Potential Therapeutic Benefit of NAD+ Supplementation for Glaucoma and Age-Related Macular Degeneration

 ,
,  and
and
Abstract
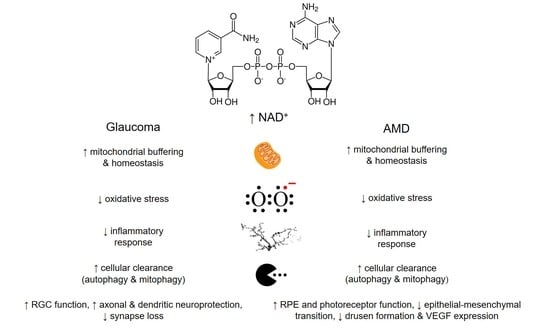
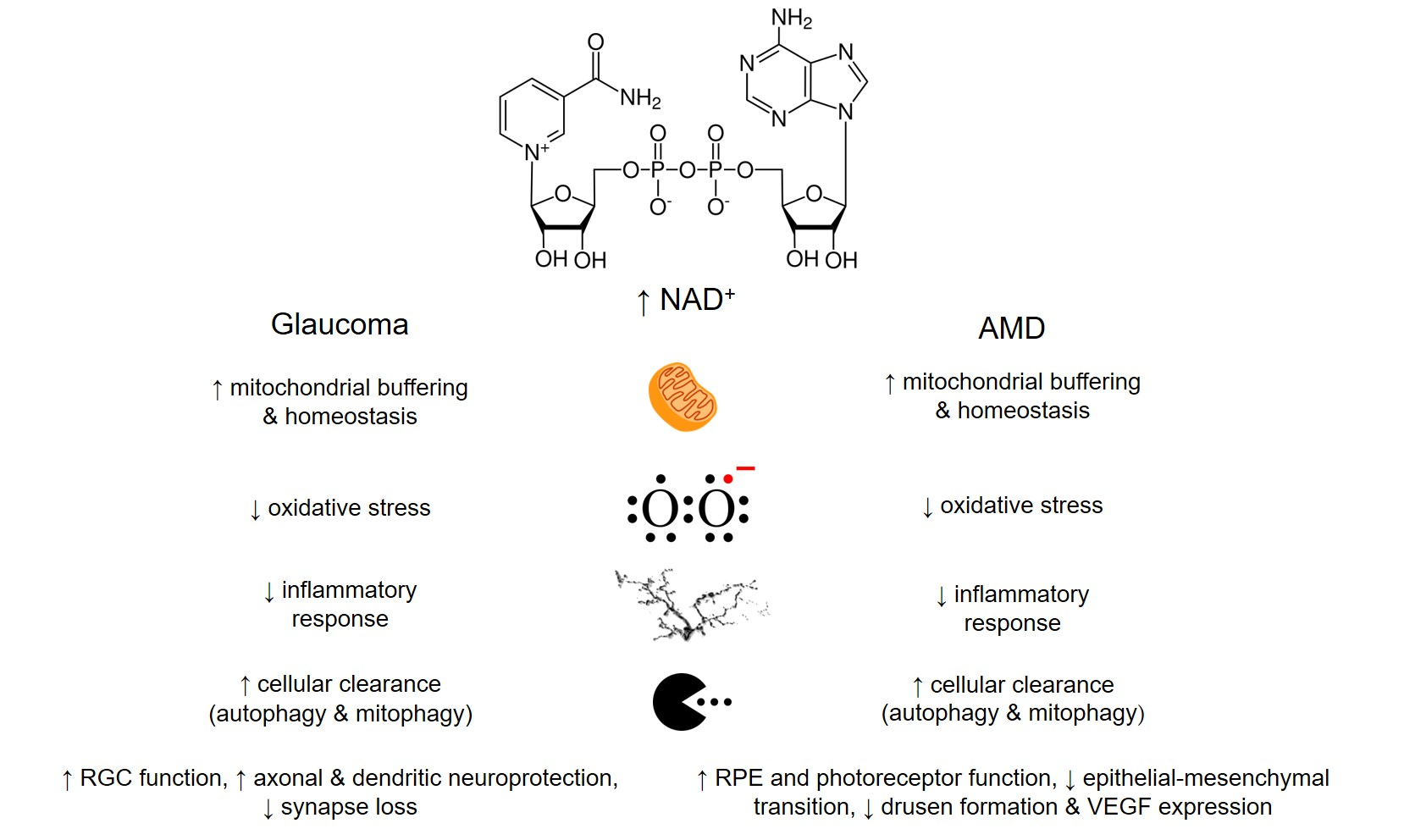{kind=link}
1. Introduction
2. Nicotinamide Adenine Dinucleotide
2.1. NAD Biosynthetic Pathways
2.2. NAD+ in Aging and Neurodegeneration
2.3. NAD+ and Eye Disease
3. Glaucoma
Available Therapies
4. Age-Related Macular Degeneration
4.1. Available Therapies
4.1.1. Wet AMD
4.1.2. Dry AMD
5. Supplementing NAD+ as a Therapeutic Approach/Strategy in Glaucoma and AMD
5.1. Potentials of NAD+ Intervention in Glaucoma
5.2. Potentials of NAD+ Intervention in Age-Related Macular Degeneration
5.3. Safety of NAD+ Supplementation
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tham, Y.C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: A systematic review and meta-analysis. Ophthalmology 2014, 121, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.G.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef]
- Jonas, J.B.; Aung, T.; Bourne, R.R.; Bron, A.M.; Ritch, R.; Panda-Jonas, S. Glaucoma. Lancet 2017, 390, 2183–2193. [Google Scholar] [CrossRef]
- Almasieh, M.; Wilson, A.M.; Morquette, B.; Cueva Vargas, J.L.; Di Polo, A. The molecular basis of retinal ganglion cell death in glaucoma. Prog. Retin. Eye Res. 2012, 31, 152–181. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.A.; Higginbotham, E.J. Glaucoma and its treatment: A review. Am. J. Health Syst. Pharm. 2005, 62, 691–699. [Google Scholar] [CrossRef]
- Hoyng, P.F.J.; Kitazawa, Y. Medical treatment of normal tension glaucoma. Surv. Ophthalmol. 2002, 47, S116–S124. [Google Scholar] [CrossRef]
- Al-Zamil, W.M.; Yassin, S.A. Recent developments in age-related macular degeneration: A review. Clin. Interv. Aging 2017, 12, 1313–1330. [Google Scholar] [CrossRef]
- Algvere, P.V.; Kvanta, A.; Seregard, S. Drusen maculopathy: A risk factor for visual deterioration. Acta Ophthalmol. 2016, 94, 427–433. [Google Scholar] [CrossRef]
- Hernandez-Zimbron, L.F.; Zamora-Alvarado, R.; Ochoa-De la Paz, L.; Velez-Montoya, R.; Zenteno, E.; Gulias-Canizo, R.; Quiroz-Mercado, H.; Gonzalez-Salinas, R. Age-related macular degeneration: New paradigms for treatment and management of AMD. Oxid. Med. Cell Longev. 2018, 2018, 8374647. [Google Scholar] [CrossRef]
- Velez-Montoya, R.; Oliver, S.C.N.; Olson, J.L.; Fine, S.L.; Mandava, N.; Quiroz-Mercado, H. Current knowledge and trends in age-related macular degeneration: Today’s and future treatments. Retina 2013, 33, 1487–1502. [Google Scholar] [CrossRef]
- Velez-Montoya, R.; Oliver, S.C.N.; Olson, J.L.; Fine, S.L.; Quiroz-Mercado, H.; Naresh, M. Current knowledge and trends in age-related macular degeneration: Genetics, epidemiology, and prevention. Retina 2014, 34, 423–441. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov (accessed on 18 September 2020).
