Age-Related Changes and Sex-Related Differences in Brain Iron Metabolism



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Systemic Iron Metabolism
2.1. Iron Absorption
2.2. Iron Transport and Distribution
2.3. Iron Storage and Recycling
2.4. Regulation of Systemic Iron Homeostasis
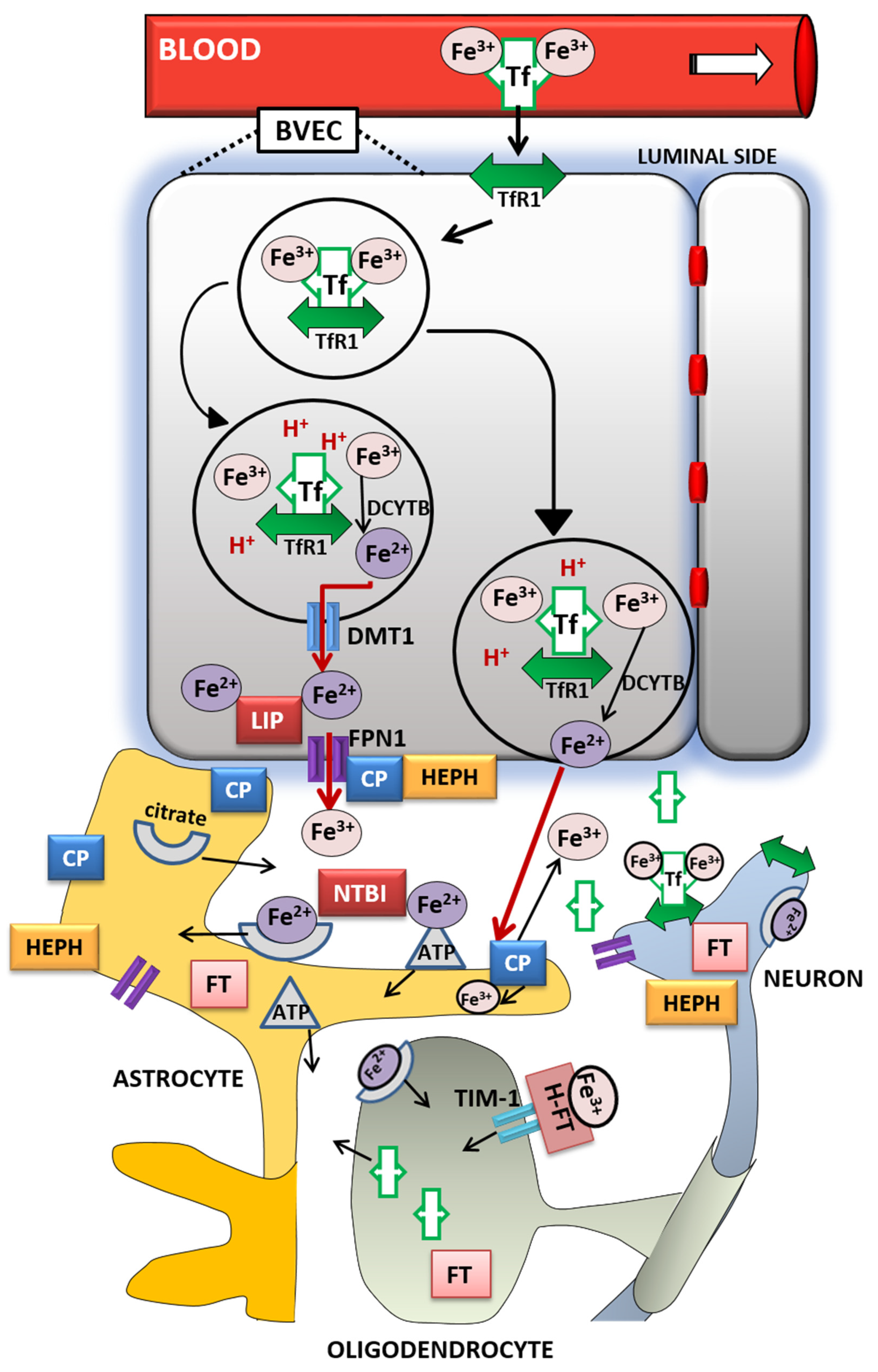
3. Brain Iron Metabolism
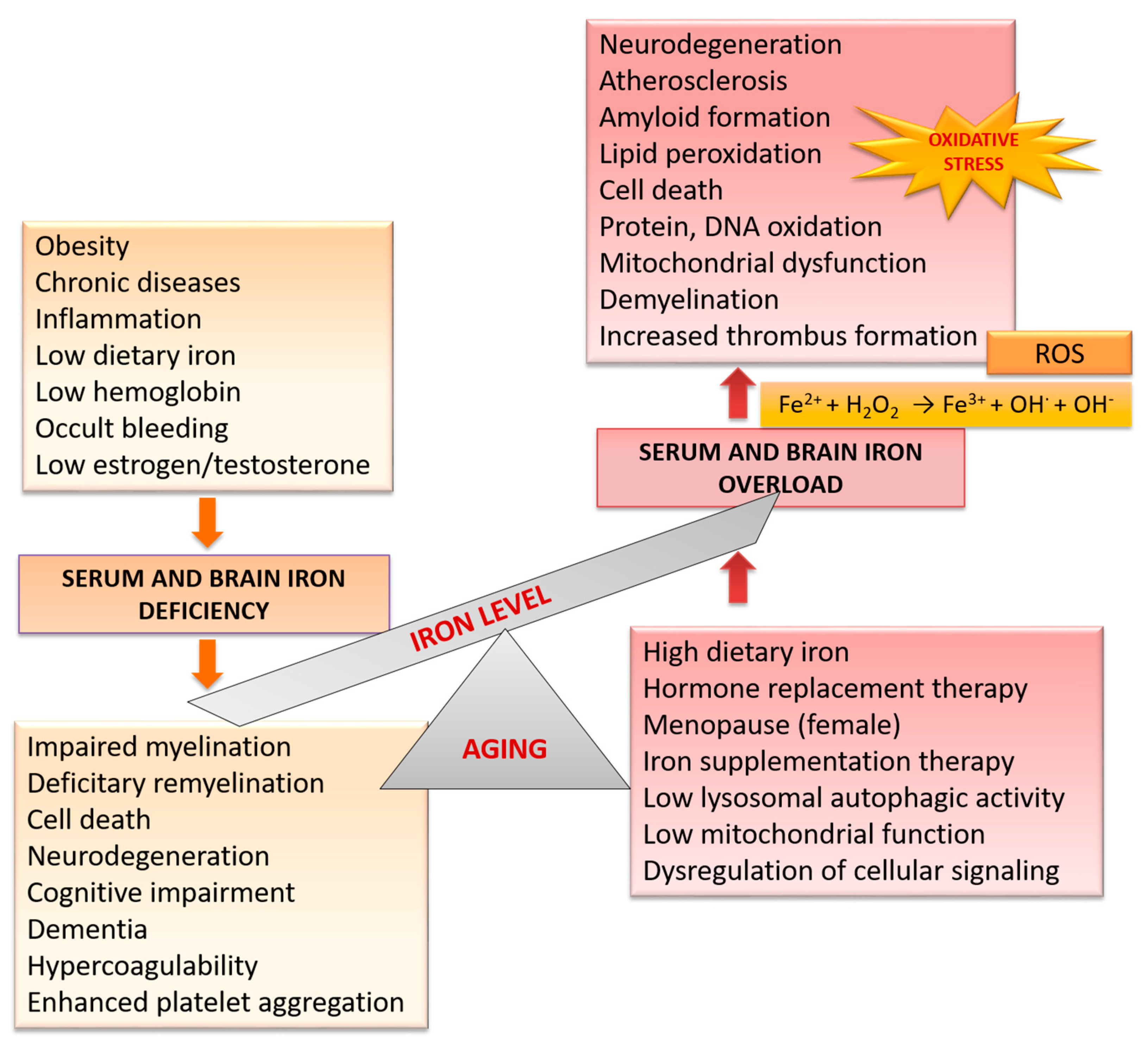
4. Age-Related Iron Dyshomeostasis
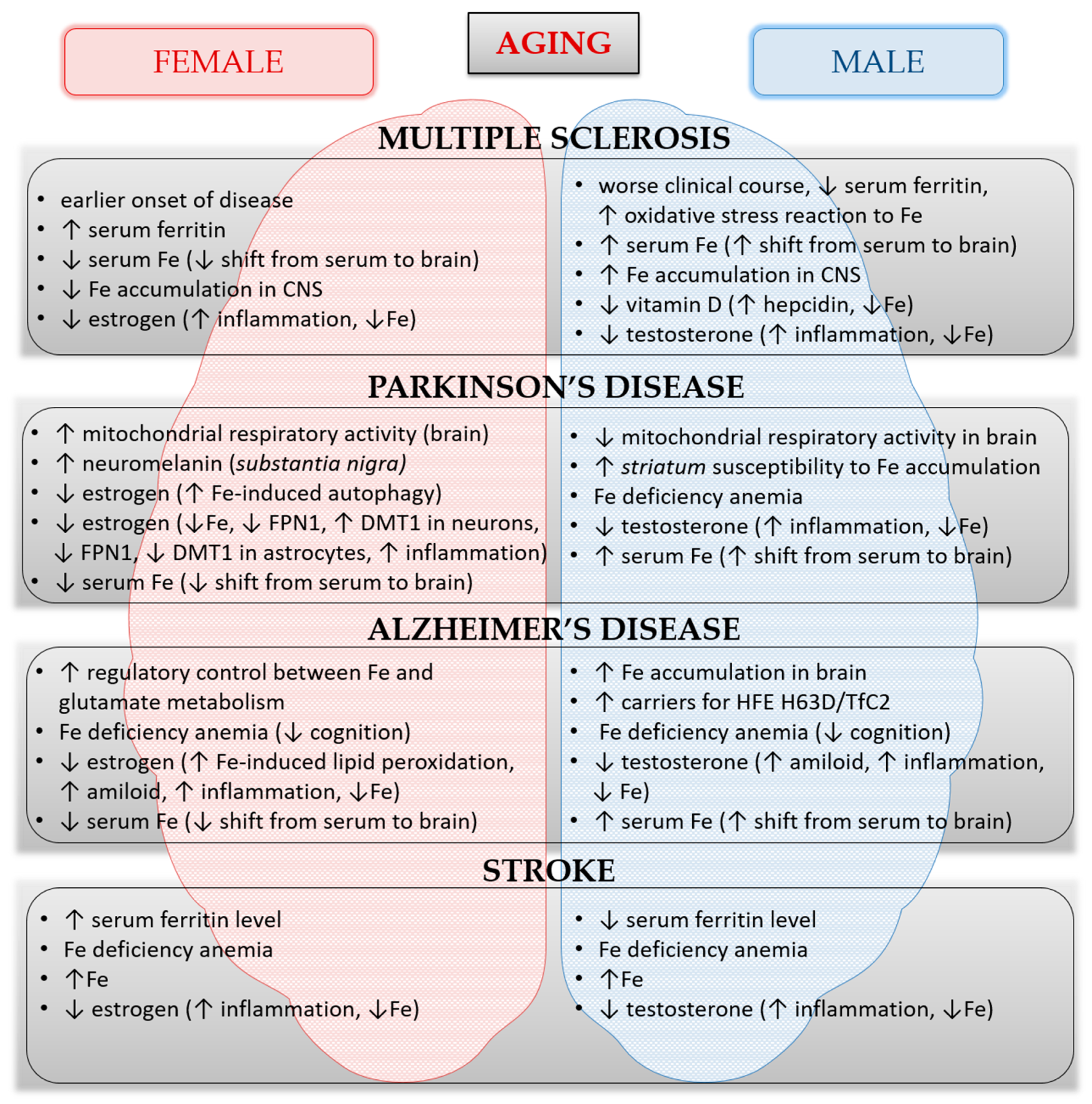
5. Sex-Related Differences in Iron Homeostasis during Healthy Aging and in Neurological Disorders
5.1. Iron and Multiple Sclerosis
5.2. Iron and Stroke
5.3. Iron and Parkinson’s Disease
5.4. Iron and Alzheimer’s Disease
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Munoz, M.; Villar, I.; Garcia-Erce, J.A. An Update on Iron Physiology. World J. Gastroenterol. 2009, 15, 4617–4626. [Google Scholar] [CrossRef] [PubMed]
- Darshan, D.; Frazer, D.M.; Anderson, G.J. Molecular Basis of Iron-Loading Disorders. Expert Rev. Mol. Med. 2010, 12, e36. [Google Scholar] [CrossRef] [PubMed]
- Sheftel, A.; Stehling, O.; Lill, R. Iron-Sulfur Proteins in Health and Disease. Trends Endocrinol. Metab. 2010, 21, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Paw, B.H. Cellular and Mitochondrial Iron Homeostasis in Vertebrates. Biochim. Biophys. Acta 2012, 1823, 1459–1467. [Google Scholar] [CrossRef]
- Siah, C.W.; Ombiga, J.; Adams, L.A.; Trinder, D.; Olynyk, J.K. Normal Iron Metabolism and the Pathophysiology of Iron Overload Disorders. Clin. Biochem. Rev. 2006, 27, 5–16. [Google Scholar]
- Gammella, E.; Recalcati, S.; Cairo, G. Dual Role of ROS as Signal and Stress Agents: Iron Tips the Balance in Favor of Toxic Effects. Oxid. Med. Cell. Longev. 2016, 2016, 8629024. [Google Scholar] [CrossRef]
- Zhao, Z. Iron and Oxidizing Species in Oxidative Stress and Alzheimer’s Disease. Aging Med. 2019, 2, 82–87. [Google Scholar] [CrossRef]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to Tango: Regulation of Mammalian Iron Metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef]
- Ye, H.; Rouault, T.A. Human Iron-Sulfur Cluster Assembly, Cellular Iron Homeostasis, and Disease. Biochemistry 2010, 49, 4945–4956. [Google Scholar] [CrossRef]
- Musallam, K.M.; Taher, A.T. Iron Deficiency Beyond Erythropoiesis: Should We Be Concerned? Curr. Med. Res. Opin. 2018, 34, 81–93. [Google Scholar] [CrossRef]
- Van Rensburg, S.J.; Kotze, M.J.; van Toorn, R. The Conundrum of Iron in Multiple Sclerosis—Time for an Individualised Approach. Metab. Brain Dis. 2012, 27, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Real, J.M.; Manco, M. Effects of Iron Overload on Chronic Metabolic Diseases. Lancet Diabetes Endocrinol. 2014, 2, 513–526. [Google Scholar] [CrossRef]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The Role of Iron in Brain Ageing and Neurodegenerative Disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef]
- Ashraf, A.; Clark, M.; So, P.-W. The Aging of Iron Man. Front. Aging Neurosci. 2018, 10, 65. [Google Scholar] [CrossRef] [PubMed]
- Gill, D.; Monori, G.; Tzoulaki, I.; Dehghan, A. Iron Status and Risk of Stroke. A Mendelian Randomization Study. Stroke 2018, 49, 2815–2821. [Google Scholar] [CrossRef]
- Ćurko-Cofek, B.; Grubić Kezele, T.; Marinić, J.; Tota, M.; Starčević Čizmarević, N.; Milin, Č.; Ristić, S.; Radošević-Stašić, B.; Barac-Latas, V. Chronic Iron Overload Induces Gender-Dependent Changes in Iron Homeostasis, Lipid Peroxidation and Clinical Course of Experimental Autoimmune Encephalomyelitis. Neurotoxicology 2016, 27, 1–12. [Google Scholar] [CrossRef]
