
Early-Life Iron Deficiency Anemia Programs the Hippocampal Epigenomic Landscape



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Main Text
2.1. Early-Life Iron Deficiency Modifies Gene Regulation and Epigenetic Landscape in the Adult Rat Hippocampus
2.2. Early-Life Iron Deficiency Reprograms Gene Regulation
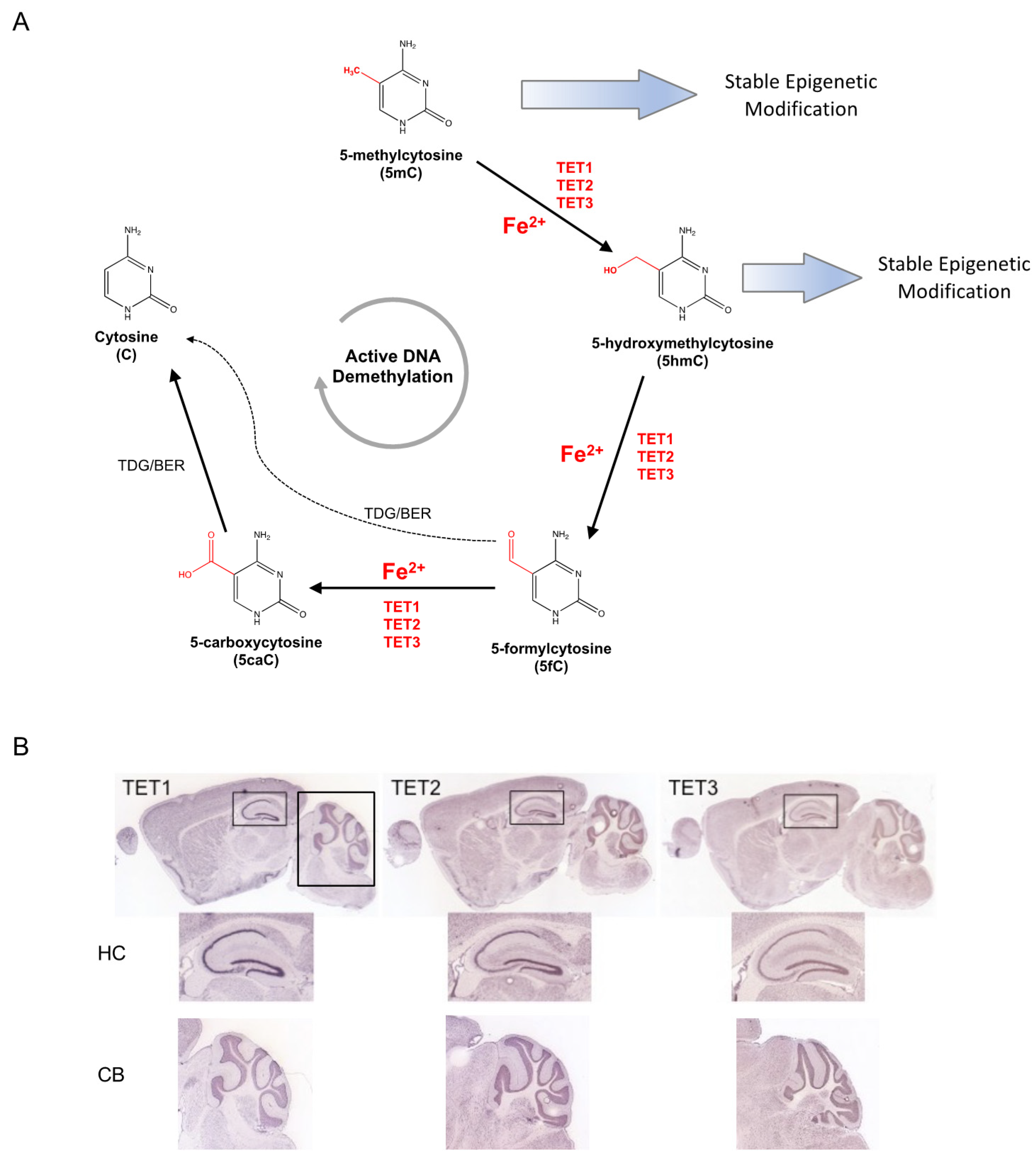
2.3. DNA Methylation and Hydroxymethylation
2.4. Histone Methylation
2.5. Prenatal Choline Supplementation and Iron Deficiency Interact to Regulate the Rat Hippocampal Epigenomic Landscape
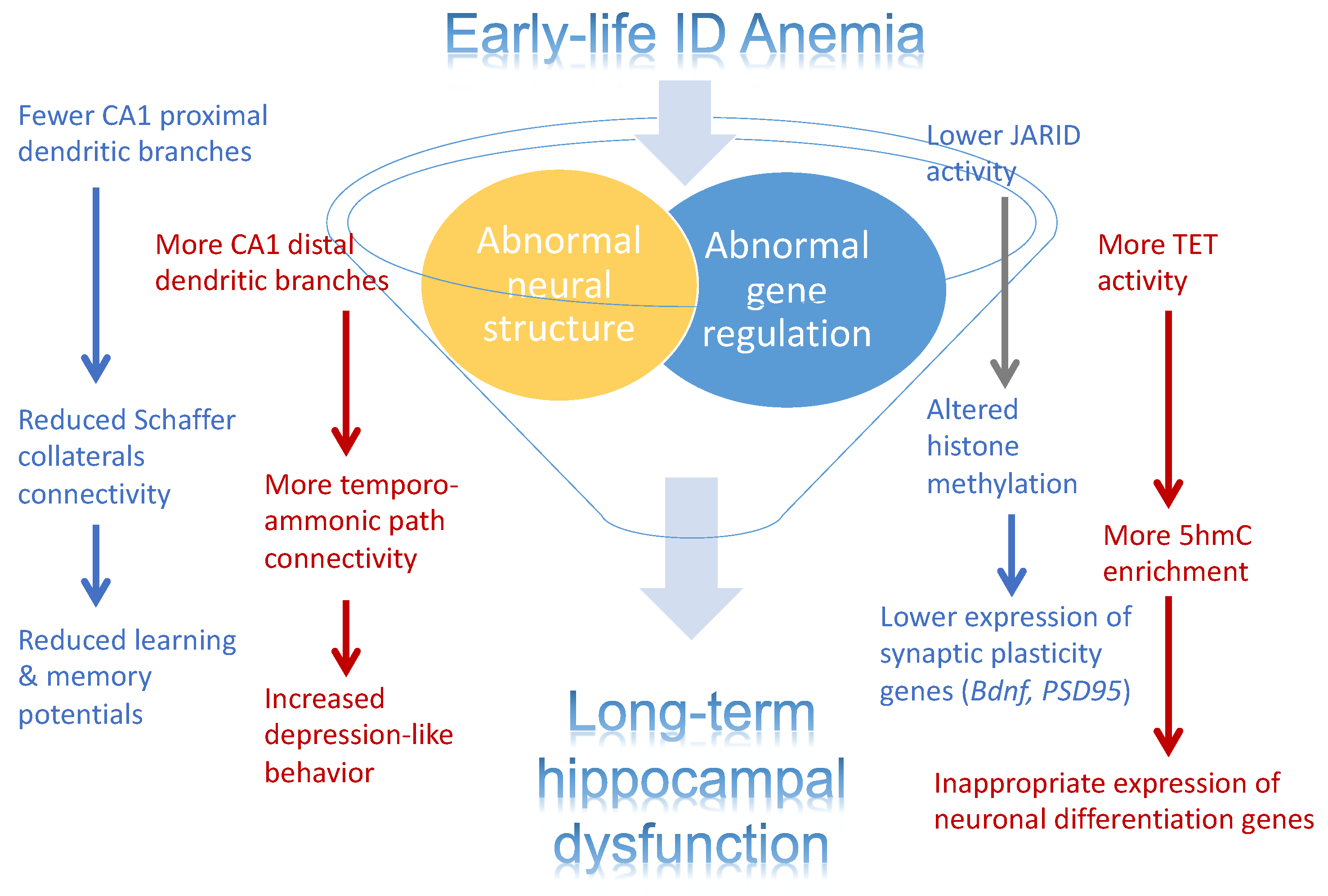
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, Y.; Baram, T.Z. Toward Understanding How Early-Life Stress Reprograms Cognitive and Emotional Brain Networks. Neuropsychopharmacology 2016, 41, 197–206. [Google Scholar] [CrossRef]
- Vaiserman, A.M.; Koliada, A.K. Early-life adversity and long-term neurobehavioral outcomes: Epigenome as a bridge? Hum. Genom. 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- McLean, E.; Cogswell, M.; Egli, I.; Wojdyla, D.; De Benoist, B. Worldwide prevalence of anaemia, WHO Vitamin and Mineral Nutrition Information System, 1993-2005. Public Health Nutr. 2009, 12, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Stevens, G.A.; Finucane, M.M.; De-Regil, L.M.; Paciorek, C.J.; Flaxman, S.R.; Branca, F.; Peña-Rosas, J.P.; Bhutta, Z.A.; Ezzati, M. Global, regional, and national trends in haemoglobin concentration and prevalence of total and severe anaemia in children and pregnant and non-pregnant women for 1995-2011: A systematic analysis of population-representative data. Lancet Glob. Health 2013, 1, e16–e25. [Google Scholar] [CrossRef]
- Lozoff, B.; Smith, J.B.; Kaciroti, N.; Clark, K.M.; Guevara, S.; Jimenez, E. Functional significance of early-life iron deficiency: Outcomes at 25 years. J. Pediatr. 2013, 163, 1260–1266. [Google Scholar] [CrossRef]
- Lukowski, A.F.; Koss, M.; Burden, M.J.; Jonides, J.; Nelson, C.A.; Kaciroti, N.; Jimenez, E.; Lozoff, B. Iron deficiency in infancy and neurocognitive functioning at 19 years: Evidence of long-term deficits in executive function and recognition memory. Nutr. Neurosci. 2010, 13, 54–70. [Google Scholar] [CrossRef] [PubMed]
- Insel, B.J.; Schaefer, C.A.; McKeague, I.W.; Susser, E.S.; Brown, A.S. Maternal iron deficiency and the risk of schizophrenia in offspring. Arch. Gen. Psychiatry 2008, 65, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.J.; Tancredi, D.J.; Krakowiak, P.; Hansen, R.L.; Ozonoff, S. Maternal intake of supplemental iron and risk of autism spectrum disorder. Am. J. Epidemiol. 2014, 180, 890–900. [Google Scholar] [CrossRef]
