C.V. Humble beginnings with big goals: Small molecule soluble epoxide hydrolase inhibitors for treating CNS disorders. Prog. Neurobiol. 2019, 172, 23–39. [Google Scholar] [CrossRef]
- Ren, Q.; Ma, M.; Yang, J.; Nonaka, R.; Yamaguchi, A.; Ishikawa, K.I.; Kobayashi, K.; Murayama, S.; Hwang, S.H.; Saiki, S.; et al. Soluble epoxide hydrolase plays a key role in the pathogenesis of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, E5815–E5823. [Google Scholar] [CrossRef] [Green Version]
- Nelson, J.W.; Young, J.M.; Borkar, R.N.; Woltjer, R.L.; Quinn, J.F.; Silbert, L.C.; Grafe, M.R.; Alkayed, N.J. Role of soluble epoxide hydrolase in age-related vascular cognitive decline. Prostaglandins Other Lipid Mediat. 2014, 113–115, 30–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, D.; Hennebelle, M.; Sahlas, D.J.; Ramirez, J.; Gao, F.; Masellis, M.; Cogo-Moreira, H.; Swartz, R.H.; Herrmann, N.; Chan, P.C.; et al. Soluble Epoxide Hydrolase-Derived Linoleic Acid Oxylipins in Serum Are Associated with Periventricular White Matter Hyperintensities and Vascular Cognitive Impairment. Transl. Stroke Res. 2019, 10, 522–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griñán-Ferré, C.; Codony, S.; Pujol, E.; Yang, J.; Leiva, R.; Escolano, C.; Puigoriol-Illamola, D.; Companys-Alemany, J.; Corpas, R.; Sanfeliu, C.; et al. Pharmacological Inhibition of Soluble Epoxide Hydrolase as a New Therapy for Alzheimer’s Disease. Neurotherapeutics 2020, 17, 1825–1835. [Google Scholar] [CrossRef]
- Simpkins, A.N.; Rudic, R.D.; Schreihofer, D.A.; Roy, S.; Manhiani, M.; Tsai, H.J.; Hammock, B.D.; Imig, J.D. Soluble epoxide inhibition is protective against cerebral ischemia via vascular and neural protection. Am. J. Pathol. 2009, 174, 2086–2095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaik, J.S.; Ahmad, M.; Li, W.; Rose, M.E.; Foley, L.M.; Hitchens, T.K.; Graham, S.H.; Hwang, S.H.; Hammock, B.D.; Poloyac, S.M. Soluble epoxide hydrolase inhibitor trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid is neuroprotective in rat model of ischemic stroke. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1605–H1613. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Tian, H.; Yao, E.; Tian, Y.; Zhang, H.; Xu, L.; Yu, Z.; Fang, Y.; Wang, W.; Du, P.; et al. Soluble epoxide hydrolase inhibition Promotes White Matter Integrity and Long-Term Functional Recovery after chronic hypoperfusion in mice. Sci. Rep. 2017, 7, 7758. [Google Scholar] [CrossRef] [Green Version]
- Hao, J.; Chen, Y.; Yao, E.; Liu, X. Soluble epoxide hydrolase inhibition alleviated cognitive impairments via NRG1/ErbB4 signaling after chronic cerebral hypoperfusion induced by bilateral carotid artery stenosis in mice. Brain Res. 2018, 1699, 89–99. [Google Scholar] [CrossRef]
- Matin, N.; Fisher, C.; Lansdell, T.A.; Hammock, B.D.; Yang, J.; Jackson, W.F.; Dorrance, A.M. Soluble epoxide hydrolase inhibition improves cognitive function and parenchymal artery dilation in a hypertensive model of chronic cerebral hypoperfusion. Microcirculation 2021, 28, e12653. [Google Scholar] [CrossRef] [PubMed]
- Minaz, N.; Razdan, R.; Hammock, B.D.; Goswami, S.K. An inhibitor of soluble epoxide hydrolase ameliorates diabetes-induced learning and memory impairment in rats. Prostaglandins Other Lipid Mediat. 2018, 136, 84–89. [Google Scholar] [CrossRef]