- Persson, N.; Wu, J.; Zhang, Q.; Liu, T.; Shen, J.; Bao, R.; Ni, M.; Liu, T.; Wang, Y.; Spincemaille, P. Age and Sex Related Differences in Subcortical Brain Iron Concentrations Among Healthy Adults. Neuroimage 2015, 122, 385–398. [Google Scholar] [CrossRef]
- Koellhoffer, E.C.; McCullough, L.D. The Effects of Estrogen in Ischemic Stroke. Transl. Stroke Res. 2013, 4, 390–401. [Google Scholar] [CrossRef]
- Zeller, T.; Schnabel, R.B.; Appelbaum, S.; Ojeda, F.; Berisha, F.; Schulte-Steinberg, B.; Brueckmann, B.E.; Kuulasmaa, K.; Jousilahti, P.; Blankenberg, S.; et al. Low Testosterone Levels Are Predictive for Incident Atrial Fibrillation and Ischaemic Stroke in Men, but Protective in Women—Results from the FINRISK Study. Eur. J. Prev. Cardiol. 2018, 25, 1133–1139. [Google Scholar] [CrossRef]
- Khalifa, A.R.M.; Abdel-Rahman, E.A.; Mahmoud, A.M.; Ali, M.H.; Noureldin, M.; Saber, S.H.; Mohsen, M.; Ali, S.S. Sex-Specific Differences in Mitochondria Biogenesis, Morphology, Respiratory Function, and ROS Homeostasis in Young Mouse Heart and Brain. Physiol. Rep. 2017, 5, e13125. [Google Scholar] [CrossRef]
- Jurado-Coronel, J.C.; Cabezas, R.; Rodríguez, M.F.A.; Echeverria, V.; García-Segura, L.M.; Barreto, G.E. Sex Differences in Parkinson’s Disease: Features on Clinical Symptoms, Treatment Outcome, Sexual Hormones and Genetics. Front. Neuroendocrinol. 2018, 50, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Bartzokis, G.; Lu, P.H.; Tingus, K.; Peters, D.G.; Amar, C.P.; Tishler, T.A.; Finn, J.P.; Villablanca, P.; Altshuler, L.L.; Mintz, J. Gender and Iron Genes may Modify Associations Between Brain Iron and Memory in Healthy Aging. Neuropsychopharmacology 2011, 36, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Fairweather-Tait, S.J.; Wawer, A.A.; Gillings, R.; Jennings, A.; Myint, P.K. Iron Status in the Elderly. Mech. Ageing Dev. 2014, 136, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Belaidi, A.A.; Bush, A.I. Iron Neurochemistry in Alzheimer’s Disease and Parkinson’s Disease: Targets for Therapeutics. J. Neurochem. 2016, 139, 179–197. [Google Scholar] [CrossRef]
- Camaschella, C.; Nai, A.; Silvestri, L. Iron Metabolism and Iron Disorders Revisited in the Hepcidin Era. Haematologica 2020, 105, 260–272. [Google Scholar] [CrossRef]
- Miret, S.; Simpson, R.J.; McKie, A.T. Physiology and Molecular Biology of Dietary Iron Absorption. Annu. Rev. Nutr. 2003, 23, 283–301. [Google Scholar] [CrossRef]
- Fuqua, B.K.; Vulpe, C.D.; Anderson, G.J. Intestinal Iron Absorption. J. Trace Elem. Med. Biol. 2012, 26, 115–119. [Google Scholar] [CrossRef]
- Lane, D.J.R.; Richardson, D.R. The Active Role of Vitamin C in Mammalian Iron Metabolism: Much More than just Enhanced Iron Absorption! Free Radic. Biol. Med. 2014, 75, 69–83. [Google Scholar] [CrossRef]
- McKie, A.T.; Barrow, D.; Latunde-Dada, G.O.; Rolfs, A.; Sager, G.; Mudaly, M.; Richardson, C.; Barlow, D.; Bomford, A.; Peters, T.J.; et al. An Iron-Regulated Ferric Reductase Associated with the Absorption of Dietary Iron. Science 2001, 291, 1755–1759. [Google Scholar] [CrossRef]
- Ems, T.; St Lucia, K.; Huecker, M.R. Biochemistry, Iron Absorption. In: StatPearls [Internet]. (Updated 30 April 2020). Available online: http://www.ncbi.nlm.nih.gov/books/NBK448204/ (accessed on 15 May 2020).
