Obese Adipose Tissue Secretion Induces Inflammation in Preadipocytes: Role of Toll-Like Receptor-4

 , , and
, , and
Abstract
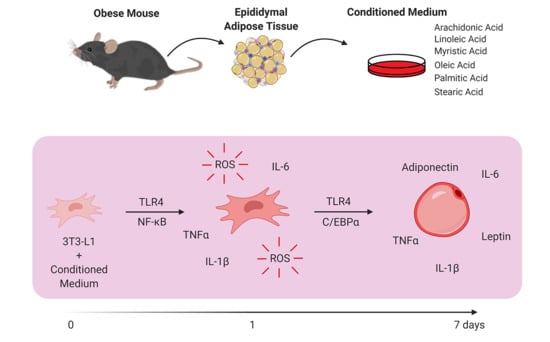
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Obesity Animal Model
2.3. AT Explant Culture
2.4. Gas Chromatography-Mass Spectrometry (GC–MS)
2.5. Cell Culture
2.6. Reactive Oxygen Species (ROS) Production
2.7. NF-ĸB Activation
2.8. Immunofluorescence Microscopy
2.9. ELISA
2.10. Cellular Extract
2.11. Western Blot Analysis
2.12. Adipogenesis Quantification
2.13. Statistical Analysis
3. Results
3.1. AT from Obese Animals Released Increased Amounts of FFAs
3.2. FFAs Released by Obese AT Induce ROS Production and NF-ĸB Translocation in Preadipocytes in a TLR4-Dependent Manner
3.3. Stimulation of Preadipocytes with CM Derived from Obese AT Increased Inflammatory Cytokines Released via TLR4 Signaling
3.4. CM Derived from Obese AT Increased C/EBPα Expression in Preadipocytes via TLR4 Signaling
3.5. Preadipocytes Stimulated with the CM Derived from Obese AT Underwent Morphologic Changes and Lipid Accumulation in a TLR4-Dependent Manner
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hotamisligil, G.S. Inflammation and Metabolic Disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity Is Associated with Macrophage Accumulation in Adipose Tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic Inflammation in Fat Plays a Crucial Role in the Development of Obesity-Related Insulin Resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Blüher, M. Adipose Tissue Dysfunction in Obesity. Exp. Clin. Endocrinol. Diabetes. 2009, 117, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Wong, S.; Xie, W.; Lei, T.; Luo, Z. Palmitate Modulates Intracellular Signaling, Induces Endoplasmic Reticulum Stress, and Causes Apoptosis in Mouse 3t3-l1 and Rat Primary Preadipocytes. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E576–E586. [Google Scholar] [CrossRef] [PubMed]
- Makki, K.P.; Wolowczuk, I. Adipose Tissue in Obesity-Related Inflammation and Insulin Resistance: Cells, Cytokines, and Chemokines. ISRN Inflamm. 2013, 139239. [Google Scholar] [CrossRef]
- Bartelt, A.; Merkel, M.; Heeren, J. A New, Powerful Player in Lipoprotein Metabolism: Brown Adipose Tissue. J. Mol. Med. 2012, 90, 887–893. [Google Scholar] [CrossRef]
- Bjorntorp, P.; Bergman, H.; Varnauskas, E. Plasma Free Fatty Acid Turnover in Obesity. Acta Med. Scand. 1969, 185, 351–356. [Google Scholar] [CrossRef]
- Karpe, F.; Dickmann, J.R.; Frayn, K.N. Fatty Acids, Obesity, and Insulin Resistance: Time for a Reevaluation. Diabetes 2011, 60, 2441–2449. [Google Scholar] [CrossRef]
- Ferrante, A.W., Jr. Obesity-induced inflammation: A metabolic dialogue in the language of inflammation. J. Intern. Med. 2007, 262, 408–414. [Google Scholar] [CrossRef]
- Carlsen, H.; Haugen, F.; Zadelaar, S.; Kleemann, R.; Kooistra, T.; Drevon, C.A.; Blomhoff, R. Diet-Induced Obesity Increases NF-κB Signaling in Reporter Mice. Genes Nutr. 2009, 4, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Dasu, M.R.; Jialal, I. Free fatty acids in the presence of high glucose amplify monocyte inflammation via Toll-like receptors. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E145–E154. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Kim, Y.J.; Fillmore, J.J.; Chen, Y.; Moore, I.; Lee, J.; Yuan, M.; Li, Z.W.; Karin, M.; Perret, P.; et al. Prevention of fat-induced insulin resistance by salicylate. J. Clin. Investig. 2001, 108, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Konstantopoulos, N.; Lee, J.; Hansen, L.; Li, Z.W.; Karin, M.; Shoelson, S.E. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science 2001, 293, 1673–1677. [Google Scholar] [CrossRef]