- Chew, E.Y. The age-related eye disease study (AREDS) and AREDS 2 supplements in 2018. Retin. Physician 2018, 15, 31–34. [Google Scholar]
- Grunwald, J.E.; Pistilli, M.; Daniel, E.; Ying, G.S.; Pan, W.; Jaffe, G.J.; Toth, C.A.; Hagstrom, S.A.; Maguire, M.G.; Martin, D.F. Incidence and growth of geographic atrophy during 5 years of comparison of age-related macular degeneration treatments trials. Ophthalmology 2017, 124, 97–104. [Google Scholar] [CrossRef]
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Mattson, M.P.; Arumugam, T.V. Hallmarks of brain aging: Adaptive and pathological modification by metabolic states. Cell Metab. 2018, 27, 1176–1199. [Google Scholar] [CrossRef] [PubMed]
- Boatright, J. NAD+/NADH precursor treatment protects against retinal degeneration. Acta Ophthalmol. 2019, 97. [Google Scholar] [CrossRef]
- Di Marco, S.; Carnicelli, V.; Franceschini, N.; Di Paolo, M.; Piccardi, M.; Bisti, S.; Falsini, B. Saffron: A multitask neuroprotective agent for retinal degenerative diseases. Antioxidants 2019, 8, 224. [Google Scholar] [CrossRef]
- Heitmar, R.; Brown, J.; Kyrou, I. Saffron (Crocus sativus L.) in ocular diseases: A narrative review of the existing evidence from clinical studies. Nutrients 2019, 11, 649. [Google Scholar] [CrossRef]
- Demmig-Adams, B.; Lopez-Pozo, M.; Stewart, J.J.; Adams, W.W., III. Zeaxanthin and lutein: Photoprotectors, anti-inflammatories, and brain food. Molecules 2020, 25, 3607. [Google Scholar] [CrossRef]
- Murillo, A.G.; Hu, S.; Fernandez, M.L. Zeaxanthin: Metabolism, properties, and antioxidant protection of eyes, heart, liver, and skin. Antioxidants 2019, 8, 390. [Google Scholar] [CrossRef]
- Williams, P.A.; Harder, J.M.; Guymer, C.; Wood, J.P.M.; Daskalaki, E.; Chidlow, G.; Cardozo, B.H.; Foxworth, N.E.; Cochran, K.E.; Ouellette, T.B.; et al. Oral pyruvate prevents glaucomatous neurodegeneration. bioRxiv 2020. [Google Scholar] [CrossRef]
- Harden, A.; Young, W.J.; Martin, C.J. The alcoholic ferment of yeast-juice. Proc. R. Soc. Lond. Ser. B Contain. Pap. Biol. Character 1906, 77, 405–420. [Google Scholar] [CrossRef]
- Harden, A.; Young, W.J.; Martin, C.J. The alcoholic ferment of yeast-juice. Part II.—The coferment of yeast-juice. Proc. R. Soc. Lond. Ser. B Contain. Pap. Biol. Character 1906, 78, 369–375. [Google Scholar] [CrossRef]
- Morabia, A. Joseph Goldberger’s research on the prevention of pellagra. J. R. Soc. Med. 2008, 101, 566–568. [Google Scholar] [CrossRef]
- Lautrup, S.; Sinclair, D.A.; Mattson, M.P.; Fang, E.F. NAD(+) in Brain Aging and Neurodegenerative Disorders. Cell Metab. 2019, 30, 630–655. [Google Scholar] [CrossRef] [PubMed]
- Felicitas Berger, C.L.; Dahlmann, M.; Ziegler, M. Subcellular Compartmentation and Differential Catalytic Properties of the Three Human Nicotinamide Mononucleotide Adenylyltransferase Isoforms. J. Biol. Chem. 2005, 280, 36334–36341. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.L. Why is NMNAT protective against neuronal cell death and axon degeneration, but inhibitory of axon regeneration? Cells 2019, 8, 267. [Google Scholar] [CrossRef]
- Gilley, J.; Mayer, P.R.; Yu, G.; Coleman, M.P. Low levels of NMNAT2 compromise axon development and survival. Hum. Mol. Genet. 2018, 28, 448–458. [Google Scholar] [CrossRef]
- Gilley, J.; Coleman, M.P. Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 2010, 8, e1000300. [Google Scholar] [CrossRef]
- Hicks, A.N.; Lorenzetti, D.; Gilley, J.; Lu, B.; Andersson, K.E.; Miligan, C.; Overbeek, P.A.; Oppenheim, R.; Bishop, C.E. Nicotinamide mononucleotide adenylyltransferase 2 (Nmnat2) regulates axon integrity in the mouse embryo. PLoS ONE 2012, 7, e47869. [Google Scholar] [CrossRef]
- Di Stefano, M.; Loreto, A.; Orsomando, G.; Mori, V.; Zamporlini, F.; Hulse, R.P.; Webster, J.; Donaldson, L.F.; Gering, M.; Raffaelli, N.; et al. NMN deamidase delays wallerian degeneration and rescues axonal defects caused by NMNAT2 deficiency in vivo. Curr. Biol. 2017, 27, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Loreto, A.; Hill, C.S.; Hewitt, V.L.; Orsomando, G.; Angeletti, C.; Gilley, J.; Lucci, C.; Sanchez-Martinez, A.; Whitworth, A.J.; Conforti, L.; et al. Mitochondrial impairment activates the Wallerian pathway through depletion of NMNAT2 leading to SARM1-dependent axon degeneration. Neurobiol. Dis. 2019, 134, 104678. [Google Scholar] [CrossRef] [PubMed]
- Sauve, A.A. NAD+ and Vitamin B3: From metabolism to therapies. J. Pharmacol. Exp. Ther. 2008, 324, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Opitz, C.A.; Heiland, I. Dynamics of NAD-metabolism: Everything but constant. Biochem. Soc. Trans. 2015, 43, 1127–1132. [Google Scholar] [CrossRef] [PubMed]
- Castro-Portuguez, R.; Sutphin, G.L. Kynurenine pathway, NAD+ synthesis, and mitochondrial function: Targeting tryptophan metabolism to promote longevity and healthspan. Exp. Gerontol. 2020, 132, 110841. [Google Scholar] [CrossRef]
- Preiss, J.; Handler, P. Biosynthesis of diphosphopyridine nucleotide I. Identification of intermediates. J. Biol. Chem. 1958, 233, 488–492. [Google Scholar]
- Preiss, J.; Handler, P. Biosynthesis of diphosphopyridine nucleotide II. Enzymatic aspects. J. Biol. Chem. 1958, 233, 493–500. [Google Scholar]
- Song, S.B.; Park, J.S.; Chung, G.J.; Lee, I.H.; Hwang, E.S. Diverse therapeutic efficacies and more diverse mechanisms of nicotinamide. Metabolomics 2019, 15, 137. [Google Scholar] [CrossRef]
- Brenner, P.B.A.C. Discoveries of Nicotinamide Riboside as a Nutrient and Conserved NRK Genes Establish a Preiss-Handler Independent Route to NAD+ in Fungi and Humans. Cell 2004, 117, 495–502. [Google Scholar]
- Poddar, S.K.; Sifat, A.E.; Haque, S.; Nahid, N.A.; Chowdhury, S.; Mehedi, I. Nicotinamide Mononucleotide: Exploration of Diverse Therapeutic Applications of a Potential Molecule. Biomolecules 2019, 9, 34. [Google Scholar] [CrossRef]
- Imai, S.-I.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403, 795–800. [Google Scholar] [CrossRef]
- Quarona, V.; Zaccarello, G.; Chillemi, A.; Brunetti, E.; Singh, V.K.; Ferrero, E.; Funaro, A.; Horenstein, A.L.; Malavasi, F. CD38 and CD157: A long journey from activation markers to multifunctional molecules. Cytom. Part B Clin. Cytom. 2013, 84B, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Zhang, T.; Kraus, W.L. Poly(ADP-ribosyl)ation by PARP-1: ‘PAR-laying’ NAD+ into a nuclear signal. Genes Dev. 2005, 19, 1951–1967. [Google Scholar] [CrossRef] [PubMed]