- Fleming, R.E.; Bacon, B.R. Orchestration of Iron Homeostasis. N. Engl. J. Med. 2005, 352, 1741–1744. [Google Scholar] [CrossRef]
- Duck, K.A.; Connor, J.R. Iron Uptake and Transport Across Physiological Barriers. Biometals 2016, 29, 573–591. [Google Scholar] [CrossRef] [PubMed]
- Abboud, S.; Haile, D.J. A Novel Mammalian Iron-Regulated Protein Involved in Intracellular Iron Metabolism. J. Biol. Chem. 2000, 275, 19906–19912. [Google Scholar] [CrossRef]
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem. Sci. 2016, 41, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The Iron Exporter Ferroportin/Slc40a1 Is Essential for Iron Homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef] [PubMed]
- De Domenico, I.; Ward, D.M.; Bonaccorsi di Patti, M.C.; Jeong, S.Y.; David, S.; Musci, G.; Kaplan, J. Ferroxidase Activity Is Required for the Stability of Cell Surface Ferroportin in Cells Expressing GPI-ceruloplasmin. EMBO J. 2007, 26, 2823–2831. [Google Scholar] [CrossRef]
- Vulpe, C.D.; Kuo, Y.M.; Murphy, T.L.; Cowley, L.; Askwith, C.; Libina, N.; Gitschier, J.; Anderson, G.J. Hephaestin, a Ceruloplasmin Homologue Implicated in Intestinal Iron Transport, Is Defective in the SLA Mouse. Nat. Genet. 1999, 21, 195–199. [Google Scholar] [CrossRef]
- Coffey, R.; Ganz, T. Iron Homeostasis: An Anthropocentric Perspective. J. Biol. Chem. 2017, 292, 12727–12734. [Google Scholar] [CrossRef]
- Gunshin, H.; Fujiwara, Y.; Custodio, A.O.; Direnzo, C.; Robine, S.; Andrews, N.C. Slc11a2 Is Required for Intestinal Iron Absorption and Erythropoiesis but Dispensable in Placenta and Liver. J. Clin. Investig. 2005, 115, 1258–1266. [Google Scholar] [CrossRef]
- Liuzzi, J.P.; Aydemir, F.; Nam, H.; Knutson, M.D.; Cousins, R.J. Zip14 (Slc39a14) Mediates Non-Transferrin-Bound Iron Uptake into Cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13612–13617. [Google Scholar] [CrossRef]
- Krishnamurthy, P.; Xie, T.; Schuetz, J.D. The Role of Transporters in Cellular Heme and Porphyrin Homeostasis. Pharmacol. Ther. 2007, 114, 345–358. [Google Scholar] [CrossRef]
- Le Blanc, S.; Garrick, M.D.; Arredondo, M. Heme Carrier Protein 1 Transports Heme and Is Involved in Heme-Fe Metabolism. Am. J. Physiol. Cell Physiol. 2012, 302, C1780–C1785. [Google Scholar] [CrossRef] [PubMed]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [PubMed]
- Crichton, R.R.; Danielsson, B.G.; Geisser, P. Iron Therapy with Special Emphasis on Intravenous Administration, 4th ed.; Crichton, R.R., Danielsson, B.G., Geisser, P., Eds.; UNI-MED Verlag AG: Bremen, Germany, 2008; pp. 14–24. [Google Scholar]
- Peyrin-Biroulet, L.; Williet, N.; Cacoub, P. Guidelines on the Diagnosis and Treatment of Iron Deficiency Across Indications: A Systematic Review. Am. J. Clin. Nutr. 2015, 102, 1585–1594. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, H. Transferrin and Transferrin Receptors Update. Free Radic. Biol. Med. 2019, 133, 46–54. [Google Scholar] [CrossRef]
- Lambert, L.A.; Mitchell, S.L. Molecular Evolution of the Transferrin Receptor/Glu-tamate Carboxypeptidase II Family. J. Mol. Evol. 2007, 64, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Ohgami, R.S.; Campagna, D.R.; Greer, E.L.; Antiochos, B.; McDonald, A.; Chen, J.; Sharp, J.J.; Fujiwara, Y.; Barker, J.E.; Fleming, M.D. Identification of a Ferrireductase Required for Efficien Ttransferrin-Dependent Iron Uptake in Erythroid Cells. Nat. Genet. 2005, 37, 1264–1269. [Google Scholar] [CrossRef]
- Rishi, G.; Subramaniam, V.N. The Liver in Regulation of Iron Homeostasis. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 313, G157–G165. [Google Scholar] [CrossRef]
- Xu, M.M.; Wang, J.; Xie, J.X. Regulation of Iron Metabolism by Hypoxia-Inducible Factors. Sheng Li Xue Bao 2017, 69, 598–610. [Google Scholar]
- Rouault, T.A. The Role of Iron Regulatory Proteins in Mammalian Iron Homeostasis and Disease. Nat. Chem. Biol. 2006, 2, 406–414. [Google Scholar] [CrossRef]
- Yikilmaz, E.; Rouault, T.A.; Schuck, P. Self-Association and Ligand-Induced Conformational Changes of Iron Regulatory Proteins 1 and 2. Biochemistry 2005, 44, 8470–8478. [Google Scholar] [CrossRef]
- Iwai, K.; Drake, S.K.; Wehr, N.B.; Weissman, A.M.; LaVaute, T.; Minato, N.; Klausner, R.D.; Levine, R.L.; Rouault, T.A. Iron-Dependent Oxidation, Ubiquitination, and Degradation of Iron Regulatory Protein 2: Implications for Degradation of Oxidized Proteins. Proc. Natl. Acad. Sci. USA 1998, 95, 4924–4928. [Google Scholar] [CrossRef] [PubMed]
- Vashisht, A.A.; Zumbrennen, K.B.; Huang, X.; Powers, D.N.; Durazo, A.; Sun, D.; Bhaskaran, N.; Persson, A.; Uhlen, M.; Sangfelt, O.; et al. Control of Iron Homeostasis by an Iron-Regulated Ubiquitin Ligase. Science 2009, 326, 718–721. [Google Scholar] [CrossRef] [PubMed]
- Worthen, C.A.; Enns, C.A. The Role of Hepatic Transferrin Receptor 2 in the Regulation of Iron Homeostasis in the Body. Front. Pharmacol. 2014, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Bennett, M.J.; Sellers, V.M.; Andrews, N.C.; Enns, C.A.; Bjorkman, P.J. Comparison of the Interactions of Transferrin Receptor And transferrin Receptor 2 with Transferrin and the Hereditary Hemochromatosis Protein HFE. J. Biol. Chem. 2000, 275, 38135–38138. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.M.; Reutens, G.M.; Herbison, C.E.; Delima, R.D.; Chua, A.C.; Olynyk, J.K.; Trinder, D. Transferrin Receptor 2 Mediates Uptake of Transferrin-Bound and Non-Transferrin-Bound Iron. J. Hepatol. 2008, 48, 327–334. [Google Scholar] [CrossRef]
- Anderson, E.R.; Shah, Y.M. Iron Homeostasis in the Liver. Compr. Physiol. 2013, 3, 315–330. [Google Scholar] [CrossRef]
- Leidgens, S.; Bullough, K.Z.; Shi, H.; Li, F.; Shakoury-Elizeh, M.; Yabe, T.; Subramanian, P.; Hsu, E.; Natarajan, N.; Nandal, A.; et al. Each Member of the Poly-r(C)-binding Protein 1 (PCBP) Family Exhibits Iron Chaperone Activity toward Ferritin. J. Biol. Chem. 2013, 288, 17791–17802. [Google Scholar] [CrossRef]
- Bondi, A.; Valentino, P.; Daraio, F.; Porporato, P.; Gramaglia, E.; Canturan, S.; Gottardi, E.; Camaschella, C.; Roetto, A. Hepatic Expression of Hemochromatosis Genes in Two Mouse Strains After Phleotomy and Iron Overload. Haematologica 2005, 90, 1161–1167. [Google Scholar]
- Theil, E.C. Ferritin Protein Nanocages-The Story. Nanotechnol. Percept. 2012, 8, 7–16. [Google Scholar] [CrossRef]
- Torti, F.M.; Torti, S.V. Regulation of Ferritin Genes and Protein. Blood 2002, 99, 3505–3516. [Google Scholar] [CrossRef]
- Waldvogel-Abramowski, S.; Waeber, G.; Gassner, C.; Buser, A.; Frey, B.M.; Favrat, B.; Tissot, J.-D. Physiology of Iron Metabolism. Transfus. Med. Hemother. 2014, 41, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Theil, E.C. Ferritin: The Protein Nanocage and Iron Biomineral in Health and in Disease. Inorg. Chem. 2013, 52, 12223–12233. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.J.; Frazer, D.M. Current Understanding of Iron Homeostasis. Am. J. Clin. Nutr. 2017, 106, 1559S–1566S. [Google Scholar] [CrossRef] [PubMed]