- Wu, J.; Fan, Z.; Zhao, Y.; Chen, Q.; Xiao, Q. Inhibition of soluble epoxide hydrolase (sEH) protects hippocampal neurons and reduces cognitive decline in type 2 diabetic mice. Eur. J. Neurosci. 2021, 53, 2532–2540. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Comerota, M.M.; Wan, D.; Chen, F.; Propson, N.E.; Hwang, S.H.; Hammock, B.D.; Zheng, H. An epoxide hydrolase inhibitor reduces neuroinflammation in a mouse model of Alzheimer’s disease. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Lee, H.T.; Lee, K.I.; Chen, C.H.; Lee, T.S. Genetic deletion of soluble epoxide hydrolase delays the progression of Alzheimer’s disease. J. Neuroinflamm. 2019, 16, 267. [Google Scholar] [CrossRef] [Green Version]
- Knierim, J.J. The hippocampus. Curr. Biol. 2015, 25, R1116–R1121. [Google Scholar] [CrossRef] [Green Version]
- Han, F. Cerebral microvascular dysfunction and neurodegeneration in dementia. Stroke Vasc. Neurol. 2019, 4, 105–107. [Google Scholar] [CrossRef] [Green Version]
- Gui, Y.J.; Yang, T.; Liu, Q.; Liao, C.X.; Chen, J.Y.; Wang, Y.T.; Hu, J.H.; Xu, D.Y. Soluble epoxide hydrolase inhibitors, t-AUCB, regulated microRNA-1 and its target genes in myocardial infarction mice. Oncotarget 2017, 8, 94635–94649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ball, H.J.; McParland, B.; Driussi, C.; Hunt, N.H. Isolating vessels from the mouse brain for gene expression analysis using laser capture microdissection. Brain Res. Brain Res. Protoc. 2002, 9, 206–213. [Google Scholar] [CrossRef]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Licursi, V.; Conte, F.; Fiscon, G.; Paci, P. MIENTURNET: An interactive web tool for microRNA-target enrichment and network-based analysis. BMC Bioinform. 2019, 20, 545. [Google Scholar] [CrossRef] [Green Version]
- Fukunaga, T.; Iwakiri, J.; Ono, Y.; Hamada, M. LncRRIsearch: A Web Server for lncRNA-RNA Interaction Prediction Integrated With Tissue-Specific Expression and Subcellular Localization Data. Front. Genet. 2019, 10, 462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rtools CBRC. Available online: http://rtools.cbrc.jp/cgi-bin/RNARNA/index.pl (accessed on 25 April 2021).
- GeneTrail2. Available online: https://genetrail2.bioinf.uni-sb.de (accessed on 2 June 2019).
- Stockel, D.; Kehl, T.; Trampert, P.; Schneider, L.; Backes, C.; Ludwig, N.; Gerasch, A.; Kaufmann, M.; Gessler, M.; Graf, N.; et al. Multi-omics enrichment analysis using the GeneTrail2 web service. Bioinformatics 2016, 32, 1502–1508. [Google Scholar] [CrossRef]
- Cytoscape. Available online: https://cytoscape.org/ (accessed on 2 June 2019).
- Cytoscape Network Analyzer Application. Available online: http://apps.cytoscape.org/apps/networkanalyzer (accessed on 2 March 2020).
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Su, G.; Morris, J.H.; Demchak, B.; Bader, G.D. Biological network exploration with Cytoscape 3. Curr. Protoc. Bioinform. 2014, 47, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Splitstackshape. Available online: https://github.com/mrdwab/splitstackshape (accessed on 2 March 2020).
- Rdata.table. Available online: https://github.com/Rdatatable/data.table (accessed on 2 March 2020).
- Dplyr Tidyverse. Available online: http://dplyr.tidyverse.org (accessed on 2 March 2020).
- Dplyr Tidyverse Github. Available online: https://github.com/tidyverse/dplyr (accessed on 2 March 2020).
- String Tidyverse. Available online: http://stringr.tidyverse.org (accessed on 2 March 2020).