- Rasti, G.; Simonet, N.G.; Vaquero, A. Niacin. In Principles of Nutrigenetics and Nutrigenomics; Academic press: Cambridge, MA, USA, 2020; pp. 287–293. [Google Scholar]
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD+ homeostasis in health and disease. Nat. Metab. 2020, 2, 9–31. [Google Scholar] [CrossRef] [PubMed]
- Audrito, V.; Manago, A.; Gaudino, F.; Sorci, L.; Messana, V.G.; Raffaelli, N.; Deaglio, S. NAD-Biosynthetic and Consuming Enzymes as Central Players of Metabolic Regulation of Innate and Adaptive Immune Responses in Cancer. Front. Immunol. 2019, 10, 1720. [Google Scholar] [CrossRef] [PubMed]
- Tissenbaum, H.A.; Guarente, L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 2001, 410, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Rogina, B.; Helfand, S.L.; Frankel, S. Longevity Regulation by Drosophila Rpd3 Deacetylase and Caloric Restriction. Science 2002, 298, 1745. [Google Scholar] [CrossRef]
- Verdin, E. The Many Faces of Sirtuins: Coupling of NAD metabolism, sirtuins and lifespan. Nat. Med. 2014, 20, 25–27. [Google Scholar] [CrossRef]
- Van der Veer, E.; Ho, C.; O’Neil, C.; Barbosa, N.; Scott, R.; Cregan, S.P.; Pickering, J.G. Extension of human cell lifespan by nicotinamide phosphoribosyltransferase. J. Biol. Chem. 2007, 282, 10841–10845. [Google Scholar] [CrossRef]
- Hegyi, J.; Schwartz, R.A.; Hegyi, V. Pellagra: Dermatitis, dementia, and diarrhea. Int. J. Dermatol. 2004, 43, 1–5. [Google Scholar] [CrossRef]
- Schultz, M.B.; Sinclair, D.A. Why NAD(+) Declines during Aging: It’s Destroyed. Cell Metab. 2016, 23, 965–966. [Google Scholar] [CrossRef] [PubMed]
- Gasperi, V.; Sibilano, M.; Savini, I.; Catani, M.V. Niacin in the Central Nervous System: An Update of Biological Aspects and Clinical Applications. Int. J. Mol. Sci. 2019, 20, 974. [Google Scholar] [CrossRef] [PubMed]
- Fricker, R.A.; Green, E.L.; Jenkins, S.I.; Griffin, S.M. The Influence of Nicotinamide on Health and Disease in the Central Nervous System. Int. J. Tryptophan. Res. 2018, 11, 1178646918776658. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Menzies, K.J.; Auwerx, J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001, 15, 2922–2933. [Google Scholar] [PubMed]
- Fang, E.F.; Lautrup, S.; Hou, Y.; Demarest, T.G.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. NAD(+) in Aging: Molecular Mechanisms and Translational Implications. Trends Mol. Med. 2017, 23, 899–916. [Google Scholar] [CrossRef] [PubMed]
- Chaban, Y.; Boekema, E.J.; Dudkina, N.V. Structures of mitochondrial oxidative phosphorylation supercomplexes and mechanisms for their stabilisation. Biochim. Biophys. Acta 2014, 1837, 418–426. [Google Scholar] [CrossRef]
- Reinecke, F.; Smeitink, J.A.; van der Westhuizen, F.H. Oxphos gene expression and control in mitochondrial disorders. Biochim. Biophys. Acta 2009, 1792, 1113–1121. [Google Scholar] [CrossRef]
- Ito, Y.A.; Di Polo, A. Mitochondrial dynamics, transport, and quality control: A bottleneck for retinal ganglion cell viability in optic neuropathies. Mitochondrion 2017, 36, 186–192. [Google Scholar] [CrossRef]
- Srivastava, S. The Mitochondrial Basis of Aging and Age-Related Disorders. Genes 2017, 8, 398. [Google Scholar] [CrossRef]
- Ermak, G.; Davies, K.J.A. Calcium and oxidative stress: From cell signaling to cell death. Mol. Immunol. 2002, 38, 713–721. [Google Scholar] [CrossRef]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Calcium and mitochondria in the regulation of cell death. Biochem. Biophys. Res. Commun. 2015, 460, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Solleiro-Villavicencio, H.; Rivas-Arancibia, S. Effect of Chronic Oxidative Stress on Neuroinflammatory Response Mediated by CD4(+)T Cells in Neurodegenerative Diseases. Front. Cell Neurosci. 2018, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Eter, N.; Heiduschka, P. The microglia in healthy and diseased retina. Exp. Eye Res. 2015, 136, 116–130. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, B.N.; Thackray, J.K.; Serrano, L. Sirtuins and DNA damage repair: SIRT7 comes to play. Nucleus 2017, 8, 107–115. [Google Scholar] [CrossRef]
- Lee, C.F.; Caudal, A.; Abell, L.; Nagana Gowda, G.A.; Tian, R. Targeting NAD(+) Metabolism as Interventions for Mitochondrial Disease. Sci. Rep. 2019, 9, 3073. [Google Scholar] [CrossRef] [PubMed]
- Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M. Salvaging hope: Is increasing NAD(+) a key to treating mitochondrial myopathy? EMBO Mol. Med. 2014, 6, 705–707. [Google Scholar] [CrossRef]
- Khan, N.A.; Auranen, M.; Paetau, I.; Pirinen, E.; Euro, L.; Forsström, S.; Pasila, L.; Velagapudi, V.; Carroll, C.J.; Auwerx, J.; et al. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol. Med. 2014, 6, 721–731. [Google Scholar] [CrossRef]
- Shpilka, T.; Haynes, C.M. The mitochondrial UPR: Mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 2018, 19, 109–120. [Google Scholar] [CrossRef]
- Pellegrino, M.W.; Nargund, A.M.; Haynes, C.M. Signaling the mitochondrial unfolded protein response. Biochim. Biophys. Acta 2013, 1833, 410–416. [Google Scholar] [CrossRef]
- Papa, L.; Germain, D. SirT3 regulates the mitochondrial unfolded protein response. Mol. Cell Biol. 2014, 34, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Yanez, M.; Jhanji, M.; Murphy, K.; Gower, R.M.; Sajish, M.; Jabbarzadeh, E. Nicotinamide Augments the Anti-Inflammatory Properties of Resveratrol through PARP1 Activation. Sci. Rep. 2019, 9, 10219. [Google Scholar] [CrossRef] [PubMed]
- Jadeja, R.N.; Thounaojam, M.C.; Bartoli, M.; Martin, P.M. Implications of NAD(+) Metabolism in the Aging Retina and Retinal Degeneration. Oxid. Med. Cell Longev. 2020, 2020, 2692794. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Margolin, Z.; Borgo, B.; Havranek, J.J.; Milbrandt, J. Characterization of Leber Congenital Amaurosis-associated NMNAT1 Mutants. J. Biol. Chem. 2015, 290, 17228–17238. [Google Scholar] [CrossRef]
- Koenekoop, R.K.; Wang, H.; Majewski, J.; Wang, X.; Lopez, I.; Ren, H.; Chen, Y.; Li, Y.; Fishman, G.A.; Genead, M.; et al. Mutations in NMNAT1 cause Leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nat. Genet. 2012, 44, 1035–1039. [Google Scholar] [CrossRef]