- Langlois d’Estaintot, B.; Santambrogio, P.; Granier, T.; Gallois, B.; Chevalier, J.M.; Precigoux, G.; Levi, S.; Arosio, P. Crystal Structure and Biochemical Properties of the Human Mitochondrial Ferritin and its Mutant Ser144Ala. J. Mol. Biol. 2004, 340, 277–293. [Google Scholar] [CrossRef]
- Ward, D.M.; Cloonan, S.M. Mitochondrial Iron in Human Health and Disease. Annu. Rev. Physiol. 2019, 81, 453–482. [Google Scholar] [CrossRef]
- Finazzi, D.; Arosio, P. Biology of Ferritin in Mammals: An Update on Iron Storage, Oxidative Damage and Neurodegeneration. Arch. Toxicol. 2014, 88, 1787–1802. [Google Scholar] [CrossRef]
- Hentze, M.W.; Muckenthaler, M.U.; Andrews, N.C. Balancing Acts: Molecular Control of Mammalian Iron. Cell 2004, 117, 285–297. [Google Scholar] [CrossRef]
- Ganz, T. Macrophages and Systemic Iron Homeostasis. J. Innate Immun. 2012, 4, 446–453. [Google Scholar] [CrossRef]
- Sukhbaatar, N.; Weichhart, T. Iron Regulation: Macrophages in Control. Pharmaceuticals 2018, 11, 137. [Google Scholar] [CrossRef]
- Kong, W.N.; Zhao, S.E.; Duan, X.L.; Yang, Z.; Qian, Z.M.; Chang, Y.Z. Decreased DMT1 and Increased Ferroportin 1 Expression Is the Mechanisms of Reduced Iron Retention in Macrophages by Erythropoietin in Rats. J. Cell. Biochem. 2008, 104, 629–641. [Google Scholar] [CrossRef]
- Singh, A.K. Erythropoiesis: The Roles of Erythropoietin and Iron. In Textbook of Nephro-Endocrinology, 2nd ed.; Singh, A.K., Williams, G.H., Eds.; Academic Press: Bucharest, Romania, 2017; Volume 13, pp. 207–215. [Google Scholar]
- Zhang, D.L.; Ghosh, M.C.; Rouault, T.A. The Physiological Functions of Iron Regulatory Proteins in Iron Homeostasis—An Update. Front. Pharmacol. 2014, 5, 124. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T.; Nemeth, E. Hepcidin and Iron Homeostasis. Biochim. Biophys. Acta 2012, 1823, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Shah, Y.M.; Matsubara, T.; Ito, S.; Yim, S.H.; Gonzales, E.J. Intestinal Hypoxia-Inducible Transcription Factors are Essential for Iron Absorption Following Iron Deficiency. Cell Metab. 2009, 9, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.K.; Yee, J. Hepcidin. Adv. Chronic Kidney Dis. 2019, 26, 298–305. [Google Scholar] [CrossRef]
- Rochette, R.; Gudjoncik, A.; Guenancia, C.; Zeller, M.; Cottin, Y.; Vergely, C. The Iron-Regulator Hormone Hepcidin: A Possible Therapeutic Target? Pharmacol. Ther. 2015, 146, 35–52. [Google Scholar] [CrossRef]
- Canali, S.; Zumbrennen-Bullough, K.B.; Core, A.B.; Wang, C.-Y.; Nairz, M.; Bouley, R.; Swirski, F.K.; Babitt, J.L. Endothelial Cells Produce Bone Morphogenetic Protein 6 Required for Iron Homeostasis in Mice. Blood 2017, 129, 405–414. [Google Scholar] [CrossRef]
- Liu, X.B.; Nguyen, N.B.; Marquess, K.D.; Yang, F.; Haile, D.J. Regulation of Hepcidin and Ferroportin Expression by Lipopolysaccharide in Splenic Macrophages. Blood Cells Mol. Dis. 2005, 35, 47–56. [Google Scholar] [CrossRef]
- Bekri, S.; Gual, P.; Anty, R.; Luciani, N.; Dahman, M.; Ramesh, B.; Iannelli, A.; Staccini-Myx, A.; Casanova, D.; Amor, I.B.; et al. Increased Adipose Tissue Expression of Hepcidin in Severe Obesity Is Independent from Diabetes and NASH. Gastroenterology 2006, 131, 788–796. [Google Scholar] [CrossRef]
- Darshan, D.; Anderson, G.J. Interacting Signals in the Control of Hepcidin Expression. BioMetals 2009, 22, 77–87. [Google Scholar] [CrossRef]
- Nicolas, G.; Chauvet, C.; Viatte, L.; Danan, J.L.; Bigard, X.; Devaux, I.; Beaumont, C.; Kahn, A.; Vaulont, S. The Gene Encoding the Iron Regulatory Peptide Hepcidin Is Regulated by Anemia, Hypoxia, and Inflammation. J. Clin. Investig. 2002, 110, 1037–1044. [Google Scholar] [CrossRef]
- Rishi, G.; Wallace, D.F.; Subramaniam, V.N. Hepcidin: Regulation of the Master Iron Regulator. Biosci. Rep. 2015, 35, e00192. [Google Scholar] [CrossRef] [PubMed]
- Babitt, J.L.; Huang, F.W.; Wrighting, D.M.; Xia, Y.; Sidis, Y.; Samad, T.A.; Campagna, J.A.; Chung, R.T.; Schneyer, A.L.; Woolf, C.J.; et al. Bone Morphogenetic Protein Signaling by Hemojuvelin Regulates Hepcidin Expression. Nat. Genet. 2006, 38, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron Homeostasisand Oxidative Stress: An Intimate Relationship. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118535. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Maxson, J.E.; Zhang, R.H.; Wahedi, M.; Enns, C.A.; Zhang, A.-S. Neogenin Facilitates the Induction of Hepcidin Expression by Hemojuvelin in the Liver. J. Biol. Chem. 2016, 291, 12322–12335. [Google Scholar] [CrossRef]
- Wu, H.; Yung, L.M.; Cheng, W.H.; Yu, P.B.; Babitt, J.L.; Lin, H.Y.; Xia, Y. Hepcidin Regulation by BMP Signaling in Macrophages Is Lipopolysaccharide Dependent. PLoS ONE 2012, 7, e44622. [Google Scholar] [CrossRef]
- Tangudu, N.K.; Vujić Spasić, M. Heme Activates Macrophage Hepcidin Expression via Toll like Receptor 4 and Extracellular Signal-Regulated Kinases Signaling Pathway. Clin. Pharmacol. Biopharm. 2017, 6, 166. [Google Scholar] [CrossRef]
- Barton, J.C.; Edwards, C.Q.; Acton, R.T. HFE Gene: Structure, Function, Mutations, and Associated Iron Abnormalities. Gene 2015, 574, 179–192. [Google Scholar] [CrossRef]
- Lebron, J.A.; Bennett, M.J.; Vaughn, D.E.; Chirino, A.J.; Snow, P.M.; Mintier, G.A.; Feder, J.N.; Bjorkman, P.J. Crystal Structure of the Hemochromatosis Protein HFE and Characterization of its Interaction with Transferrin Receptor. Cell 1998, 93, 111–123. [Google Scholar] [CrossRef]
- Camaschella, C.; Silvestri, L. New and Old Players in the Hepcidin Pathway. Haematologica 2008, 93, 1441–1444. [Google Scholar] [CrossRef][Green Version]
- Pagani, A.; Nai, A.; Silvestri, L.; Camaschella, C. Hepcidin and Anemia: A Tight Relationship. Front. Physiol. 2019, 10, 1294. [Google Scholar] [CrossRef]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; McVey Ward, D.; Ganz, T.; Kaplan, J. Hepcidin Regulates Cellular Iron Efflux by Binding to Ferroportin and Inducing Its Internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 Mediates Hypoferremia of Inflammation by Inducing the Synthesis of the Iron Regulatory Hormone Hepcidin. J. Clin. Investig. 2004, 113, 1271–1276. [Google Scholar] [CrossRef]
- Verga Falzacappa, M.V.; Vujic Spasic, M.; Kessler, R.; Stolte, J.; Hentze, M.W.; Muckenthaler, M.U. STAT3 Mediates Hepatic Hepcidin Expression and Its Inflammatory Stimulation. Blood 2007, 109, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I. A Cellular Perspective on Brain Energy Metabolism and Functional Imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [PubMed]
- Salvador, G.A. Iron in Neuronal Function and Dysfunction. Biofactors 2010, 36, 103–110. [Google Scholar] [CrossRef]
- Moos, T.; Nielsen, T.R.; Skjørringe, T.; Morgan, E.H. Iron Trafficking Inside the Brain. J. Neurochem. 2007, 103, 1730–1740. [Google Scholar] [CrossRef]