- String Tidyverse Github. Available online: https://github.com/tidyverse/stringr (accessed on 2 March 2020).
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]
- PermutMatrix. Available online: http://www.atgc-montpellier.fr/permutmatrix/ (accessed on 2 June 2019).
- Caraux, G.; Pinloche, S. PermutMatrix: A graphical environment to arrange gene expression profiles in optimal linear order. Bioinformatics 2005, 21, 1280–1281. [Google Scholar] [CrossRef] [Green Version]
- ggpubr R package. Available online: http://www.sthda.com/english/articles/24-ggpubr-publication-ready-plots/78-perfect-scatter-plots-with-correlation-and-marginal-histograms/ (accessed on 25 July 2021).
- NCBI GEO2R. Available online: https://www.ncbi.nlm.nih.gov/geo/info/geo2r.html (accessed on 25 July 2021).
- Van Schothorst, E.M.; Bunschoten, A.; Schrauwen, P.; Mensink, R.P.; Keijer, J. Effects of a high-fat, low-versus high-glycemic index diet: Retardation of insulin resistance involves adipose tissue modulation. FASEB J. 2009, 23, 1092–1101. [Google Scholar] [CrossRef]
- Zhu, Y.; Smith, K.; Rowan, S.; Greenberg, A. The Short-Term Effect of a High-Glycemic Diet on Mouse Obesity and Intestinal Microbiota Composition. Curr. Dev. Nutr. 2020, 4 (Suppl. 2), 1602. [Google Scholar] [CrossRef]
- Sousa, L.G.O.d.; Marshall, A.G.; Norman, J.E.; Fuqua, J.D.; Lira, V.A.; Rutledge, J.C.; Bodine, S.C. The effects of diet composition and chronic obesity on muscle growth and function. J. Appl. Physiol. 2021, 130, 124–138. [Google Scholar] [CrossRef]
- Cho, K.A.; Ryu, S.J.; Oh, Y.S.; Park, J.H.; Lee, J.W.; Kim, H.P.; Kim, K.T.; Jang, I.S.; Park, S.C. Morphological adjustment of senescent cells by modulating caveolin-1 status. J. Biol. Chem. 2004, 279, 42270–42278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flentje, A.; Kalsi, R.; Monahan, T.S. Small GTPases and Their Role in Vascular Disease. Int. J. Mol. Sci. 2019, 20, 917. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.K.; Yokoyama, M.; Yoshida, Y.; Nojima, A.; Kassai, H.; Oishi, K.; Okada, S.; Kinoshita, D.; Kobayashi, Y.; Fruttiger, M.; et al. A crucial role for CDC42 in senescence-associated inflammation and atherosclerosis. PLoS ONE 2014, 9, e102186. [Google Scholar] [CrossRef] [Green Version]
- Zouein, F.A.; Booz, G.W.; Altara, R. STAT3 and Endothelial Cell-Cardiomyocyte Dialog in Cardiac Remodeling. Front. Cardiovasc. Med. 2019, 6, 50. [Google Scholar] [CrossRef]
- Bourgeais, J.; Gouilleux-Gruart, V.; Gouilleux, F. Oxidative metabolism in cancer: A STAT affair? Jak-Stat 2013, 2, e25764. [Google Scholar] [CrossRef] [Green Version]
- Millot, P.; San, C.; Bennana, E.; Porte, B.; Vignal, N.; Hugon, J.; Paquet, C.; Hosten, B.; Mouton-Liger, F. STAT3 inhibition protects against neuroinflammation and BACE1 upregulation induced by systemic inflammation. Immunol. Lett. 2020, 228, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Faurobert, E.; Rome, C.; Lisowska, J.; Manet-Dupé, S.; Boulday, G.; Malbouyres, M.; Balland, M.; Bouin, A.P.; Kéramidas, M.; Bouvard, D.; et al. CCM1-ICAP-1 complex controls β1 integrin-dependent endothelial contractility and fibronectin remodeling. J. Cell Biol. 2013, 202, 545–561. [Google Scholar] [CrossRef] [Green Version]