- Falk, M.J.; Zhang, Q.; Nakamaru-Ogiso, E.; Kannabiran, C.; Fonseca-Kelly, Z.; Chakarova, C.; Audo, I.; Mackay, D.S.; Zeitz, C.; Borman, A.D.; et al. NMNAT1 mutations cause Leber congenital amaurosis. Nat. Genet. 2012, 44, 1040–1045. [Google Scholar] [CrossRef]
- Lin, J.B.; Kubota, S.; Ban, N.; Yoshida, M.; Santeford, A.; Sene, A.; Nakamura, R.; Zapata, N.; Kubota, M.; Tsubota, K.; et al. NAMPT-Mediated NAD+ Biosynthesis Is Essential for Vision In Mice. Cell Rep. 2016, 17, 69–85. [Google Scholar] [CrossRef]
- Kuribayashi, H.; Baba, Y.; Iwagawa, T.; Arai, E.; Murakami, A.; Watanabe, S. Roles of Nmnat1 in the survival of retinal progenitors through the regulation of pro-apoptotic gene expression via histone acetylation. Cell Death Dis. 2018, 9, 891. [Google Scholar] [CrossRef]
- Bai, S.; Sheline, C.T. NAD(+) maintenance attenuates light induced photoreceptor degeneration. Exp. Eye Res. 2013, 108, 76–83. [Google Scholar] [CrossRef]
- Sheline, C.T.; Zhou, Y.; Bai, S. Light-induced photoreceptor and RPE degeneration involve zinc toxicity and are attenuated by pyruvate, nicotinamide, or cyclic light. Mol. Vis. 2010, 16, 2639–2652. [Google Scholar] [PubMed]
- Fisher, C.R.; Ferrington, D.A. Perspective on AMD Pathobiology: A Bioenergetic Crisis in the RPE. Investig. Ophthalmol. Vis. Sci. 2018, 59, AMD41–AMD47. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhao, K.-K.; Tong, Y.; Zhou, Y.-L.; Wang, Y.-X.; Zhao, P.-Q.; Wang, Z.-Y. Exogenous NAD+ decreases oxidative stress and protects H2O2-treated RPE cells against necrotic death through the up-regulation of autophagy. Sci. Rep. 2016, 6, 26322. [Google Scholar] [CrossRef] [PubMed]
- Bergen, A.A. Nicotinamide, iRPE-in-a dish, and age-related macular degeneration therapy development. Stem Cell Investig. 2017, 4, 81. [Google Scholar] [CrossRef]
- Zabka, T.S.; Singh, J.; Dhawan, P.; Liederer, B.M.; Oeh, J.; Kauss, M.A.; Xiao, Y.; Zak, M.; Lin, T.; McCray, B.; et al. Retinal Toxicity, in vivo and in vitro, Associated with Inhibition of Nicotinamide Phosphoribosyltransferase. Toxicol. Sci. 2014, 144, 163–172. [Google Scholar] [CrossRef]
- Zhao, G.; Green, C.F.; Hui, Y.-H.; Prieto, L.; Shepard, R.; Dong, S.; Wang, T.; Tan, B.; Gong, X.; Kays, L.; et al. Discovery of a Highly Selective NAMPT Inhibitor That Demonstrates Robust Efficacy and Improved Retinal Toxicity with Nicotinic Acid Coadministration. Mol. Cancer Ther. 2017, 16, 2677–2688. [Google Scholar] [CrossRef]
- Sasaki, Y.; Araki, T.; Milbrandt, J. Stimulation of Nicotinamide Adenine Dinucleotide Biosynthetic Pathways Delays Axonal Degeneration after Axotomy. J. Neurosci. 2006, 26, 8484–8491. [Google Scholar] [CrossRef]
- Sasaki, Y.; Vohra, B.P.S.; Lund, F.E.; Milbrandt, J. Nicotinamide mononucleotide adenylyl transferase-mediated axonal protection requires enzymatic activity but not increased levels of neuronal nicotinamide adenine dinucleotide. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 5525–5535. [Google Scholar] [CrossRef]
- Mi, W.; Beirowski, B.; Gillingwater, T.H.; Adalbert, R.; Wagner, D.; Grumme, D.; Osaka, H.; Conforti, L.; Arnhold, S.; Addicks, K.; et al. The slow Wallerian degeneration gene, WldS, inhibits axonal spheroid pathology in gracile axonal dystrophy mice. Brain 2005, 128, 405–416. [Google Scholar] [CrossRef]
- Sasaki, Y.; Vohra, B.P.S.; Baloh, R.H.; Milbrandt, J. Transgenic mice expressing the Nmnat1 protein manifest robust delay in axonal degeneration in vivo. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 6526–6534. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, L.; Sasaki, Y.; Milbrandt, J.; Gidday, J.M. Protection of Mouse Retinal Ganglion Cell Axons and Soma from Glaucomatous and Ischemic Injury by Cytoplasmic Overexpression of Nmnat1. Investig. Ophthalmol. Vis. Sci. 2013, 54, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Kitaoka, Y.; Munemasa, Y.; Kojima, K.; Hirano, A.; Ueno, S.; Takagi, H. Axonal protection by Nmnat3 overexpression with involvement of autophagy in optic nerve degeneration. Cell Death Dis. 2013, 4, e860. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Harder, J.M.; Foxworth, N.E.; Cochran, K.E.; Philip, V.M.; Porciatti, V.; Smithies, O.; John, S.W.M. Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science 2017, 355, 756–760. [Google Scholar] [CrossRef]
- Williams, P.A.; Harder, J.M.; Foxworth, N.E.; Cardozo, B.H.; Cochran, K.E.; John, S.W.M. Nicotinamide and WLD(S) Act Together to Prevent Neurodegeneration in Glaucoma. Front. Neurosci. 2017, 11, 232. [Google Scholar] [CrossRef]
- Williams, P.A.; Harder, J.M.; Cardozo, B.H.; Foxworth, N.E.; John, S.W.M. Nicotinamide treatment robustly protects from inherited mouse glaucoma. Commun. Integr. Biol. 2018, 11, e1356956. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Parrish, J.Z.; He, R.; Zhai, R.G.; Kim, M.D. Nmnat exerts neuroprotective effects in dendrites and axons. Mol. Cell Neurosci. 2011, 48, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, R.N.; Aung, T.; Medeiros, F.A. The pathophysiology and treatment of glaucoma: A review. JAMA 2014, 311, 1901–1911. [Google Scholar] [CrossRef]
- Marshak, D.W. Retinal ganglion cells: Anatomy. In Encyclopedia of Neuroscience; Squire, L.R., Ed.; Academic Press: Oxford, UK, 2009; pp. 211–218. [Google Scholar]
- Feher, J. Quantitative Human Physiology; Academic press: Cambridge, MA, USA, 2012; pp. 456–470. [Google Scholar]
- Weinreb, R.N.; Khaw, P.T. Primary open-angle glaucoma. Lancet 2004, 363, 1711–1720. [Google Scholar] [CrossRef]
- Morgan, J.E. Retina ganglion cell degeneration in glaucoma: An opportunity missed? A review. Clin. Exp. Ophthalmol. 2012, 40, 364–368. [Google Scholar] [CrossRef]
- Whitmore, A.V.; Libby, R.T.; John, S.W.M. Glaucoma: Thinking in new ways—A rôle for autonomous axonal self-destruction and other compartmentalised processes? Prog. Retin. Eye Res. 2005, 24, 639–662. [Google Scholar] [CrossRef]
- Williams, P.A.; Howell, G.R.; Barbay, J.M.; Braine, C.E.; Sousa, G.L.; John, S.W.; Morgan, J.E. Retinal ganglion cell dendritic atrophy in DBA/2J glaucoma. PLoS ONE 2013, 8, e72282. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.E.; Tribble, J.; Fergusson, J.; White, N.; Erchova, I. The optical detection of retinal ganglion cell damage. Eye 2017, 31, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Binley, K.E.; Ng, W.S.; Barde, Y.-A.; Song, B.; Morgan, J.E.; Kirik, D. Brain-derived neurotrophic factor prevents dendritic retraction of adult mouse retinal ganglion cells. Eur. J. Neurosci. 2016, 44, 2028–2039. [Google Scholar] [CrossRef] [PubMed]