- Renovato-Martins, M.; Matheus, M.E.; de Andrade, I.R.; Moraes, J.A.; da Silva, S.V.; Citelli dos Reis, M.; de Souza, A.A.; Bouskela, E.; Barja-Fidalgo, C. Microparticles derived from obese adipose tissue elicit a pro-inflammatory phenotype of CD16 +, CCR5 + and TLR8 + monocytes. Biochim. Biophys. Acta Mol. Bas. Dis. 2017, 1863, 139–151. [Google Scholar] [CrossRef]
- Christie, W.W. Gas Chromatography and Lipids: A Practical Guide. Bridgwater; The Oily Press Ltd.: Dundee, Scotland, UK, 1989. [Google Scholar]
- Han, C.J.; Kargi, A.Y.; Omer, M.; Chan, C.K.; Wabitsch, M.; O’Brien, K.D.; Wight, T.N.; Chait, A. Differential effect of saturated and unsaturated free fatty acids on the generation of monocyte adhesion and chemotactic factors by adipocytes: Dissociation of adipocyte hypertrophy from inflammation. Diabetes 2010, 59, 386–396. [Google Scholar] [CrossRef]
- Schaeffler, A.; Gross, P.; Buettner, R.; Bollheimer, C.; Buechler, C.; Neumeier, M.; Kopp, A.; Schoelmerich, J.; Falk, W. Fatty acid-induced induction of Toll-like receptor-4/nuclear factor-kappaB pathway in adipocytes links nutritional signalling with innate immunity. Immunology 2009, 126, 233–245. [Google Scholar] [CrossRef]
- Palmer, J.D.; Rifkind, D. Neutralization of the hemodynamic effects of endotoxin by polymyxin B. Surg. Gynecol. Obs. 1974, 138, 755–759. [Google Scholar]
- Lindemann, R.A. Bacterial activation of human natural killer cells: Role of cell surface lipopolysaccharide. Infect. Immun. 1988, 56, 1301–1308. [Google Scholar] [CrossRef]
- Longo, M.; Zatterale, F.; Naderi, J.; Parrillo, L.; Formisano, P.; Alexander, G.; Beguinot, F.; Miele, C. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. Int. J. Mol. Sci. 2019, 20, 2358. [Google Scholar] [CrossRef] [PubMed]
- MacDougald, A.O.; Lane, M.D. Transcriptional regulation of gene expression during adipocyte differentiation. Annu. Rev. Biochem. 1995, 64, 345–373. [Google Scholar] [CrossRef]
- Madsen, L.; Petersen, R.K.; Kristiansen, K. Regulation of adipocytes differentiation and function by polyunsaturated fatty acids. Biochim. Biophys. Acta 2005, 1740, 266–286. [Google Scholar] [CrossRef] [PubMed]
- Unamuno, X.; Gómez-Ambrosi, J.; Rodríguez, A.; Becerril, S.; Frühbeck, G.; Catalán, V. Adipokine dysregulation and adipose tissue inflammation in human obesity. Eur. J. Clin. Investig. 2018, e1299. [Google Scholar] [CrossRef] [PubMed]
- Cousin, B.; André, M.; Casteilla, L.; Pénicaud, L. Altered macrophage-like functions of preadipocytes in inflammation and genetic obesity. J. Cell. Physiol. 2001, 186, 380–386. [Google Scholar] [CrossRef]
- O’Hara, A.; Lim, F.L.; Mazzattim, D.J. Trayhurn P Stimulation of inflammatory gene expression in human preadipocytes by macrophage-conditioned medium: Upregulation of IL-6 production by macrophage-derived IL-1beta. Mol. Cell. Endocrinol. 2012, 349, 239–247. [Google Scholar] [CrossRef]
- Chung, S.; Lapoint, K.; Martinez, K.; Kennedy, A.; Boysen Sandberg, M.B.; McIntosh, M.K. Preadipocytes mediate lipopolysaccharide-induced inflammation and insulin resistance in primary cultures of newly differentiated human adipocytes. Endocrinology 2006, 147, 5340–5351. [Google Scholar] [CrossRef]