- Brunette, K.E.; Tran, P.V.; Wobken, J.D.; Carlson, E.S.; Georgieff, M.K. Gestational and Neonatal Iron Deficiency Alters Apical Dendrite Structure of CA1 Pyramidal Neurons in Adult Rat Hippocampus. Dev. Neurosci. 2010, 32, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Carlson, E.S.; Stead, J.D.H.; Neal, C.R.; Petryk, A.; Georgieff, M.K. Perinatal iron deficiency results in altered developmental expression of genes mediating energy metabolism and neuronal morphogenesis in hippocampus. Hippocampus 2007, 17, 679–691. [Google Scholar] [CrossRef]
- Carlson, E.S.; Tkac, I.; Magid, R.; O’Connor, M.B.; Andrews, N.C.; Schallert, T.; Gunshin, H.; Georgieff, M.K.; Petryk, A. Iron Is Essential for Neuron Development and Memory Function in Mouse Hippocampus. J. Nutr. 2009, 139, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Fretham, S.J.B.; Carlson, E.S.; Georgieff, M.K. Neuronal-specific iron deficiency dysregulates mammalian target of rapamycin signaling during hippocampal development in nonanemic genetic mouse Models1,2. J. Nutr. 2013, 143, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Fretham, S.J.B.; Carlson, E.S.; Wobken, J.; Tran, P.V.; Petryk, A.; Georgieff, M.K. Temporal manipulation of transferrin-receptor-1-dependent iron uptake identifies a sensitive period in mouse hippocampal neurodevelopment. Hippocampus 2012, 22, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Jorgenson, L.A.; Sun, M.; O’Connor, M.; Georgieff, M.K. Fetal iron deficiency disrupts the maturation of synaptic function and efficacy in area CA1 of the developing rat hippocampus. Hippocampus 2005, 15, 1094–1102. [Google Scholar] [CrossRef]
- Jorgenson, L.A.; Wobken, J.D.; Georgieff, M.K. Perinatal Iron Deficiency Alters Apical Dendritic Growth in Hippocampal CA1 Pyramidal Neurons. Dev. Neurosci. 2003, 25, 412–420. [Google Scholar] [CrossRef]
- Tran, P.V.; Carlson, E.S.; Fretham, S.J.B.; Georgieff, M.K. Early-Life Iron Deficiency Anemia Alters Neurotrophic Factor Expression and Hippocampal Neuron Differentiation in Male Rats. J. Nutr. 2008, 138, 2495. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.V.; Fretham, S.J.B.; Carlson, E.S.; Georgieff, M.K. Long-term reduction of hippocampal brain-derived neurotrophic factor activity after fetal-neonatal iron deficiency in adult rats. Pediatr. Res. 2009, 65, 493–498. [Google Scholar] [CrossRef]
- Tran, P.V.; Kennedy, B.C.; Pisansky, M.T.; Won, K.-J.; Gewirtz, J.C.; Simmons, R.A.; Georgieff, M.K. Prenatal Choline Supplementation Diminishes Early-Life Iron Deficiency–Induced Reprogramming of Molecular Networks Associated with Behavioral Abnormalities in the Adult Rat Hippocampus. J. Nutr. 2016, 146, 484–493. [Google Scholar] [CrossRef]
- Dolinoy, D.C.; Jirtle, R.L. Environmental epigenomics in human health and disease. Environ. Mol. Mutagen. 2008, 49, 4–8. [Google Scholar] [CrossRef]