- Chong, R.S.; Martin, K.R. Retinal ganglion cell dendrites and glaucoma: A case of missing the wood for the trees? Expert Rev. Ophthalmol. 2014, 9, 149–152. [Google Scholar] [CrossRef]
- Zhou, L.; Chen, W.; Lin, D.; Hu, W.; Tang, Z. Neuronal apoptosis, axon damage and synapse loss occur synchronously in acute ocular hypertension. Exp. Eye Res. 2019, 180, 77–85. [Google Scholar] [CrossRef]
- Williams, P.A.; Tribble, J.R.; Pepper, K.W.; Cross, S.D.; Morgan, B.P.; Morgan, J.E.; John, S.W.; Howell, G.R. Inhibition of the classical pathway of the complement cascade prevents early dendritic and synaptic degeneration in glaucoma. Mol. Neurodegener. 2016, 11, 26. [Google Scholar] [CrossRef]
- Williams, P.A.; Morgan, J.E.; Votruba, M. Opa1 deficiency in a mouse model of dominant optic atrophy leads to retinal ganglion cell dendropathy. Brain 2010, 133, 2942–2951. [Google Scholar] [CrossRef]
- Williams, P.A.; Piechota, M.; von Ruhland, C.; Taylor, E.; Morgan, J.E.; Votruba, M. Opa1 is essential for retinal ganglion cell synaptic architecture and connectivity. Brain 2012, 135, 493–505. [Google Scholar] [CrossRef]
- Conforti, L.; Adalbert, R.; Coleman, M.P. Neuronal death: Where does the end begin? Trends Neurosci. 2007, 30, 159–166. [Google Scholar] [CrossRef]
- Morgan, J.E. Optic nerve head structure in glaucoma: Astrocytes as mediators of axonal damage. Eye 2000, 14, 437–444. [Google Scholar] [CrossRef]
- Vrabec, J.P.; Levin, L.A. The neurobiology of cell death in glaucoma. Eye 2007, 21 (Suppl S1), S11–S14. [Google Scholar] [CrossRef] [PubMed]
- Howell, G.R.; Libby, R.T.; Jakobs, T.C.; Smith, R.S.; Phalan, F.C.; Barter, J.W.; Barbay, J.M.; Marchant, J.K.; Mahesh, N.; Porciatti, V.; et al. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J. Cell Biol. 2007, 179, 1523–1537. [Google Scholar] [CrossRef] [PubMed]
- Libby, R.T.; Anderson, M.G.; Pang, I.-H.; Robinson, Z.H.; Savinova, O.V.; Cosma, I.M.; Snow, A.M.Y.; Wilson, L.A.; Smith, R.S.; Clark, A.F.; et al. Inherited glaucoma in DBA/2J mice: Pertinent disease features for studying the neurodegeneration. Vis. Neurosci. 2005, 22, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Libby, R.T.; Li, Y.; Savinova, O.V.; Barter, J.; Smith, R.S.; Nickells, R.W.; John, S.W. Susceptibility to neurodegeneration in a glaucoma is modified by Bax gene dosage. PLoS Genet. 2005, 1, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Wang, D.; Grosskreutz, C.L. Mechanisms of retinal ganglion cell injury and defense in glaucoma. Exp. Eye Res. 2010, 91, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Kong, G.Y.X.; Van Bergen, N.J.; Trounce, I.A.; Crowston, J.G. Mitochondrial Dysfunction and Glaucoma. J. Glaucoma 2009, 18, 93–100. [Google Scholar] [CrossRef]
- Levin, L.A.; Crowe, M.E.; Quigley, H.A.; The Lasker/IRRF Initiative on Astrocytes and Glaucomatous Neurodegeneration Participants. Neuroprotection for glaucoma: Requirements for clinical translation. Exp. Eye Res. 2017, 157, 34–37. [Google Scholar] [CrossRef]
- Tamm, E.R.; Ethier, C.R.; Lasker, I.I.O.A.; Glaucomatous Neurodegeneration, P. Biological aspects of axonal damage in glaucoma: A brief review. Exp. Eye Res. 2017, 157, 5–12. [Google Scholar] [CrossRef]
- Pfeiffer, R.L.; Marc, R.E.; Jones, B.W. Persistent remodeling and neurodegeneration in late-stage retinal degeneration. Prog. Retin. Eye Res. 2020, 74, 100771. [Google Scholar] [CrossRef]
- Kamel, K.; Farrell, M.; O’Brien, C. Mitochondrial dysfunction in ocular disease: Focus on glaucoma. Mitochondrion 2017, 35, 44–53. [Google Scholar] [CrossRef]
- Carelli, V.; La Morgia, C.; Valentino, M.L.; Barboni, P.; Ross-Cisneros, F.N.; Sadun, A.A. Retinal ganglion cell neurodegeneration in mitochondrial inherited disorders. Biochim. Biophys. Acta 2009, 1787, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Osborne, N.N. Mitochondria: Their role in ganglion cell death and survival in primary open angle glaucoma. Exp. Eye Res. 2010, 90, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Cell biology. Metabolic control of cell death. Science 2014, 345, 1250256. [Google Scholar] [CrossRef] [PubMed]
- Alqawlaq, S.; Flanagan, J.G.; Sivak, J.M. All roads lead to glaucoma: Induced retinal injury cascades contribute to a common neurodegenerative outcome. Exp. Eye Res. 2019, 183, 88–97. [Google Scholar] [CrossRef]
- Tezel, G. TNF-α signaling in glaucomatous neurodegeneration. In Progress in Brain Research; Nucci, C., Cerulli, L., Osborne, N.N., Bagetta, G., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; Volume 173, pp. 409–421. [Google Scholar]
- Johnson, E.C.; Guo, Y.; Cepurna, W.O.; Morrison, J.C. Neurotrophin roles in retinal ganglion cell survival: Lessons from rat glaucoma models. Exp. Eye Res. 2009, 88, 808–815. [Google Scholar] [CrossRef]
- Johnson, E.C.; Morrison, J.C. Friend or foe? Resolving the impact of glial responses in glaucoma. J. Glaucoma 2009, 18, 341–353. [Google Scholar] [CrossRef]
- Eastlake, K.; Luis, J.; Limb, G.A. Potential of Müller Glia for Retina Neuroprotection. Curr. Eye Res. 2020, 45, 339–348. [Google Scholar] [CrossRef]
- Seitz, R.; Ohlmann, A.; Tamm, E.R. The role of Müller glia and microglia in glaucoma. Cell Tissue Res. 2013, 353, 339–345. [Google Scholar] [CrossRef]
- Lambiase, A.; Aloe, L.; Centofanti, M.; Parisi, V.; Mantelli, F.; Colafrancesco, V.; Manni, G.L.; Bucci, M.G.; Bonini, S.; Levi-Montalcini, R. Experimental and clinical evidence of neuroprotection by nerve growth factor eye drops: Implications for glaucoma. Proc. Natl. Acad. Sci. USA 2009, 106, 13469–13474. [Google Scholar] [CrossRef]
- Di Polo, A.; Aigner, L.J.; Dunn, R.J.; Bray, G.M.; Aguayo, A.J. Prolonged delivery of brain-derived neurotrophic factor by adenovirus-infected Müller cells temporarily rescues injured retinal ganglion cells. Proc. Natl. Acad. Sci. USA 1998, 95, 3978–3983. [Google Scholar] [CrossRef]