- McCarthy, R.C.; Kosman, D.J. Iron Transport Across the Blood-Brain Barrier: Development, Neurovascular Regulation and Cerebral Amyloid Angiopathy. Cell. Mol. Life Sci. 2015, 72, 709–727. [Google Scholar] [CrossRef]
- Moos, T.; Morgan, E.H. Transferrin and Transferrin Receptor Function in Brain Barrier Systems. Cell. Mol. Neurobiol. 2000, 20, 77–95. [Google Scholar] [CrossRef]
- Montagne, A.; Zhao, Z.; Zlokovic, B.V. Alzheimer’s Disease: A Matter of Blood-Brain Barrier Dysfunction? J. Exp. Med. 2017, 214, 3151–3169. [Google Scholar] [CrossRef]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and Dysfunction of the Blood-Brain Barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. Neurovascular Pathways to Neurodegeneration in Alzheimer’s Disease and Other Disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey Through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Qian, Z.M. Brain Iron Metabolism: Neurobiology and Neurochemistry. Prog. Neurobiol. 2007, 83, 149–173. [Google Scholar] [CrossRef] [PubMed]
- Simpson, I.A.; Ponnuru, P.; Klinger, M.E.; Myers, R.L.; Devraj, K.; Coe, C.L.; Lubach, G.R.; Carruthers, A.; Connor, J.R. A Novel Model for Brain Iron Uptake: Introducing the Concept of Regulation. J. Cereb. Blood Flow Metab. 2015, 35, 48–57. [Google Scholar] [CrossRef]
- Khan, A.I.; Liu, J.; Dutta, P. Iron Transport Kinetics Through Blood-Brain Barrier Endothelial Cells. Biochim. Biophys. Acta 2018, 1862, 1168–1179. [Google Scholar] [CrossRef]
- Rouault, T.A.; Cooperman, S. Brain Iron Metabolism. Semin. Pediatr. Neurol. 2006, 13, 142–148. [Google Scholar] [CrossRef]
- De Domenico, I.; McVey Ward, D.; Kaplan, J. Regulation of Iron Acquisition and Storage: Consequences for Iron-Linked Disorders. Nat. Rev. Mol. Cell Biol. 2008, 9, 72–81. [Google Scholar] [CrossRef]
- Benarroch, E.E. Brain Iron Homeostasis and Neurodegenerative Disease. Neurology 2009, 72, 1436–1440. [Google Scholar] [CrossRef]
- Burkhart, A.; Skjørringe, T.; Johnsen, K.B.; Siupka, P.; Thomsen, L.B.; Nielsen, M.S.; Thomsen, L.L.; Moos, T. Expression of Iron-Related Proteins at the Neurovascular Unit Supports Reduction and Reoxidation of Iron for Transport Through the Blood-Brain Barrier. Mol. Neurobiol. 2016, 53, 7237–7253. [Google Scholar] [CrossRef]
- Zecca, L.; Stroppolo, A.; Gatti, A.; Tampellini, D.; Toscani, M.; Gallorini, M.; Giaveri, G.; Arosio, P.; Santambrogio, P.; Fariello, R.G.; et al. The Role of Iron and Copper Molecules in the Neuronal Vulnerability of Locus Coeruleus and Substantia Nigra during Aging. Proc. Natl. Acad. Sci. USA 2004, 101, 9843–9848. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, M.P.; Greenamyre, J.T. Mitochondrial Iron Metabolism and Its Role in Neurodegeneration. J. Alzheimers Dis. 2010, 20, S551–S568. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.J.; Leenders, A.G.; Cooperman, S.; Meyron-Holtz, E.; Smith, S.; Land, W.; Tsai, R.Y.; Berger, U.V.; Sheng, Z.H.; Rouault, T.A. Expression of the Iron Transporter Ferroportin in Synaptic Vesicles and the Blood–Brain Barrier. Brain Res. 2004, 1001, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.M.; Chang, Y.Z.; Du, J.R.; Ho, K.P.; Zhu, L.; Xu, Y.J.; Li, L.Z.; Wang, C.Y.; Wang, Q.; Ge, X.H.; et al. Development and Iron-Dependent Expression of Hephaestin in Different Brain Regions of Rats. J. Cell. Biochem. 2007, 102, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- De Arriba Zerpa, G.A.; Saleh, M.C.; Fernandez, P.M.; Guillou, F.; Espinosa de los Monteros, A.; De Vellis, J.; Zakin, M.M.; Baron, B. Alternative Splicing Prevents Transferrin Secretion During Differentiation of a Human Oligodendrocyte Cell Line. J. Neurosci. Res. 2000, 61, 388–395. [Google Scholar] [CrossRef]
- Singh, N.; Haldar, S.; Tripathi, A.K.; Horback, K.; Wong, J.; Sharma, D.; Beserra, A.; Suda, S.; Anbalagan, C.; Dev, S.; et al. Brain Iron Homeostasis: From Molecular Mechanisms to Clinical Significance and Therapeutic Opportunities. Antioxid. Redox Signal. 2014, 20, 1324–1363. [Google Scholar] [CrossRef]
- Nnah, I.C.; Wessling-Resnick, M. Brain Iron Homeostasis: A Focus on Microglial Iron. Pharmaceuticals 2018, 11, 129. [Google Scholar] [CrossRef]
- Rouault, T.A.; Zhang, D.L.; Jeong, S.Y. Brain Iron Homeostasis, the Choroid Plexus, and Localization of Iron Transport Proteins. Metab. Brain Dis. 2009, 24, 673–684. [Google Scholar] [CrossRef]
- Brown, P.D.; Davies, S.L.; Speake, T.; Millar, I.D. Molecular Mechanisms of Cerebrospinal Fluid Production. Neuroscience 2004, 129, 957–970. [Google Scholar] [CrossRef]
- Leitner, D.F.; Connor, J.R. Functional Roles of Transferrin in the Brain. Biochim. Biophys. Acta 2012, 1820, 393–402. [Google Scholar] [CrossRef]
- Burdo, J.R.; Menzies, S.L.; Simpson, I.A.; Garrick, L.M.; Garrick, M.D.; Dolan, K.G.; Haile, D.J.; Beard, J.L.; Connor, J.R. Distribution of Divalent Metal Transporter 1 and Metal Transport Protein 1 in the Normal and Belgrade Rat. J. Neurosci. Res. 2001, 66, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Seaman, W.E.; Di, X.; Wang, W.; Willingham, M.; Torti, F.M.; Torti, S.V. Iron Uptake Mediated by Binding of H-ferritin to the Tim-2 Receptor in Mouse Cells. PLoS ONE 2011, 6, e23800. [Google Scholar] [CrossRef] [PubMed]
- Bishop, G.M.; Scheiber, I.F.; Dringen, R.; Robinson, S.R. Synergistic Accumulation of Iron and Zinc by Cultured Astrocytes. J. Neural Transm. 2010, 117, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Vidal, R.; Miravalle, L.; Gao, X.; Barbeito, A.G.; Baraibar, M.A.; Hekmatyar, S.K.; Widel, M.; Bansal, N.; Delisle, M.B.; Ghetti, B. Expression of a Mutant Form of the Ferritin Light Chain Gene Induces Neurodegeneration and Iron Overload in Transgenic Mice. J. Neurosci. 2008, 28, 60–67. [Google Scholar] [CrossRef]
- Zucca, F.A.; Segura-Aguilar, J.; Ferrari, E.; Muñoz, P.; Paris, I.; Sulzer, D.; Sarna, T.; Casella, L.; Zecca, L. Interactions of Iron, Dopamine and Neuromelanin Pathways in Brain Aging and Parkinson’s Disease. Prog. Neurobiol. 2017, 155, 96–119. [Google Scholar] [CrossRef]
- MacKenzie, E.L.; Iwasaki, K.; Tsuji, Y. Intracellular Iron Transport and Storage: From Molecular Mechanisms to Health Implications. Antioxid. Redox Signal. 2008, 10, 997–1030. [Google Scholar] [CrossRef]
- Irace, C.; Scorziello, A.; Maffettone, C.; Pignataro, G.; Matrone, C.; Adornetto, A.; Santamaria, R.; Annunziato, L.; Colonna, A. Divergent Modulation of Iron Regulatory Proteins and Ferritin Biosynthesis by Hypoxia/Reoxygenation in Neurons and Glial Cells. J. Neurochem. 2005, 95, 1321–1331. [Google Scholar] [CrossRef]
- Asano, T.; Komatsu, M.; Yamaguchi, Y.; Ishikawa-Iwai, F.; Mizushima, N.; Iwai, K. Distinct Mechanisms of Ferritin Delivery to Lysosomes in Iron-Depleted and Iron-Replete Cells. Mol. Cell. Biol. 2011, 31, 2040–2052. [Google Scholar] [CrossRef]
- Biasiotto, G.; Di Lorenzo, D.; Archetti, S.; Zanella, I. Iron and Neurodegeneration: Is Ferritinophagy the Link? Mol. Neurobiol. 2016, 53, 5542–5574. [Google Scholar] [CrossRef]