- Feil, R. Environmental and nutritional effects on the epigenetic regulation of genes. Mutat. Res. Mol. Mech. Mutagen. 2006, 600, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Verjan, J.C.; Barrera-Vázquez, O.S.; García-Velázquez, L.; Samper-Ternent, R.; Arroyo, P. Epigenetic variations due to nutritional status in early-life and its later impact on aging and disease. Clin. Genet. 2020, 98, 313–321. [Google Scholar] [CrossRef]
- Stevens, A.J.; Rucklidge, J.J.; Kennedy, M.A. Epigenetics, nutrition and mental health. Is there a relationship? Nutr. Neurosci. 2018, 21, 602–613. [Google Scholar] [CrossRef]
- Blegen, M.B.; Kennedy, B.C.; Thibert, K.A.; Gewirtz, J.C.; Tran, P.V.; Georgieff, M.K. Multigenerational effects of fetal-neonatal iron deficiency on hippocampal BDNF signaling. Physiol. Rep. 2013, 1, e00096. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.V.; Kennedy, B.C.; Lien, Y.-C.; Simmons, R.A.; Georgieff, M.K. Fetal iron deficiency induces chromatin remodeling at the Bdnf locus in adult rat hippocampus. Am. J. Physiol. Integr. Comp. Physiol. 2015, 308, R276–R282. [Google Scholar] [CrossRef] [PubMed]
- Lien, Y.C.; Condon, D.E.; Georgieff, M.K.; Simmons, R.A.; Tran, P.V. Dysregulation of neuronal genes by fetal-neonatal iron deficiency anemia is associated with altered DNA methylation in the rat hippocampus. Nutrients 2019, 11, 1191. [Google Scholar] [CrossRef]
- Hu, L.; Lu, J.; Cheng, J.; Rao, Q.; Li, Z.; Hou, H.; Lou, Z.; Zhang, L.; Li, W.; Gong, W.; et al. Structural insight into substrate preference for TET-mediated oxidation. Nature 2015, 527, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Kriaucionis, S.; Heintz, N. The Nuclear DNA Base 5-Hydroxymethylcytosine Is Present in Purkinje Neurons and the Brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef]
- Kennedy, B.C.; Dimova, J.G.; Siddappa, A.J.M.; Tran, P.V.; Gewirtz, J.C.; Georgieff, M.K. Prenatal choline supplementation ameliorates the long-term neurobehavioral effects of fetal-neonatal iron deficiency in rats. J. Nutr. 2014, 144, 1858–1865. [Google Scholar] [CrossRef] [PubMed]
- Nag, N.; Mellott, T.J.; Berger-Sweeney, J.E. Effects of postnatal dietary choline supplementation on motor regional brain volume and growth factor expression in a mouse model of Rett syndrome. Brain Res. 2008, 1237, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Schaevitz, L.; Berger-Sweeney, J.; Ricceri, L. One-carbon metabolism in neurodevelopmental disorders: Using broad-based nutraceutics to treat cognitive deficits in complex spectrum disorders. Neurosci. Biobehav. Rev. 2014, 46, 270–284. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.D.; Abou, E.J.; Dominguez, H.D. Prenatal choline supplementation mitigates the adverse effects of prenatal alcohol exposure on development in rats. Neurotoxicol. Teratol. 2009, 31, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.D.; La Fiette, M.H.; Quinn, V.R.E.; Riley, E.P. Neonatal choline supplementation ameliorates the effects of prenatal alcohol exposure on a discrimination learning task in rats. Neurotoxicol. Teratol. 2000, 22, 703–711. [Google Scholar] [CrossRef]