- Ji, J.-Z.; Elyaman, W.; Yip, H.K.; Lee, V.W.H.; Yick, L.-W.; Hugon, J.; So, K.-F. CNTF promotes survival of retinal ganglion cells after induction of ocular hypertension in rats: The possible involvement of STAT3 pathway. Eur. J. Neurosci. 2004, 19, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, N.; Peinado-Ramónn, P.; Vidal-Sanz, M.; Hallböök, F. GDNF, Ret, GFRα1 and 2 in the adult rat retino-tectal system after optic nerve transection. Exp. Neurol. 2004, 187, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Quigley, H.A.; Pease, M.E.; Yang, Y.; Qian, J.; Valenta, D.; Zack, D.J. Changes in Gene Expression in Experimental Glaucoma and Optic Nerve Transection: The Equilibrium between Protective and Detrimental Mechanisms. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5539–5548. [Google Scholar] [CrossRef]
- Maes, M.E.; Schlamp, C.L.; Nickells, R.W. BAX to basics: How the BCL2 gene family controls the death of retinal ganglion cells. Prog. Retin. Eye Res. 2017, 57, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G. Oxidative stress in glaucomatous neurodegeneration: Mechanisms and consequences. Prog. Retin. Eye Res. 2006, 25, 490–513. [Google Scholar] [CrossRef]
- Attar, M.; Shen, J.; Ling, K.-H.J.; Tang-Liu, D. Ophthalmic drug delivery considerations at the cellular level: Drug-metabolising enzymes and transporters. Expert Opin. Drug Deliv. 2005, 2, 891–908. [Google Scholar] [CrossRef]
- Nocentini, A.; Supuran, C.T. Adrenergic agonists and antagonists as antiglaucoma agents: A literature and patent review (2013–2019). Expert Opin. Ther. Pat. 2019, 29, 805–815. [Google Scholar] [CrossRef]
- Berrino, E.; Supuran, C.T. Rho-kinase inhibitors in the management of glaucoma. Expert Opin. Ther. Pat. 2019, 29, 817–827. [Google Scholar] [CrossRef]
- Naik, S.; Pandey, A.; Lewis, S.A.; Sadashiva, S.R.B.; Mutalik, S. Neuroprotection: A versatile approach to combat glaucoma. Eur. J. Pharmacol. 2020, 881, 173208. [Google Scholar] [CrossRef]
- Schwartz, K.; Budenz, D. Current management of glaucoma. Curr. Opin. Ophthalmol. 2004, 15, 119–126. [Google Scholar] [CrossRef]
- Conlon, R.; Saheb, H.; Ahmed, I.I.K. Glaucoma treatment trends: A review. Can. J. Ophthalmol. 2017, 52, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Ambati, J.; Fowler, B.J. Mechanisms of Age-Related Macular Degeneration. Neuron 2012, 75, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Jager, R.D.; Mieler, W.F.; Miller, J.W. Age-Related Macular Degeneration. N. Engl. J. Med. 2008, 358, 2606–2617. [Google Scholar] [CrossRef] [PubMed]
- Ferris, F.L.; Wilkinson, C.P.; Bird, A.; Chakravarthy, U.; Chew, E.; Csaky, K.; Sadda, S.R. Clinical Classification of Age-related Macular Degeneration. Ophthalmology 2013, 120, 844–851. [Google Scholar] [CrossRef]
- Lamin, A.; Dubis, A.M.; Sivaprasad, S. Changes in macular drusen parameters preceding the development of neovascular age-related macular degeneration. Eye 2019, 33, 910–916. [Google Scholar] [CrossRef]
- Buschini, E.; Piras, A.; Nuzzi, R.; Vercelli, A. Age related macular degeneration and drusen: Neuroinflammation in the retina. Prog. Neurobiol. 2011, 95, 14–25. [Google Scholar] [CrossRef]
- Casson, R.J.; Chidlow, G.; Crowston, J.G.; Williams, P.A.; Wood, J.P.M. Retinal energy metabolism in health and glaucoma. Prog. Retin. Eye Res. 2020, 100881. [Google Scholar] [CrossRef]
- Jarrett, S.G.; Boulton, M.E. Consequences of oxidative stress in age-related macular degeneration. Mol. Asp. Med. 2012, 33, 399–417. [Google Scholar] [CrossRef]
- Pennesi, M.E.; Neuringer, M.; Courtney, R.J. Animal models of age related macular degeneration. Mol. Asp. Med. 2012, 33, 487–509. [Google Scholar] [CrossRef]
- Jones, M.; Mitchell, P.; Wang, J.J.; Sue, C. MELAS A3243G mitochondrial DNA mutation and age related maculopathy. Am. J. Ophthalmol. 2004, 138, 1051–1053. [Google Scholar] [CrossRef]
- Jones, M.M.; Manwaring, N.; Wang, J.J.; Rochtchina, E.; Mitchell, P.; Sue, C.M. Mitochondrial DNA Haplogroups and Age-Related Maculopathy. Arch. Ophthalmol. 2007, 125, 1235–1240. [Google Scholar] [CrossRef] [PubMed]
- Ebeling, M.C.; Polanco, J.R.; Qu, J.; Tu, C.; Montezuma, S.R.; Ferrington, D.A. Improving retinal mitochondrial function as a treatment for age-related macular degeneration. Redox. Biol. 2020, 34, 101552. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res. 2020, 100858. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye Res. 2017, 60, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Patel, M.; Chan, C.C. Molecular pathology of age-related macular degeneration. Prog. Retin. Eye Res. 2009, 28, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Tarallo, V.; Hirano, Y.; Gelfand, B.D.; Dridi, S.; Kerur, N.; Kim, Y.; Cho, W.G.; Kaneko, H.; Fowler, B.J.; Bogdanovich, S.; et al. DICER1 Loss and Alu RNA Induce Age-Related Macular Degeneration via the NLRP3 Inflammasome and MyD88. Cell 2012, 149, 847–859. [Google Scholar] [CrossRef]
- Liu, J.; Copland, D.A.; Theodoropoulou, S.; Chiu, H.A.A.; Barba, M.D.; Mak, K.W.; Mack, M.; Nicholson, L.B.; Dick, A.D. Impairing autophagy in retinal pigment epithelium leads to inflammasome activation and enhanced macrophage-mediated angiogenesis. Sci. Rep. 2016, 6, 20639. [Google Scholar] [CrossRef]
- Luo, C.; Zhao, J.; Madden, A.; Chen, M.; Xu, H. Complement expression in retinal pigment epithelial cells is modulated by activated macrophages. Exp. Eye Res. 2013, 112, 93–101. [Google Scholar] [CrossRef]
- Vecino, E.; Rodriguez, F.D.; Ruzafa, N.; Pereiro, X.; Sharma, S.C. Glia–neuron interactions in the mammalian retina. Prog. Retin. Eye Res. 2016, 51, 1–40. [Google Scholar] [CrossRef]
- Sivaprasad, S.; Chong, N.V. The complement system and age-related macular degeneration. Eye 2006, 20, 867–872. [Google Scholar] [CrossRef]
- Mammadzada, P.; Corredoira, P.M.; Andre, H. The role of hypoxia-inducible factors in neovascular age-related macular degeneration: A gene therapy perspective. Cell Mol. Life Sci. 2020, 77, 819–833. [Google Scholar] [CrossRef] [PubMed]
- Stefansson, E.; Geirsdottir, A.; Sigurdsson, H. Metabolic physiology in age related macular degeneration. Prog. Retin. Eye Res. 2011, 30, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.O.; Ninchoji, T.; Gordon, E.; André, H.; Dejana, E.; Vestweber, D.; Kvanta, A.; Claesson-Welsh, L. Vascular permeability in retinopathy is regulated by VEGFR2 Y949 signaling to VE-cadherin. eLife 2020, 9, e54056. [Google Scholar] [CrossRef] [PubMed]
- Arjamaa, O.; Nikinmaa, M.; Salminen, A.; Kaarniranta, K. Regulatory role of HIF-1alpha in the pathogenesis of age-related macular degeneration (AMD). Ageing Res. Rev. 2009, 8, 349–358. [Google Scholar] [CrossRef]