- Vela, D. The Dual Role of Hepcidin in Brain Iron Load and Inflammation. Front. Neurosci. 2018, 12, 740. [Google Scholar] [CrossRef]
- Zechel, S.; Huber-Wittmer, K.; Von Bohlen und Halbach, O. Distribution of the Iron-Regulating Protein Hepcidin in the Murine Central Nervous System. J. Neurol. Res. 2006, 84, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Du, F.; Qian, Z.M.; Ge, X.H.; Zhu, L.; Yung, W.H.; Yang, L.; Ke, Y. Lipopolysaccharide Induces a Significant Increase in Expression of Iron Regulatory Hormone Hepcidin in the Cortex and Substantia Nigra in Rat Brain. Endocrinology 2008, 149, 3920–3925. [Google Scholar] [CrossRef] [PubMed]
- Beard, J.L.; Connor, J.R. Iron Status and Neural Functioning. Annu. Rev. Nutr. 2003, 23, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.; Neves, P.; Gozzelino, R. Multilevel Impacts of Iron in the Brain: The Cross Talk between Neurophysiological Mechanisms, Cognition, and Social Behavior. Pharmaceuticals 2019, 12, 126. [Google Scholar] [CrossRef] [PubMed]
- Petranovic, D.; Batinac, T.; Petranovic, D.; Ruzic, A.; Ruzic, T. Iron Deficiency Anaemia Influences Cognitive Functions. Med. Hypotheses 2008, 70, 70–72. [Google Scholar] [CrossRef]
- Bourre, J.M. Effects of Nutrients (in Food) on the Structure and Function of the Nervous System: Update on Dietary Requirements for Brain. Part 1: Micronutrients. J. Nutr. Health Aging 2006, 10, 377–385. [Google Scholar]
- Kuhn, S.; Gritti, L.; Crooks, D.; Dombrowski, Y. Oligodendrocytes in Development, Myelin Generation and Beyond. Cells 2019, 8, 1424. [Google Scholar] [CrossRef]
- Andro, M.; Le Squere, P.; Estivin, S.; Gentric, A. Anaemia and Cognitive Performances in the Elderly: A Systematic Review. Eur. J. Neurol. 2013, 20, 1234–1240. [Google Scholar] [CrossRef]
- Fleming, D.J.; Tucker, K.L.; Jacques, P.F.; Dallal, G.E.; Wilson, P.W.; Wood, R.J. Dietary Factors Associated with the Risk of High Iron Stores in the Elderly Framingham Heart Study Cohort. Am. J. Clin. Nutr. 2002, 76, 1375–1384. [Google Scholar] [CrossRef]
- Hunnicutt, J.; He, K.; Xun, P. Dietary Iron Intake and Body Iron Stores Are Associated with Risk of Coronary Heart Disease in a Meta-Analysis of Prospective Cohort Studies. J. Nutr. 2013, 144, 359–366. [Google Scholar] [CrossRef]
- Bao, W.; Rong, Y.; Rong, S.; Liu, L. Dietary Iron Intake, Body Iron Stores, and the Risk of Type 2 Diabetes: A Systematic Review and Meta-Analysis. BMC Med. 2012, 10, 119. [Google Scholar] [CrossRef] [PubMed]
- Penke, L.; Valdés Hernandéz, M.C.; Maniega, S.M.; Gow, A.J.; Murray, C.; Starr, J.M.; Bastin, M.E.; Deary, I.J.; Wardlaw, J.M. Brain Iron Deposits Are Associated with General Cognitive Ability and Cognitive Aging. Neurobiol. Aging 2012, 33, 510–517.e512. [Google Scholar] [CrossRef] [PubMed]
- Hosking, D.E.; Ayton, S.; Beckett, N.; Booth, A.; Peters, R. More Evidence Is Needed. Iron, Incident Cognitive Decline and Dementia: A Systematic Review. Ther. Adv. Chronic. Dis. 2018, 9, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Poon, H.F.; Calabrese, V.; Calvani, M.; Butterfield, D.A. Proteomics Analyses of Specific Protein Oxidation and Protein Expression in Aged Rat Brain and its Modulation by L-Acetylcarnitine: Insights into the Mechanisms of Action of this Proposed Therapeutic Agent for CNS Disorders Associated with Oxidative Stress. Antioxid. Redox Signal. 2006, 8, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.-N.; Qian, Z.-M.; Wu, K.-C.; Yung, W.-H.; Ke, Y. Expression of Iron Transporters and Pathological Hallmarks of Parkinson’s and Alzheimer’s Diseases in the Brain of Young, Adult, and Aged Rats. Mol. Neurobiol. 2016, 54, 5213–5224. [Google Scholar] [CrossRef] [PubMed]
- Connor, J.R.; Menzies, S.L.; St Martin, S.M.; Mufson, E.J. Cellular Distribution of Transferrin, Ferritin and Iron in Normal and Aged Human Brains. J. Neurosci. Res. 1990, 27, 595–611. [Google Scholar] [CrossRef] [PubMed]
- Hare, D.J.; Gerlach, M.; Riederer, P. Considerations for Measuring Iron in Post-Mortem Tissue of Parkinson’s Disease Patients. J. Neural Transm. 2012, 119, 1515–1521. [Google Scholar] [CrossRef]
- Zecca, L.; Youdim, M.B.; Riederer, P.; Connor, J.R.; Crichton, R.R. Iron, Brain Ageing and Neurodegenerative Disorders. Nat. Rev. Neurosci. 2004, 5, 863–873. [Google Scholar] [CrossRef]
- Yan, S.-Q.; Sun, J.-Z.; Yan, Y.-Q.; Wang, H.; Lou, M. Evaluation of Brain Iron Content Based on Magnetic Resonance Imaging (MRI): Comparison among Phase Value, R2* and Magnitude Signal Intensity. PLoS ONE 2012, 7, e31748. [Google Scholar] [CrossRef]
- Xu, X.; Wang, Q.; Zhang, M. Age, Gender, and Hemispheric Differences in Iron Deposition in the Human Brain: An in Vivo MRI Study. Neuroimage 2008, 40, 35–42. [Google Scholar] [CrossRef]
- Iadecola, C. Neurovascular Regulation in the Normal Brain and in Alzheimer’s Disease. Nat. Rev. Neurosci. 2004, 5, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Conde, J.R.; Streit, W.J. Microglia in the Aging Brain. J. Neuropathol. Exp. Neurol. 2006, 65, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Farrall, A.J.; Wardlaw, J.M. Blood-Brain Barrier: Ageing and Microvascular Disease–Systematic Review and Meta-Analysis. Neurobiol. Aging 2009, 30, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Crichton, R.R. Inorganic Biochemistry of Iron Metabolism: From Molecular Mechanisms to Clinical Consequences, 2nd ed.; John Wiley & Sons: Chichester, UK, 2001; pp. 1–21. [Google Scholar]
- Weinreb, O.; Amit, T.; Youdim, M.B.H. Targeting Dysregulation of Brain Iron Homeostasis in Ageing. Nutr. Aging 2012, 1, 27–39. [Google Scholar] [CrossRef]
- Kumar, M.J.; Andersen, J.K. Perspectives on MAO-B in Aging and Neurological Disease: Where Do We Go from Here? Mol. Neurobiol. 2004, 30, 77–89. [Google Scholar] [CrossRef]
- Shemyakov, S.E. Monoamine Oxidase Activity, Lipid Peroxidation, and Morphological Changes in Human Hypothalamus During Aging. Bull. Exp. Biol. Med. 2001, 131, 586–588. [Google Scholar] [CrossRef]
- Carrasquilla, G.D.; Frumento, P.; Berglund, A.; Borgfeldt, C.; Bottai, M.; Chiavenna, C.; Eliasson, M.; Engström, G.; Hallmans, G.; Jansson, J.H.; et al. Postmenopausal hormone therapy and risk of stroke: A pooled analysis of data from population-based cohort studies. PLoS Med. 2017, 14, e1002445. [Google Scholar] [CrossRef]
- Yu, J.; Liu, H.; He, S.; Li, P.; Ma, C.; Ma, M.; Liu, Y.; Lv, L.; Ping, F.; Zhang, H.; et al. Sex-Specific Negative Association between Iron Intake and Cellular Aging Markers: Mediation Models Involving TNFα. Oxid. Med. Cell. Longev. 2019, 2019, 4935237. [Google Scholar] [CrossRef]
- Ćurko-Cofek, B.; Grubić Kezele, T.; Barac-Latas, V. Hepcidin and Metallothioneins as Molecular Base for Sex-Dependent Differences in Clinical Course of Experimental Autoimmune Encephalomyelitis in Chronic Iron Overload. Med. Hypotheses 2017, 107, 51–54. [Google Scholar] [CrossRef]