- Cooney, C.A.; Dave, A.A.; Wolff, G.L. Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring. J. Nutr. 2002, 132, 2393S–2400S. [Google Scholar] [CrossRef]
- Davison, J.M.; Mellott, T.J.; Kovacheva, V.P.; Blusztajn, J.K. Gestational choline supply regulates methylation of histone H3, expression of histone methyltransferases G9a (Kmt1c) and Suv39h1 (Kmt1a), and DNA methylation of their genes in rat fetal liver and brain. J. Biol. Chem. 2009, 284, 1982–1989. [Google Scholar] [CrossRef]
- Mehedint, M.G.; Niculescu, M.D.; Craciunescu, C.N.; Zeisel, S.H. Choline deficiency alters global histone methylation and epigenetic marking at the Rel site of the calbindin 1 gene. FASEB J. 2010, 24, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Yip, R. Iron Deficiency: Contemporary Scientific Issues and International Programmatic Approaches. J. Nutr. 1994, 124, 1479S–1490S. [Google Scholar] [CrossRef]
- Mei, Z.; Cogswell, M.E.; Looker, A.C.; Pfeiffer, C.M.; Cusick, S.E.; Lacher, D.A.; Grummer-Strawn, L.M. Assessment of iron status in US pregnant women from the National Health and Nutrition Examination Survey (NHANES), 1999–2006. Am. J. Clin. Nutr. 2011, 93, 1312–1320. [Google Scholar] [CrossRef]
- Cogswell, M.E.; Looker, A.C.; Pfeiffer, C.M.; Cook, J.D.; Lacher, D.A.; Beard, J.L.; Lynch, S.R.; Grummer-Strawn, L.M. Assessment of iron deficiency in US preschool children and nonpregnant females of childbearing age: National Health and Nutrition Examination Survey 2003–2006. Am. J. Clin. Nutr. 2009, 89, 1334–1342. [Google Scholar] [CrossRef]
- Auerbach, M.; Abernathy, J.; Juul, S.; Short, V.; Derman, R. Prevalence of iron deficiency in first trimester, nonanemic pregnant women. J. Matern. Fetal. Neonatal Med. 2021, 34, 1002–1005. [Google Scholar] [CrossRef] [PubMed]
- Barton, J.C.; Wiener, H.H.; Acton, R.T.; Adams, P.C.; Eckfeldt, J.H.; Gordeuk, V.R.; Harris, E.L.; McLaren, C.E.; Harrison, H.; McLaren, G.D.; et al. Prevalence of iron deficiency in 62,685 women of seven race/ethnicity groups: The HEIRS Study. PLoS ONE 2020, 15, e0232125. [Google Scholar] [CrossRef]
- Lozoff, B.; Jimenez, E.; Wolf, A.W. Long-Term Developmental Outcome of Infants with Iron Deficiency. N. Engl. J. Med 1991, 325, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Goldenberg, R.L.; Hou, J.; Johnston, K.E.; Cliver, S.P.; Ramey, S.L.; Nelson, K.G. Cord serum ferritin concentrations and mental and psychomotor development of children at five years of age. J. Pediatr. 2002, 140, 165–170. [Google Scholar] [CrossRef]