- Penn, J.S.; Madan, A.; Caldwell, R.B.; Bartoli, M.; Caldwell, R.W.; Hartnett, M.E. Vascular endothelial growth factor in eye disease. Prog. Retin. Eye Res. 2008, 27, 331–371. [Google Scholar] [CrossRef]
- Lin, M.; Hu, Y.; Chen, Y.; Zhou, K.K.; Jin, J.; Zhu, M.; Le, Y.-Z.; Ge, J.; Ma, J.-X. Impacts of Hypoxia-Inducible Factor-1 Knockout in the Retinal Pigment Epithelium on Choroidal Neovascularization. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6197–6206. [Google Scholar] [CrossRef]
- Andre, H.; Tunik, S.; Aronsson, M.; Kvanta, A. Hypoxia-Inducible Factor-1alpha Is Associated With Sprouting Angiogenesis in the Murine Laser-Induced Choroidal Neovascularization Model. Investig. Ophthalmol. Vis. Sci. 2015, 56, 6591–6604. [Google Scholar] [CrossRef]
- Wang, H.; Geisen, P.; Wittchen, E.S.; King, B.; Burridge, K.; D’Amore, P.A.; Hartnett, M.E. The Role of RPE Cell-Associated VEGF189 in Choroidal Endothelial Cell Transmigration across the RPE. Investig. Ophthalmol. Vis. Sci. 2011, 52, 570–578. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, Y.-S.; Hui, Y.-N.; Zhu, J.; Zhang, P.; Li, X.; Dou, G.-R. Inhibition of proliferation, migration and tube formation of choroidal microvascular endothelial cells by targeting HIF-1α with short hairpin RNA-expressing plasmid DNA in human RPE cells in a coculture system. Graefe’s Arch. Clin. Exp. Ophthalmol. 2008, 246, 1413–1422. [Google Scholar] [CrossRef]
- Hanus, J.; Anderson, C.; Wang, S. RPE necroptosis in response to oxidative stress and in AMD. Ageing Res. Rev. 2015, 24, 286–298. [Google Scholar] [CrossRef]
- Rozing, M.P.; Durhuus, J.A.; Krogh Nielsen, M.; Subhi, Y.; Kirkwood, T.B.; Westendorp, R.G.; Sorensen, T.L. Age-related macular degeneration: A two-level model hypothesis. Prog. Retin. Eye Res. 2020, 76, 100825. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.Y.; Cipi, J.; Ma, S.; Hafler, B.P.; Kanadia, R.N.; Brush, R.S.; Agbaga, M.P.; Punzo, C. Altered photoreceptor metabolism in mouse causes late stage age-related macular degeneration-like pathologies. Proc. Natl. Acad. Sci. USA 2020, 117, 13094–13104. [Google Scholar] [CrossRef] [PubMed]
- Leveillard, T.; Philp, N.J.; Sennlaub, F. Is Retinal Metabolic Dysfunction at the Center of the Pathogenesis of Age-related Macular Degeneration? Int. J. Mol. Sci. 2019, 20, 762. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.M.; Kaiser, P.K.; Michels, M.; Soubrane, G.; Heier, J.S.; Kim, R.Y.; Sy, J.P.; Schneider, S. Ranibizumab versus Verteporfin for Neovascular Age-Related Macular Degeneration. N. Engl. J. Med. 2006, 355, 1432–1444. [Google Scholar] [CrossRef]
- Rosenfeld, P.J.; Brown, D.M.; Heier, J.S.; Boyer, D.S.; Kaiser, P.K.; Chung, C.Y.; Kim, R.Y. Ranibizumab for Neovascular Age-Related Macular Degeneration. N. Engl. J. Med. 2006, 355, 1419–1431. [Google Scholar] [CrossRef]
- Mantel, I. Optimizing the Anti-VEGF Treatment Strategy for Neovascular Age-Related Macular Degeneration: From Clinical Trials to Real-Life Requirements. Transl. Vis. Sci. Technol. 2015, 4, 6. [Google Scholar] [CrossRef]
- Costa, R.A.; Jorge, R.; Calucci, D.; Melo, L.A.S.; Cardillo, J.A.; Scott, I.U. Intravitreal bevacizumab (Avastin) in combination with verteporfin photodynamic therapy for choroidal neovascularization associated with age-related macular degeneration (IBeVe Study). Graefes Arch. Clin. Exp. Ophthalmol. 2007, 245, 1273–1280. [Google Scholar] [CrossRef]
- Kaiser, P.K. Verteporfin photodynamic therapy and anti-angiogenic drugs: Potential for combination therapy in exudative age-related macular degeneration. Curr. Med. Res. Opin. 2007, 23, 477–487. [Google Scholar] [CrossRef]
- Jaffe, G.J.; Eliott, D.; Wells, J.A.; Prenner, J.L.; Papp, A.; Patel, S. A Phase 1 Study of Intravitreous E10030 in Combination with Ranibizumab in Neovascular Age-Related Macular Degeneration. Ophthalmology 2016, 123, 78–85. [Google Scholar] [CrossRef]
- Kertes, P.J. Massive Peripapillary Subretinal Neovascularization: An Indication for Submacular Surgery. Retina 2004, 24, 219–225. [Google Scholar] [CrossRef]
- Giansanti, F.; Eandi, C.M.; Virgili, G. Submacular surgery for choroidal neovascularisation secondary to age-related macular degeneration. Cochrane Database Syst. Rev. 2009. [Google Scholar] [CrossRef] [PubMed]
- Rhoades, W.; Dickson, D.; Do, D.V. Potential role of lampalizumab for treatment of geographic atrophy. Clin. Ophthalmol. 2015, 9, 1049–1056. [Google Scholar] [CrossRef] [PubMed]
- Holz, F.G.; Sadda, S.R.; Busbee, B.; Chew, E.Y.; Mitchell, P.; Tufail, A.; Brittain, C.; Ferrara, D.; Gray, S.; Honigberg, L.; et al. Efficacy and Safety of Lampalizumab for Geographic Atrophy Due to Age-Related Macular Degeneration: Chroma and Spectri Phase 3 Randomized Clinical Trials. JAMA Ophthalmol. 2018, 136, 666–677. [Google Scholar] [CrossRef] [PubMed]
- Yates, P.A.; Holbrook, K.; Reichel, E.; Waheed, N.K.; Patrie, J.; Investigators, T. Designing a Clinical Study to Evaluate Potential Therapeutics for Geographic Atrophy Secondary to Non-Exudative Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2835. [Google Scholar]
- Di Caprio, R.; Lembo, S.; Di Costanzo, L.; Balato, A.; Monfrecola, G. Anti-Inflammatory Properties of Low and High Doxycycline Doses: An In Vitro Study. Mediat. Inflamm. 2015, 2015, 329418. [Google Scholar] [CrossRef]
- Schwartz, S.D.; Regillo, C.D.; Lam, B.L.; Eliott, D.; Rosenfeld, P.J.; Gregori, N.Z.; Hubschman, J.-P.; Davis, J.L.; Heilwell, G.; Spirn, M.; et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: Follow-up of two open-label phase 1/2 studies. Lancet 2015, 385, 509–516. [Google Scholar] [CrossRef]
- Lin, J.B.; Apte, R.S. NAD(+) and sirtuins in retinal degenerative diseases: A look at future therapies. Prog. Retin. Eye Res. 2018, 67, 118–129. [Google Scholar] [CrossRef]
- Syc-Mazurek, S.B.; Libby, R.T. Axon injury signaling and compartmentalized injury response in glaucoma. Prog. Retin. Eye Res. 2019, 73, 100769. [Google Scholar] [CrossRef]
- Kouassi Nzoughet, J.; Chao de la Barca, J.M.; Guehlouz, K.; Leruez, S.; Coulbault, L.; Allouche, S.; Bocca, C.; Muller, J.; Amati-Bonneau, P.; Gohier, P.; et al. Nicotinamide Deficiency in Primary Open-Angle Glaucoma. Invest. Ophthalmol. Vis. Sci. 2019, 60, 2509–2514. [Google Scholar] [CrossRef]