- Naruse, K.; Rask-Madsen, C.; Takahara, N.; Ha, S.W.; Suzuma, K.; Way, K.J.; Jacobs, J.R.; Clermont, A.C.; Ueki, K.; Ohshiro, Y.; et al. Activation of vascular protein kinase C-beta inhibits Akt-dependent endothelial nitric oxide synthase function in obesity-associated insulin resistance. Diabetes 2006, 55, 691–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Soroush, F.; Sun, S.; Liverani, E.; Langston, J.C.; Yang, Q.; Kilpatrick, L.E.; Kiani, M.F. Protein kinase C-delta inhibition protects blood-brain barrier from sepsis-induced vascular damage. J. Neuroinflamm. 2018, 15, 309. [Google Scholar] [CrossRef] [Green Version]
- Shen, Q.; Rigor, R.R.; Pivetti, C.D.; Wu, M.H.; Yuan, S.Y. Myosin light chain kinase in microvascular endothelial barrier function. Cardiovasc. Res. 2010, 87, 272–280. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.M.; Huang, Q.; Yuan, Y.; Granger, H.J. VEGF induces NO-dependent hyperpermeability in coronary venules. Am. J. Physiol. 1996, 271, H2735–H2739. [Google Scholar] [CrossRef]
- Manczak, M.; Park, B.S.; Jung, Y.; Reddy, P.H. Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease: Implications for early mitochondrial dysfunction and oxidative damage. Neuromol. Med. 2004, 5, 147–162. [Google Scholar] [CrossRef]
- Di Francesco, A.; Arosio, B.; Falconi, A.; Micioni Di Bonaventura, M.V.; Karimi, M.; Mari, D.; Casati, M.; Maccarrone, M.; D’Addario, C. Global changes in DNA methylation in Alzheimer’s disease peripheral blood mononuclear cells. Brain Behav. Immun. 2015, 45, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Potenza, M.A.; Sgarra, L.; Desantis, V.; Nacci, C.; Montagnani, M. Diabetes and Alzheimer’s Disease: Might Mitochondrial Dysfunction Help Deciphering the Common Path? Antioxidants 2021, 10, 1257. [Google Scholar] [CrossRef] [PubMed]
- Akram, A.; Schmeidler, J.; Katsel, P.; Hof, P.R.; Haroutunian, V. Association of ApoE and LRP mRNA levels with dementia and AD neuropathology. Neurobiol. Aging 2012, 33, 628.e1–628.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruzali, W.A.; Kehoe, P.G.; Love, S. LRP1 expression in cerebral cortex, choroid plexus and meningeal blood vessels: Relationship to cerebral amyloid angiopathy and APOE status. Neurosci. Lett. 2012, 525, 123–128. [Google Scholar] [CrossRef]
- Donahue, J.E.; Flaherty, S.L.; Johanson, C.E.; Duncan, J.A., 3rd; Silverberg, G.D.; Miller, M.C.; Tavares, R.; Yang, W.; Wu, Q.; Sabo, E.; et al. RAGE, LRP-1, and amyloid-beta protein in Alzheimer’s disease. Acta Neuropathol. 2006, 112, 405–415. [Google Scholar] [CrossRef]
- Caricasole, A.; Copani, A.; Caraci, F.; Aronica, E.; Rozemuller, A.J.; Caruso, A.; Storto, M.; Gaviraghi, G.; Terstappen, G.C.; Nicoletti, F. Induction of Dickkopf-1, a negative modulator of the Wnt pathway, is associated with neuronal degeneration in Alzheimer’s brain. J. Neurosci. 2004, 24, 6021–6027. [Google Scholar] [CrossRef]
- Rosi, M.C.; Luccarini, I.; Grossi, C.; Fiorentini, A.; Spillantini, M.G.; Prisco, A.; Scali, C.; Gianfriddo, M.; Caricasole, A.; Terstappen, G.C.; et al. Increased Dickkopf-1 expression in transgenic mouse models of neurodegenerative disease. J. Neurochem. 2010, 112, 1539–1551. [Google Scholar] [CrossRef] [PubMed]
- Rebeck, G.W.; Harr, S.D.; Strickland, D.K.; Hyman, B.T. Multiple, Diverse senile plaque-associated proteins are ligands of an apolipoprotein E receptor, the alpha 2-macroglobulin receptor/low-density-lipoprotein receptor-related protein. Ann. Neurol. 1995, 37, 211–217. [Google Scholar] [CrossRef]