- Lozoff, B.; Jimenez, E.; Hagen, J.; Mollen, E.; Wolf, A.W. Poorer behavioral and developmental outcome more than 10 years after treatment for iron deficiency in infancy. Pediatrics 2000, 105, e51. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Wang, L.; Wang, Y.; Brouwer, I.D.; Kok, F.J.; Lozoff, B.; Chen, C. Iron-Deficiency Anemia in Infancy and Social Emotional Development in Preschool-Aged Chinese Children. Pediatrics 2011, 127, e927–e933. [Google Scholar] [CrossRef]
- Lozoff, B. Iron deficiency and child development. Food Nutr. Bull. 2007, 28. [Google Scholar] [CrossRef] [PubMed]
- Corapci, F.; Radan, A.E.; Lozoff, B. Iron deficiency in infancy and mother-child interaction at 5 years. J. Dev. Behav. Pediatr. 2006, 27, 371–378. [Google Scholar] [CrossRef]
- Sørensen, H.J.; Nielsen, P.R.; Pedersen, C.B.; Mortensen, P.B. Association Between Prepartum Maternal Iron Deficiency and Offspring Risk of Schizophrenia: Population-Based Cohort Study With Linkage of Danish National Registers. Schizophr. Bull. 2011, 37, 982. [Google Scholar] [CrossRef] [PubMed]
- Wiegersma, A.M.; Dalman, C.; Lee, B.K.; Karlsson, H.; Gardner, R.M. Association of Prenatal Maternal Anemia With Neurodevelopmental Disorders. JAMA Psychiatry 2019, 76, 1294. [Google Scholar] [CrossRef]
- Hensch, T.K. Critical period regulation. Annu. Rev. Neurosci. 2004, 27, 549–579. [Google Scholar] [CrossRef]
- Tyagi, E.; Zhuang, Y.; Agrawal, R.; Ying, Z.; Gomez-Pinilla, F. Interactive actions of Bdnf methylation and cell metabolism for building neural resilience under the influence of diet. Neurobiol. Dis. 2015, 73, 307–318. [Google Scholar] [CrossRef]
- Zeisel, S. Choline, Other Methyl-Donors and Epigenetics. Nutrients 2017, 9, 445. [Google Scholar] [CrossRef] [PubMed]
- Ly, A.; Ishiguro, L.; Kim, D.; Im, D.; Kim, S.E.; Sohn, K.J.; Croxford, R.; Kim, Y.I. Maternal folic acid supplementation modulates DNA methylation and gene expression in the rat offspring in a gestation period-dependent and organ-specific manner. J. Nutr. Biochem. 2016, 33, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Barks, A.; Fretham, S.J.B.; Georgieff, M.K.; Tran, P.V. Early-Life Neuronal-Specific Iron Deficiency Alters the Adult Mouse Hippocampal Transcriptome. J. Nutr. 2018, 148, 1521–1528. [Google Scholar] [CrossRef] [PubMed]
- Barks, A.; Hall, A.M.; Tran, P.V.; Georgieff, M.K. Iron as a model nutrient for understanding the nutritional origins of neuropsychiatric disease. Pediatr. Res. 2019, 85, 176–182. [Google Scholar] [CrossRef]