- Hui, F.; Tang, J.; Williams, P.A.; McGuinness, M.B.; Hadoux, X.; Casson, R.J.; Coote, M.; Trounce, I.A.; Martin, K.R.; van Wijngaarden, P.; et al. Improvement in inner retinal function in glaucoma with nicotinamide (vitamin B3) supplementation: A crossover randomized clinical trial. Clin. Exp. Ophthalmol. 2020. [Google Scholar] [CrossRef]
- McElnea, E.M.; Quill, B.; Docherty, N.; Wallace, D.M.; Irnaten, M.; Farrell, M.; O’Brien, C. Lipofuscin Accumulation and Autophagy in Human Lamina Cribrosa Cells. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2677. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.-X.; Zhang, J.-P.; Hu, J.-Y.; Huang, Y.-S. The potential regulatory roles of NAD+ and its metabolism in autophagy. Metab. Clin. Exp. 2016, 65, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Sedlackova, L.; Otten, E.G.; Scialo, F.; Shapira, D.; Kataura, T.; Carroll, B.; Seranova, E.; Rabanal-Ruiz, Y.; Kelly, G.; Stefanatos, R.; et al. Autophagy promotes cell and organismal survival by maintaining NAD(H) pools. bioRxiv 2020. [Google Scholar] [CrossRef]
- Luo, H.; Zhou, M.; Ji, K.; Zhuang, J.; Dang, W.; Fu, S.; Sun, T.; Zhang, X. Expression of Sirtuins in the Retinal Neurons of Mice, Rats, and Humans. Front. Aging Neurosci. 2017, 9, 366. [Google Scholar] [CrossRef]
- Mimura, T.; Kaji, Y.; Noma, H.; Funatsu, H.; Okamoto, S. The role of SIRT1 in ocular aging. Exp. Eye Res. 2013, 116, 17–26. [Google Scholar] [CrossRef]
- Ji, D.; Li, G.Y.; Osborne, N.N. Nicotinamide attenuates retinal ischemia and light insults to neurones. Neurochem. Int. 2008, 52, 786–798. [Google Scholar] [CrossRef]
- Aman, Y.; Frank, J.; Lautrup, S.H.; Matysek, A.; Niu, Z.; Yang, G.; Shi, L.; Bergersen, L.H.; Storm-Mathisen, J.; Rasmussen, L.J.; et al. The NAD(+)-mitophagy axis in healthy longevity and in artificial intelligence-based clinical applications. Mech. Ageing Dev. 2020, 185, 111194. [Google Scholar] [CrossRef]
- Hyttinen, J.M.T.; Viiri, J.; Kaarniranta, K.; Blasiak, J. Mitochondrial quality control in AMD: Does mitophagy play a pivotal role? Cell Mol. Life Sci. 2018, 75, 2991–3008. [Google Scholar] [CrossRef]
- Westenskow, P.D. Nicotinamide: A novel treatment for age-related macular degeneration? Stem. Cell Investig. 2017, 4, 86. [Google Scholar] [CrossRef]
- Sridevi Gurubaran, I.; Viiri, J.; Koskela, A.; Hyttinen, J.M.T.; Paterno, J.J.; Kis, G.; Antal, M.; Urtti, A.; Kauppinen, A.; Felszeghy, S.; et al. Mitophagy in the Retinal Pigment Epithelium of Dry Age-Related Macular Degeneration Investigated in the NFE2L2/PGC-1alpha(-/-) Mouse Model. Int. J. Mol. Sci. 2020, 21, 1976. [Google Scholar] [CrossRef]
- Felszeghy, S.; Viiri, J.; Paterno, J.J.; Hyttinen, J.M.T.; Koskela, A.; Chen, M.; Leinonen, H.; Tanila, H.; Kivinen, N.; Koistinen, A.; et al. Loss of NRF-2 and PGC-1alpha genes leads to retinal pigment epithelium damage resembling dry age-related macular degeneration. Redox. Biol. 2019, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Kassahun, H.; Croteau, D.L.; Scheibye-Knudsen, M.; Marosi, K.; Lu, H.; Shamanna, R.A.; Kalyanasundaram, S.; Bollineni, R.C.; Wilson, M.A.; et al. NAD(+) Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab. 2016, 24, 566–581. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Bohr, V.A. NAD(+): The convergence of DNA repair and mitophagy. Autophagy 2017, 13, 442–443. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Lautrup, S.; Jensen, M.B.; Yang, B.; SenGupta, T.; Caponio, D.; Khezri, R.; Demarest, T.G.; Aman, Y.; et al. NAD(+) augmentation restores mitophagy and limits accelerated aging in Werner syndrome. Nat. Commun. 2019, 10, 5284. [Google Scholar] [CrossRef] [PubMed]
- Saini, J.S.; Corneo, B.; Miller, J.D.; Kiehl, T.R.; Wang, Q.; Boles, N.C.; Blenkinsop, T.A.; Stern, J.H.; Temple, S. Nicotinamide Ameliorates Disease Phenotypes in a Human iPSC Model of Age-Related Macular Degeneration. Cell Stem Cell 2017, 20, 635–647. [Google Scholar] [CrossRef]
- Zhou, M.; Geathers, J.S.; Grillo, S.L.; Weber, S.R.; Wang, W.; Zhao, Y.; Sundstrom, J.M. Role of Epithelial-Mesenchymal Transition in Retinal Pigment Epithelium Dysfunction. Front. Cell Dev. Biol. 2020, 8, 501. [Google Scholar] [CrossRef]
- Hyttinen, J.M.T.; Kannan, R.; Felszeghy, S.; Niittykoski, M.; Salminen, A.; Kaarniranta, K. The Regulation of NFE2L2 (NRF2) Signalling and Epithelial-to-Mesenchymal Transition in Age-Related Macular Degeneration Pathology. Int. J. Mol. Sci. 2019, 20, 5800. [Google Scholar] [CrossRef]
- Poljsak, B.; Milisav, I. Vitamin B3 forms as precursors to NAD+: Are they safe? Trends Food Sci. Technol. 2018, 79, 198–203. [Google Scholar] [CrossRef]
- Knip, M.; Douek, I.F.; Moore, W.P.T.; Gillmor, H.A.; McLean, A.E.M.; Bingley, P.J.; Gale, E.A.M.; ENDIT Group. Safety of high-dose nicotinamide: A review. Diabetologia 2000, 43, 1337–1345. [Google Scholar] [CrossRef]
- Conze, D.; Brenner, C.; Kruger, C.L. Safety and Metabolism of Long-term Administration of NIAGEN (Nicotinamide Riboside Chloride) in a Randomized, Double-Blind, Placebo-controlled Clinical Trial of Healthy Overweight Adults. Sci. Rep. 2019, 9, 9772. [Google Scholar] [CrossRef]
- Martens, C.R.; Denman, B.A.; Mazzo, M.R.; Armstrong, M.L.; Reisdorph, N.; McQueen, M.B.; Chonchol, M.; Seals, D.R. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD(+) in healthy middle-aged and older adults. Nat. Commun. 2018, 9, 1286. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cimaglia, G.; Votruba, M.; Morgan, J.E.; André, H.; Williams, P.A. Potential Therapeutic Benefit of NAD+ Supplementation for Glaucoma and Age-Related Macular Degeneration. Nutrients 2020, 12, 2871. https://doi.org/10.3390/nu12092871
Cimaglia G, Votruba M, Morgan JE, André H, Williams PA. Potential Therapeutic Benefit of NAD+ Supplementation for Glaucoma and Age-Related Macular Degeneration. Nutrients. 2020; 12(9):2871. https://doi.org/10.3390/nu12092871
Chicago/Turabian StyleCimaglia, Gloria, Marcela Votruba, James E. Morgan, Helder André, and Pete A. Williams. 2020. "Potential Therapeutic Benefit of NAD+ Supplementation for Glaucoma and Age-Related Macular Degeneration" Nutrients 12, no. 9: 2871. https://doi.org/10.3390/nu12092871
APA StyleCimaglia, G., Votruba, M., Morgan, J. E., André, H., & Williams, P. A. (2020). Potential Therapeutic Benefit of NAD+ Supplementation for Glaucoma and Age-Related Macular Degeneration. Nutrients, 12(9), 2871. https://doi.org/10.3390/nu12092871