- Hametner, S.; Wimmer, I.; Haider, L.; Pfeifenbring, S.; Brück, W.; Lassmann, H. Iron and Neurodegeneration in the Multiple Sclerosis Brain. Ann. Neurol. 2013, 74, 848–861. [Google Scholar] [CrossRef]
- Grubić Kezele, T. Iron. In Trace Elements and Minerals in Health and Longevity, 1st ed.; Malavolta, M., Mocchegiani, E., Eds.; Springer Nature: Cham, Switzerland, 2018; Volume 8, pp. 1–34. [Google Scholar]
- Zivadinov, R.; Tavazzi, E.; Bergsland, N.; Hagemeier, J.; Lin, F.; Dwyer, M.G.; Carl, E.; Kolb, C.; Hojnacki, D.; Ramasamy, D.; et al. Brain Iron at Quantitative MRI Is Associated with Disability in Multiple Sclerosis. Radiology 2018, 289, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Rosko, L.; Smith, V.N.; Yamazaki, R.; Huang, J.K. Oligodendrocyte Bioenergetics in Health and Disease. Neuroscientist 2019, 5, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Morath, D.J.; Mayer-Pröschel, M. Iron Modulates the Differentiation of a Distinct Population of Glial Precursor Cells into Oligodendrocytes. Dev. Biol. 2001, 237, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Morath, D.J.; Mayer-Pröschel, M. Iron Deficiency During Embryogenesis and Consequences for Oligodendrocyte Generation in Vivo. Dev. Neurosci. 2002, 24, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Schonberg, D.L.; McTigue, D.M. Iron is Essential for Oligodendrocyte Genesis following Intraspinal Macrophage Activation. Exp. Neurol. 2009, 218, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Grishchuk, Y.; Peña, K.A.; Coblentz, J.; King, V.E.; Humphrey, M.D.; Wang, S.L.; Kiselyov, K.I.; Slaugenhaupt, S.A. Impaired Myelination and Reduced Brain Ferric Iron in the Mouse Model of Mucolipidosis IV. Dis. Model Mech. 2015, 8, 1591–1601. [Google Scholar] [CrossRef]
- Schulz, K.; Kroner, A.; David, S. Iron Efflux from Astrocytes Plays a Role in Remyelination. Soc. Neurosci. 2012, 32, 4841–4847. [Google Scholar] [CrossRef]
- Todorich, B.; Pasquini, J.M.; Garcia, C.I.; Paez, P.M.; Connor, J.R. Oligodendrocytes and Myelination: The Role of Iron. Glia 2009, 57, 467–478. [Google Scholar] [CrossRef]
- Morell, P.; Jurevics, H. Origin of Cholesterol in Myelin. Neurochem. Res. 1996, 21, 463–470. [Google Scholar] [CrossRef]
- Jones, G.; Prosser, D.E.; Kaufmann, M. Cytochrome P450-mediated Metabolism of Vitamin D. J. Lipid Res. 2014, 55, 13–31. [Google Scholar] [CrossRef]
- Fuente, A.G.F.; Errea, O.; Van Wijngaarden, P.; Gonzalez, G.A.; Kerninon, C.; Jarjour, A.A.; Lewis, H.J.; Jones, C.A.; Nait-Oumesmar, B.; Zhao, C. Vitamin D Receptor–Retinoid X Receptor Heterodimer Signaling Regulates Oligodendrocyte Progenitor Cell Differentiation. J. Cell Biol. 2015, 211, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Sintzel, M.B.; Rametta, M.; Reder, A.T. Vitamin D and Multiple Sclerosis: A Comprehensive Review. Neurol. Ther. 2018, 7, 59–85. [Google Scholar] [CrossRef] [PubMed]
- Abbatemarco, J.R.; Fox, R.J.; Li, H.; Ontaneda, D. Vitamin D and MRI Measures in Progressive Multiple Sclerosis. Mult. Scler. Relat. Disord. 2019, 35, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Bacchetta, J.; Zaritsky, J.J.; Sea, J.L.; Chun, R.F.; Lisse, T.S.; Zavala, K.; Nayak, A.; Wesseling-Perry, K.; Westerman, M.; Hollis, B.W.; et al. Suppression of Iron-regulatory Hepcidin by Vitamin D. J. Am. Soc. Nephrol. 2014, 25, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Kweder, H.; Eidi, H. Vitamin D Deficiency in Elderly: Risk Factors and Drugs Impact on Vitamin D Status. Avicenna J. Med. 2018, 8, 139–146. [Google Scholar] [CrossRef]
- Munger, K.L.; Chitnis, T.; Ascherio, A. Body Size and Risk of MS in Two Cohorts of US Women. Neurology 2009, 73, 1543–1550. [Google Scholar] [CrossRef]
- Gianfrancesco, M.A.; Barcellos, L.F. Obesity and Multiple Sclerosis Susceptibility: A Review. J. Neurol. Neuromed. 2016, 1, 1–5. [Google Scholar] [CrossRef]
- Low, M.S.Y.; Speedy, J.; Styles, C.E.; De-Regil, L.M.; Pasricha, S.R. Daily iron supplementation for improving anaemia, iron status and health in menstruating women. Cochrane Database Syst. Rev. 2016, 4, CD009747. [Google Scholar] [CrossRef]
- Ramos, P.; Santos, A.; Pinto, N.R.; Mendes, R.; Magalhães, T.; Agostinho Almeida, A. Iron Levels in the Human Brain: A Post-Mortem Study of Anatomical Region Differences and Age-Related Changes. J. Trace Elem. Med. Biol. 2014, 28, 13–17. [Google Scholar] [CrossRef]
- Tishler, T.A.; Raven, E.P.; Lu, P.H.; Altshuler, L.L.; Bartzokis, G. Pre-menopausal Hysterectomy Is Associated with Increased Brain Ferritin Iron. Neurobiol. Aging 2012, 33, 1950–1958. [Google Scholar] [CrossRef]
- Hallgren, B.; Sourander, P. The Effect of Age on the Non-Haemin Iron in the Human Brain. J. Neurochem. 1958, 3, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Hagemeier, J.; Weinstock-Guttman, B.; Heininen-Brown, M.; Poloni, G.U.; Bergsland, N.; Schirda, C.; Magnano, C.R.; Kennedy, C.; Carl, E.; Dwyer, M.G.; et al. Gray Matter SWI-filtered Phase and Atrophy Are Linked to Disability in MS. Front. Biosci. 2013, 5, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.J.; Rubinow, D.R. Sex Hormones and Mood in the Perimenopause. Ann. N. Y. Acad. Sci. 2009, 1179, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.; Stüve, O. Multiple Sclerosis in the Elderly Patient. Drugs Aging 2010, 27, 283–294. [Google Scholar] [CrossRef]
- Bartzokis, G.; Mintz, J.; Sultzer, D.; Marx, P.; Herzberg, J.S.; Phelan, C.K.; Marder, S.R. In Vivo MR Evaluation of Age-Related Increases in Brain Iron. Am. J. Neuroradiol. 1994, 15, 1129–1138. [Google Scholar]
- Armon-Omer, A.; Waldman, C.; Simaan, N.; Neuman, H.; Tamir, S.; Shahien, R. New Insights on the Nutrition Status and Antioxidant Capacity in Multiple Sclerosis Patients. Nutrients 2019, 11, 427. [Google Scholar] [CrossRef]
- Gill, D.; Brewer, C.F.; Monori, G.; Trégouët, D.A.; Franceschini, N.; Giambartolomei, C.; Tzoulaki, I.; Dehghan, A. Effects of Genetically Determined Iron Status on Risk of Venous Thromboembolism and Carotid Atherosclerotic Disease: A Mendelian Randomization Study. J. Am. Heart Assoc. 2019, 8, e012994. [Google Scholar] [CrossRef]
- Menon, R.G.; Khan, B.V.; Rajagopalan, S.; Fay, W.P. Chronic Iron Administration Increases Vascular Oxidative Stress and Accelerates Arterial Thrombosis. Circulation 2003, 107, 2601–2606. [Google Scholar]
- Franchini, M.; Targher, G.; Montagnana, M.; Lippi, G. Iron and Thrombosis. Ann. Hematol. 2008, 87, 167–173. [Google Scholar] [CrossRef]
- Tang, X.; Fang, M.; Cheng, R.; Zhang, Z.; Wang, Y.; Shen, C.; Han, Y.; Lu, Q.; Du, Y.; Liu, Y.; et al. Iron-Deficiency and Estrogen Are Associated with Ischemic Stroke by Up-Regulating Transferrin to Induce Hypercoagulability. Circ. Res. 2020, 127, 651–663. [Google Scholar] [CrossRef]
- Ogun, A.S.; Adeyinka, A. Biochemistry, Transferrin. In StatPearls [Internet]; StatPearls Publishing: St. Petersburg, FL, USA, 2019. Available online: https://www.ncbi.nlm.nih.gov/books/NBK532928/ (accessed on 30 May 2020).