- Schachtschneider, K.M.; Liu, Y.; Rund, L.A.; Madsen, O.; Johnson, R.W.; Groenen, M.A.M.; Schook, L.B. Impact of neonatal iron deficiency on hippocampal DNA methylation and gene transcription in a porcine biomedical model of cognitive development. BMC Genomics 2016, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lubin, F.D.; Roth, T.L.; Sweatt, J.D. Epigenetic Regulation of bdnf Gene Transcription in the Consolidation of Fear Memory. J. Neurosci. 2008, 28, 10576–10586. [Google Scholar] [CrossRef]
- McGowan, P.O.; Suderman, M.; Sasaki, A.; Huang, T.C.T.; Hallett, M.; Meaney, M.J.; Szyf, M. Broad Epigenetic Signature of Maternal Care in the Brain of Adult Rats. PLoS ONE 2011, 6, e14739. [Google Scholar] [CrossRef]
- Anderson, O.S.; Nahar, M.S.; Faulk, C.; Jones, T.R.; Liao, C.; Kannan, K.; Weinhouse, C.; Rozek, L.S.; Dolinoy, D.C. Epigenetic responses following maternal dietary exposure to physiologically relevant levels of bisphenol A. Environ. Mol. Mutagen. 2012, 53, 334–342. [Google Scholar] [CrossRef]
- McBirney, M.; King, S.E.; Pappalardo, M.; Houser, E.; Unkefer, M.; Nilsson, E.; Sadler-Riggleman, I.; Beck, D.; Winchester, P.; Skinner, M.K. Atrazine induced epigenetic transgenerational inheritance of disease, lean phenotype and sperm epimutation pathology biomarkers. PLoS ONE 2017, 12, e0184306. [Google Scholar] [CrossRef] [PubMed]
- Perkins, A.; Lehmann, C.; Lawrence, R.C.; Kelly, S.J. Alcohol exposure during development: Impact on the epigenome. Int. J. Dev. Neurosci. 2013, 31, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Ke, X.; Xing, B.; Yu, B.; Yu, X.; Majnik, A.; Cohen, S.; Lane, R.; Joss-Moore, L. IUGR disrupts the PPARγ-Setd8-H4K20me1 and Wnt signaling pathways in the juvenile rat hippocampus. Int. J. Dev. Neurosci. 2014, 38, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Waterland, R.A.; Jirtle, R.L. Transposable Elements: Targets for Early Nutritional Effects on Epigenetic Gene Regulation. Mol. Cell. Biol. 2003, 23, 5293–5300. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Szulwach, K.E.; Li, X.; Li, Y.; Song, C.-X.; Wu, H.; Dai, Q.; Irier, H.; Upadhyay, A.K.; Gearing, M.; Levey, A.I.; et al. 5-hmC–mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 2011, 14, 1607–1616. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global Epigenomic Reconfiguration During Mammalian Brain Development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef] [PubMed]
- ISH Data: Allen Brain Atlas: Mouse Brain. Available online: https://mouse.brain-map.org/ (accessed on 27 October 2021).