- Blain, J.F.; Poirier, J. Could lipoprotein lipase play a role in Alzheimer’s disease? Sci. World J. 2004, 4, 531–535. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, V.; Park, Y.; Chen, C.C.; Xu, P.Z.; Chen, M.L.; Tonic, I.; Unterman, T.; Hay, N. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell 2008, 14, 458–470. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Reyes-Irisarri, E.; Hüll, M.; Kummer, M.P. Impact and Therapeutic Potential of PPARs in Alzheimer’s Disease. Curr. Neuropharmacol. 2011, 9, 643–650. [Google Scholar] [CrossRef]
- Ortiz-Matamoros, A.; Arias, C. Chronic infusion of Wnt7a, Wnt5a and Dkk-1 in the adult hippocampus induces structural synaptic changes and modifies anxiety and memory performance. Brain Res. Bull. 2018, 139, 243–255. [Google Scholar] [CrossRef]
- Fortress, A.M.; Frick, K.M. Hippocampal Wnt Signaling: Memory Regulation and Hormone Interactions. Neuroscientist 2016, 22, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Henderson, B.W.; Gentry, E.G.; Rush, T.; Troncoso, J.C.; Thambisetty, M.; Montine, T.J.; Herskowitz, J.H. Rho-associated protein kinase 1 (ROCK1) is increased in Alzheimer’s disease and ROCK1 depletion reduces amyloid-β levels in brain. J. Neurochem. 2016, 138, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.C.; Davis, C.M.; Nelson, J.W.; Young, J.M.; Alkayed, N.J. Soluble epoxide hydrolase: Sex differences and role in endothelial cell survival. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1936–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Whitcomb, R.; MacIntyre, E.; Tran, V.; Do, Z.N.; Sabry, J.; Patel, D.V.; Anandan, S.K.; Gless, R.; Webb, H.K. Pharmacokinetics and pharmacodynamics of AR9281, an inhibitor of soluble epoxide hydrolase, in single- and multiple-dose studies in healthy human subjects. J. Clin. Pharmacol. 2012, 52, 319–328. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nuthikattu, S.; Milenkovic, D.; Norman, J.E.; Rutledge, J.; Villablanca, A. Inhibition of Soluble Epoxide Hydrolase Is Protective against the Multiomic Effects of a High Glycemic Diet on Brain Microvascular Inflammation and Cognitive Dysfunction. Nutrients 2021, 13, 3913. https://doi.org/10.3390/nu13113913
Nuthikattu S, Milenkovic D, Norman JE, Rutledge J, Villablanca A. Inhibition of Soluble Epoxide Hydrolase Is Protective against the Multiomic Effects of a High Glycemic Diet on Brain Microvascular Inflammation and Cognitive Dysfunction. Nutrients. 2021; 13(11):3913. https://doi.org/10.3390/nu13113913
Chicago/Turabian StyleNuthikattu, Saivageethi, Dragan Milenkovic, Jennifer E. Norman, John Rutledge, and Amparo Villablanca. 2021. "Inhibition of Soluble Epoxide Hydrolase Is Protective against the Multiomic Effects of a High Glycemic Diet on Brain Microvascular Inflammation and Cognitive Dysfunction" Nutrients 13, no. 11: 3913. https://doi.org/10.3390/nu13113913
APA StyleNuthikattu, S., Milenkovic, D., Norman, J. E., Rutledge, J., & Villablanca, A. (2021). Inhibition of Soluble Epoxide Hydrolase Is Protective against the Multiomic Effects of a High Glycemic Diet on Brain Microvascular Inflammation and Cognitive Dysfunction. Nutrients, 13(11), 3913. https://doi.org/10.3390/nu13113913