- Poulain-Godefroy, O.; Froguel, P. Preadipocyte response and impairment of differentiation in an inflammatory environment. Biochem. Biophys. Res. Commun. 2007, 356, 662–667. [Google Scholar] [CrossRef]
- Keophiphath, M.; Achard, V.; Henegar, C.; Rouault, C.; Clement, K.; Lacasa, D. Macrophage-secreted factors promote a profibrotic phenotype in human preadipocytes. Mol. Endocrinol. 2009, 23, 11–24. [Google Scholar] [CrossRef]
- Lacasa, D.; Taleb, S.; Keophiphath, M.; Miranville, A.; Clement, K. Macrophage-secreted factors impair human adipogenesis: Involvement of proinflammatory state in preadipocytes. Endocrinology 2007, 148, 868–877. [Google Scholar] [CrossRef]
- Dordevic, A.L.; Konstantopoulos, N.; Cameron-Smit, D. 3T3-L1 Preadipocytes Exhibit Heightened Monocyte-Chemoattractant Protein-1 Response to Acute Fatty Acid Exposure. PLoS ONE 2014, 9, e99382. [Google Scholar] [CrossRef] [PubMed]
- Shengyi, S.; Yewei, J.; Sander, K.; Ling, Q. Mechanisms of Inflammatory Responses in Obese Adipose Tissue. Annu. Rev. Nutr. 2012, 21, 261–286. [Google Scholar]
- Tsukumo, D.M.; Carvalho-Filho, M.A.; Carvalheira, J.B.; Prada, P.O.; Hirabara, S.M.; Schenka, A.A.; Araújo, E.P.; Vassallo, J.; Curi, R.; Velloso, L.A.; et al. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes 2007, 56, 1986–1998. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.L.; Bikman, B.T.; Wang, L.P.; Yuguang, G.; Sargent, K.M.; Bulchand, S.; Knotts, T.A.; Shui, G.; Clegg, D.J.; Wenk, M.R.; et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J. Clin. Investig. 2011, 121, 1858–1870. [Google Scholar] [CrossRef]
- Kramer, B.; França, L.M.; Zhang, Y.; Paes, A.M.A.; Gerdes, A.M.; Carrillo-Sepulveda, M.A. Western diet triggres toll-like receptor 4 signaling -induces endothelial discfunction in female Wistar rats. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1735–H1747. [Google Scholar] [CrossRef]
- Orr, J.S.; Puglisi, M.J.; Ellacott, K.L.; Lumeng, C.N.; Wasserman, D.H.; Hasty, A.H. Toll-like receptor 4 deficiency promotes the alternative activation of adipose tissue macrophages. Diabetes 2012, 61, 2718–2727. [Google Scholar] [CrossRef]
- Ramos-Andrade, I.; Moraes, J.; Brandão-Costa, R.M.; Vargas da Silva, S.; de Souza, A.; da Silva, C.; Renovato-Martins, M.; Barja-Fidalgo, C. Obese adipose tissue extracellular vesicles raise breast cancer cell malignancy. Endocr. Relat. Cancer 2020, 27, 571–582. [Google Scholar] [CrossRef]
- Hauck, A.K.; Huang, Y.; Hertzel, A.V.; Bernlohr, D.A. Adipose oxidative stress and protein carbonylation. J. Biol. Chem. 2019, 294, 1083–1088. [Google Scholar] [CrossRef]
- Asehnoune, K.; Strassheim, D.; Mitra, S.; Kim, J.Y.; Abraham, E. Involvement of Reactive Oxygen Species in Toll-Like Receptor 4-Dependent Activation of NF-κB. J. Immunol. 2004, 172, 2522–2529. [Google Scholar] [CrossRef]
- Lee, Y.S.; Li, P.; Huh, J.Y.; Hwang, I.J.; Lu, M.; Kim, J.I.; Ham, M.; Talukdar, S.; Chen, A.; Lu, W.J.; et al. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes 2011, 60, 2474–2483. [Google Scholar] [CrossRef]
- Hwang, D.H.; Kim, J.A.; Lee, J.Y. Mechanisms for the activation of Toll-like receptor 2/4 by saturated fatty acids and inhibition by docosahexaenoic acid. Eur. J. Pharmacol. 2016, 785, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, G.I.; Langley, K.G.; Berglund, N.A.; Kammoun, H.L.; Reibe, S.; Estevez, E.; Weir, J.; Mellett, N.A.; Pernes, G.; Conway, J.R.W.; et al. Evidence that TLR4 is not a receptor for saturated fatty acids but mediates lipid-induced inflammation by reprogramming macrophage metabolism. Cell Metab. 2018, 27, 1096–1110. [Google Scholar] [CrossRef] [PubMed]