- Chen, Z.; Zang, J.; Whetstine, J.; Hong, X.; Davrazou, F.; Kutateladze, T.G.; Simpson, M.; Mao, Q.; Pan, C.-H.; Dai, S.; et al. Structural Insights into Histone Demethylation by JMJD2 Family Members. Cell 2006, 125, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, Y.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2005, 439, 811–816. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.T.; Helin, K. Histone demethylases in development and disease. Trends Cell Biol. 2010, 20, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.; Schmitz, S.U.; Kooistra, S.M.; Malatesta, M.; Morales Torres, C.; Rekling, J.C.; Johansen, J.V.; Abarrategui, I.; Helin, K. The Histone Demethylase Jarid1b Ensures Faithful Mouse Development by Protecting Developmental Genes from Aberrant H3K4me3. PLoS Genet. 2013, 9, e1003461. [Google Scholar] [CrossRef] [PubMed]
- Fueyo, R.; García, M.A.; Martínez-Balbás, M.A. Jumonji family histone demethylases in neural development. Cell Tissue Res. 2014, 359, 87–98. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jepsen, K.; Solum, D.; Zhou, T.; McEvilly, R.J.; Kim, H.J.; Glass, C.K.; Hermanson, O.; Rosenfeld, M.G. SMRT-mediated repression of an H3K27 demethylase in progression from neural stem cell to neuron. Nature 2007, 450, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Liefke, R.; Oswald, F.; Alvarado, C.; Ferres-Marco, D.; Mittler, G.; Rodriguez, P.; Dominguez, M.; Borggrefe, T. Histone demethylase KDM5A is an integral part of the core Notch-RBP-J repressor complex. Genes Dev. 2010, 24, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, S.U.; Albert, M.; Malatesta, M.; Morey, L.; Johansen, J.V.; Bak, M.; Tommerup, N.; Abarrategui, I.; Helin, K. Jarid1b targets genes regulating development and is involved in neural differentiation. EMBO J. 2011, 30, 4586–4600. [Google Scholar] [CrossRef]
- Takahashi, M.; Kojima, M.; Nakajima, K.; Suzuki-Migishima, R.; Takeuchi, T. Functions of a jumonji-cyclin D1 pathway in the coordination of cell cycle exit and migration during neurogenesis in the mouse hindbrain. Dev. Biol. 2007, 303, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Cascante, A.; Klum, S.; Biswas, M.; Antolin-Fontes, B.; Barnabé-Heider, F.; Hermanson, O. Gene-specific methylation control of H3K9 and H3K36 on neurotrophic BDNF versus astroglial GFAP genes by KDM4A/C regulates neural stem cell differentiation. J. Mol. Biol. 2014, 426, 3467–3477. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Yan, Q.; Tothova, Z.; Yamane, K.; Erdjument-Bromage, H.; Tempst, P.; Gilliland, D.G.; Zhang, Y.; Kaelin, W.G. The Retinoblastoma Binding Protein RBP2 Is an H3K4 Demethylase. Cell 2007, 128, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Margueron, R.; Ku, M.; Chambon, P.; Bernstein, B.E.; Reinberg, D. Jarid2 and PRC2, partners in regulating gene expression. Genes Dev. 2010, 24, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Pasini, D.; Cloos, P.A.C.; Walfridsson, J.; Olsson, L.; Bukowski, J.P.; Johansen, J.V.; Bak, M.; Tommerup, N.; Rappsilber, J.; Helin, K. JARID2 regulates binding of the Polycomb repressive complex 2 to target genes in ES cells. Nature 2010, 464, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Kallin, E.M.; Zhang, Y. JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 2006, 7, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Georgieff, M.K.; Ramel, S.E.; Cusick, S.E. Nutritional influences on brain development. Acta Paediatr. 2018, 107, 1310–1321. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S.H. Nutritional Importance of Choline for Brain Development. J. Am. Coll. Nutr. 2004, 23, 621S–626S. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S.H.; Da Costa, K.A. Choline: An essential nutrient for public health. Nutr. Rev. 2009, 67, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Heindel, J.J.; Vandenberg, L.N. Developmental origins of health and disease: A paradigm for understanding disease cause and prevention. Curr. Opin. Pediatr. 2015, 27, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.A.; Qiu, R.; Wu, X.; Li, A.X.; Zhang, H.; Wang, J.; Jui, J.; Jin, S.G.; Jiang, Y.; Pfeifer, G.P.; et al. Dynamics of 5-Hydroxymethylcytosine and Chromatin Marks in Mammalian Neurogenesis. Cell Rep. 2013, 3, 291–300. [Google Scholar] [CrossRef]
- Kling, P.J. 50 Years Ago in The Journal of Pediatrics: Iron Metabolism in Premature Infants: II. Prevention of Iron Deficiency. J. Pediatr. 2014, 164, 768. [Google Scholar] [CrossRef]
- Siddappa, A.M.; Rao, R.; Long, J.D.; Widness, J.A.; Georgieff, M.K. The Assessment of Newborn Iron Stores at Birth: A Review of the Literature and Standards for Ferritin Concentrations. Neonatology 2007, 92, 73–82. [Google Scholar] [CrossRef]
- Kennedy, B.C.; Tran, P.V.; Kohli, M.; Maertens, J.J.; Gewirtz, J.C.; Georgieff, M.K. Beneficial effects of postnatal choline supplementation on long-Term neurocognitive deficit resulting from fetal-Neonatal iron deficiency. Behav. Brain Res. 2018, 336, 40–43. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barks, A.K.; Liu, S.X.; Georgieff, M.K.; Hallstrom, T.C.; Tran, P.V. Early-Life Iron Deficiency Anemia Programs the Hippocampal Epigenomic Landscape. Nutrients 2021, 13, 3857. https://doi.org/10.3390/nu13113857
Barks AK, Liu SX, Georgieff MK, Hallstrom TC, Tran PV. Early-Life Iron Deficiency Anemia Programs the Hippocampal Epigenomic Landscape. Nutrients. 2021; 13(11):3857. https://doi.org/10.3390/nu13113857
Chicago/Turabian StyleBarks, Amanda K., Shirelle X. Liu, Michael K. Georgieff, Timothy C. Hallstrom, and Phu V. Tran. 2021. "Early-Life Iron Deficiency Anemia Programs the Hippocampal Epigenomic Landscape" Nutrients 13, no. 11: 3857. https://doi.org/10.3390/nu13113857
APA StyleBarks, A. K., Liu, S. X., Georgieff, M. K., Hallstrom, T. C., & Tran, P. V. (2021). Early-Life Iron Deficiency Anemia Programs the Hippocampal Epigenomic Landscape. Nutrients, 13(11), 3857. https://doi.org/10.3390/nu13113857