- Mac Dougald, O.A.; Lane, M.D. When precursors are also regulators. Curr. Biol. 1995, 5, 618–621. [Google Scholar] [CrossRef]
- Kleemann, R.; van Erk, M.; Verschuren, L.; van den Hoek, A.M.; Koek, M.; Wielinga, P.Y.; Jie, A.; Pellis, L.; Bobeldijk-Pastorova, I.; Kelder, T.; et al. Time-resolved and tissue-specific systems analysis of the pathogenesis of insulin resistance. PLoS ONE 2010, 21, e8817. [Google Scholar] [CrossRef]
- Cullberg, K.B.; Larsen, J.Ø.; Pedersen, S.B.; Richelsen, B. Effects of LPS and dietary free fatty acids on MCP-1 in 3T3-L1 adipocytes and macrophages in vitro. Nutr. Diabetes 2014, 4, e113. [Google Scholar] [CrossRef]
- Kumar, R.; Sharma, A.; Padwad, Y.; Sharma, R. Preadipocyte secretory factors differentially modulate murine macrophage functions during aging which are reversed by the application of phytochemical EGCG. Biogerontology 2020, 21, 325–343. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkoniam, T.; Stout, M.B.; Giorgadze, N.; Wang, L.; Li, P.W.; Heppelmann, C.J.; Bouloumié, A.; Jensenm, M.D.; Bergen, H.R., 3rd; et al. Inflammation and the Depot-Specific Secretome of Human Preadipocytes. Obesity 2015, 23, 989–999. [Google Scholar] [CrossRef]
- McLaughlin, T.; Craig, C.; Liu, L.F.; Perelman, D.; Allister, C.; Spielman, D.; Cushman, S.W. Adipose Cell Size and Regional Fat Deposition as Predictors of Metabolic Response to Overfeeding in Insulin-Resistant and Insulin-Sensitive Humans. Diabetes 2016, 65, 1245–1254. [Google Scholar] [CrossRef]
- Woo, C.H.; Jang, J.E.; Lee, S.E.; Koh, E.H.; Lee, K.U. Mitochondrial Dysfunction in Adipocytes as a Primary Cause of Adipose Tissue Inflammation. Diabetes Metab. 2019, 43, 247–256. [Google Scholar] [CrossRef]
- El Hafidi, M.; Buelna-Chontal, M.; Sánchez-Muñoz, F.; Carbó, R. Adipogenesis: A Necessary but Harmful Strategy. Int. J. Mol. Sci. 2019, 20, 3657. [Google Scholar] [CrossRef]
- Frühbeck, G.; Catalán, V.; Rodríguez, A.; Gómez-Ambrosi, J. Adiponectin-leptin ratio: A promising index to estimate adipose tissue dysfunction. Relation with obesity-associated cardiometabolic risk. Adipocyte 2018, 7, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, T.; Yamauchi, T.; Kubota, N.; Hara, K.; Ueki, K.; Tobe, K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J. Clin. Investig. 2006, 116, 1784–1792. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Renovato-Martins, M.; Moreira-Nunes, C.; Atella, G.C.; Barja-Fidalgo, C.; Moraes, J.A.d. Obese Adipose Tissue Secretion Induces Inflammation in Preadipocytes: Role of Toll-Like Receptor-4. Nutrients 2020, 12, 2828. https://doi.org/10.3390/nu12092828
Renovato-Martins M, Moreira-Nunes C, Atella GC, Barja-Fidalgo C, Moraes JAd. Obese Adipose Tissue Secretion Induces Inflammation in Preadipocytes: Role of Toll-Like Receptor-4. Nutrients. 2020; 12(9):2828. https://doi.org/10.3390/nu12092828
Chicago/Turabian StyleRenovato-Martins, Mariana, Catharina Moreira-Nunes, Georgia C. Atella, Christina Barja-Fidalgo, and João Alfredo de Moraes. 2020. "Obese Adipose Tissue Secretion Induces Inflammation in Preadipocytes: Role of Toll-Like Receptor-4" Nutrients 12, no. 9: 2828. https://doi.org/10.3390/nu12092828
APA StyleRenovato-Martins, M., Moreira-Nunes, C., Atella, G. C., Barja-Fidalgo, C., & Moraes, J. A. d. (2020). Obese Adipose Tissue Secretion Induces Inflammation in Preadipocytes: Role of Toll-Like Receptor-4. Nutrients, 12(9), 2828. https://doi.org/10.3390/nu12092828