- Tang, X.; Zhang, Z.; Fang, M.; Han, Y.; Wang, G.; Wang, S.; Xue, M.; Li, Y.; Zhang, L.; Wu, J.; et al. Transferrin Plays a Central Role in Coagulation Balance by Interacting with Clotting Factors. Cell Res. 2020, 30, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Woods, H.F.; Youdim, M.B.H.; Boullin, D.; Callender, S. Monoamine Metabolism and Platelet Function in Iron-Deficiency Anaemia. In Ciba Foundation Symposium 51-Iron Metabolism; John Wiley & Sons, Ltd.: Chichester, UK, 1977; Volume 51, pp. 227–248. [Google Scholar]
- Jackson, S. The Growing Complexity of Platelet Aggregation. Blood 2007, 109, 5087–5095. [Google Scholar] [CrossRef] [PubMed]
- Shovlin, C.L.; Chamali, B.; Santhirapala, V.; Livesey, J.A.; Angus, G.; Manning, R.; Laffan, M.A.; Meek, J.; Tighe, H.C.; Jackson, J.E. Ischaemic Strokes in Patients with Pulmonary Arteriovenous Malformations and Hereditary Hemorrhagic Telangiectasia: Associations With Iron Deficiency and Platelets. PLoS ONE 2014, 9, e88812. [Google Scholar] [CrossRef] [PubMed]
- Appelros, P.; Stegmayr, B.; Terent, A. Sex Differences in Stroke Epidemiology: A Systematic Review. Stroke 2009, 40, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- Kaluza, J.; Wolk, A.; Larsson, S.C. Heme Iron Intake and Risk of Stroke: A Prospective Study of Men. Stroke 2013, 44, 334–339. [Google Scholar] [CrossRef]
- Fukuda, M.; Kanda, T.; Kamide, N.; Akutsu, T.; Sakai, F. Gender Differences in Long-term Functional Outcome after First-ever Ischemic Stroke. Intern. Med. 2009, 48, 967–973. [Google Scholar] [CrossRef]
- Petrone, A.B.; Simpkins, J.W.; Barr, T.L. 17β-Estradiol and Inflammation: Implications for Ischemic Stroke. Aging Dis. 2014, 5, 340–345. [Google Scholar] [CrossRef]
- Malutan, A.M.; Dan, M.; Nicolae, C.; Carmen, M. Proinflammatory and Anti-Inflammatory Cytokine Changes Related to Menopause. Prz. Menopauzalny 2014, 13, 162–168. [Google Scholar] [CrossRef]
- Montgomery, S.L.; Bowers, W.J. Tumor Necrosis Factor-alpha and the Roles it Plays in Homeostatic and Degenerative Processes within the Central Nervous System. J. Neuroimmune Pharmacol. 2012, 7, 42–59. [Google Scholar] [CrossRef]
- Doll, D.N.; Rellick, S.L.; Barr, T.L.; Ren, X.; Simpkins, J.W. Rapid Mitochondrial Dysfunction Mediates TNF-Alpha-Induced Neurotoxicity. J. Neurochem. 2015, 132, 443–451. [Google Scholar] [CrossRef]
- Liao, S.L.; Chen, W.Y.; Chen, C.J. Estrogen Attenuates Tumor Necrosis Factor-Alpha Expression to Provide Ischemic Neuroprotection in Female Rats. Neurosci. Lett. 2002, 330, 159–162. [Google Scholar] [CrossRef]
- Vela, D. Hepcidin, an Emerging and Important Player in Brain Iron Homeostasis. J. Transl. Med. 2018, 16, 25. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.M. Hormone Replacement Therapy Affects Iron Status More than Endometrial Bleeding in Older US Women: A Role for Estrogen in Iron Homeostasis? Maturitas 2016, 88, 46–51. [Google Scholar] [CrossRef]
- Zacharski, L.R.; Ornstein, D.L.; Woloshin, S.; Schwartz, L.M. Association of Age, Sex, and Race with Body Iron Stores in Adults: Analysis of NHANES III Data. Am. Heart J. 2000, 140, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Bajbouj, K.; Shafarin, J.; Allam, H.; Madkour, M.; Awadallah, S.; El-Serafy, A.; Sandeep, D.; Hamad, M. Elevated Levels of Estrogen Suppress Hepcidin Synthesis and Enhance Serum Iron Availability in Premenopausal Women. Exp. Clin. Endocrinol. Diabetes 2018, 126, 453–459. [Google Scholar] [CrossRef]
- Busti, F.; Campostrini, N.; Martinelli, N.; Girelli, D. Iron Deficiency in the Elderly Population, Revisited in the Hepcidin era. Front. Pharmacol. 2014, 5, 83. [Google Scholar] [CrossRef]
- Ilkovska, B.; Kotevska, B.; Trifunov, G.; Kanazirev, B. Serum Hepcidin Reference Range, Gender Differences, Menopausal Dependence and Biochemical Correlates in Healthy Subjects. J. IMAB Annu. Proc. Sci. Pap. 2016, 22, 1127–1131. [Google Scholar] [CrossRef][Green Version]
- Jian, J.; Pelle, E.; Huang, X. Iron and Menopause: Does Increased Iron Affect the Health of Postmenopausal Women? Antioxid. Redox Signal. 2009, 11, 2939–2943. [Google Scholar] [CrossRef]
- Huang, X.; Xu, Y.; Partridge, N.C. Dancing with Sex Hormones, Could Iron Contribute to the Gender Difference in Osteoporosis? Bone 2013, 55, 458–460. [Google Scholar] [CrossRef]
- Chen, B.; Li, G.F.; Shen, Y.; Huang, X.I.; Xu, Y.J. Reducing Iron Accumulation: A Potential Approach for the Prevention and Treatment of Postmenopausal Osteoporosis. Exp. Ther. Med. 2015, 10, 7–11. [Google Scholar] [CrossRef]
- Roy, C.N.; Snyder, P.J.; Stephens-Shields, A.J.; Artz, A.S.; Bhasin, S.; Cohen, H.J.; Farrar, J.T.; Gill, T.M.; Zeldow, B.; Cella, D.; et al. Association of Testosterone Levels with Anemia in Older Men: A Controlled Clinical Trial. JAMA Intern. Med. 2017, 177, 480–490. [Google Scholar] [CrossRef]
- Gabrielsen, J.S. Iron and Testosterone: Interplay and Clinical Implications. Curr. Sex. Health Rep. 2017, 9, 5–11. [Google Scholar] [CrossRef]
- Grubić Kezele, T. Androgen-driven COVID-19 Infectio—Is Testosterone an Enemy or a Friend? Horm. Mol. Biol. Clin. Investig. 2020, 41. [Google Scholar] [CrossRef]
- Grubić Kezele, T. Cryptozoospermia after Treatment with Clomiphene Citrate Following Long-Term Use of Intramuscular Testosterone Undecanoate Depot Injection (Nebido®). Horm. Mol. Biol. Clin. Investig. 2019, 39. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, L.L.; Jørgensen, H.S.; Nakayama, H.; Raaschou, H.O.; Olsen, T.S.; Winther, K. Decreased Serum Testosterone in Men with Acute Ischemic Stroke. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Sangkhae, V.; Nemeth, E. Regulation of the Iron Homeostatic Hormone Hepcidin. Adv. Nutr. 2017, 8, 126–136. [Google Scholar] [CrossRef]
- Mammi, C.; Calanchini, M.; Antelmi, A.; Cinti, F.; Rosano, G.M.C.; Lenzi, A.; Caprio, M.; Fabbri, A. Androgens and Adipose Tissue in Males: A Complex and Reciprocal Interplay. Int. J. Endocrinol. 2012, 2012, 789653. [Google Scholar] [CrossRef]
- Bianchi, V.E. The Anti-Inflammatory Effects of Testosterone. J. Endocr. Soc. 2019, 3, 91–107. [Google Scholar] [CrossRef]
- Dhindsa, S.; Ghanim, H.; Batra, M.; Kuhadiya, N.D.; Abuaysheh, S.; Green, K.; Antoine Makdissi, A.; Chaudhuri, A.; Dandona, P. Effect of Testosterone on Hepcidin, Ferroportin, Ferritin and Iron Binding Capacity in Patients with Hypogonadotropic Hypogonadism and Type 2 Diabetes. Clin. Endocrinol. 2016, 85, 772–780. [Google Scholar] [CrossRef]
- Peng, W.; Minakaki, G.; Nguyen, M.; Krainc, D. Preserving Lysosomal Function in the Aging Brain: Insights from Neurodegeneration. Neurotherapeutics 2019, 16, 611–634. [Google Scholar] [CrossRef]
- Liu, Z.; Shen, H.; Lian, T.; Mao, L.; Tang, S.; Sun, L.; Huang, X.; Guo, P.; Cao, C.; Yu, S.; et al. Iron Deposition in Substantia Nigra: Abnormal Iron Metabolism, Neuroinflammatory Mechanism and Clinical Relevance. Sci. Rep. 2017, 7, 14973. [Google Scholar] [CrossRef] [PubMed]
- Ghassaban, K.; He, N.; Sethi, S.K.; Huang, P.; Chen, S.; Yan, F.; Haacke, E.M. Regional High Iron in the Substantia Nigra Differentiates Parkinson’s Disease Patients From Healthy Controls. Front. Aging Neurosci. 2019, 11, 106. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Ge, Y.L.; Gu, C.C.; Zhang, J.R.; Jin, H.; Li, J.; Cheng, X.Y.; Yang, Y.P.; Wang, F.; Zhang, Y.C.; et al. Substantia Nigra Echogenicity is Associated with Serum Ferritin, Gender and Iron-Related Genes in Parkinson’s Disease. Sci. Rep. 2020, 10, 8660. [Google Scholar] [CrossRef] [PubMed]
- Joppe, K.; Roser, A.E.R.; Maass, F.; Lingor, P. The Contribution of Iron to Protein Aggregation Disorders in the Central Nervous System. Front. Neurosci. 2019, 13, 15. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Yang, H.; Zhang, D.; Jiang, H. Iron Concentration Does Not Differ in Blood but Tends to Decrease in Cerebrospinal Fluid in Parkinson’s Disease. Front. Neurosci. 2019, 13, 939. [Google Scholar] [CrossRef]
- Xing, Y.; Sapuan, A.; Dineen, R.A.; Auer, D.P. Life Span Pigmentation Changes of the Substantia Nigra Detected by Neuromelanin-Sensitive MRI. Mov. Disord. 2018, 33, 1792–1799. [Google Scholar] [CrossRef] [PubMed]
- Sohrabji, F. Guarding the Blood–Brain Barrier: A Role for Estrogen in the Etiology of Neurodegenerative Disease. Gene Expr. J. Liver Res. 2006, 13, 311–319. [Google Scholar] [CrossRef]
- Wang, L.F.; Yokoyama, K.K.; Chen, T.Y.; Hsiao, H.W.; Chiang, P.C.; Hsieh, Y.C.; Lo, S.; Hsu, C. Male-Specific Alleviation of Iron-Induced Striatal Injury by Inhibition of Autophagy. PLoS ONE 2015, 10, e0131224. [Google Scholar] [CrossRef]
- Mariani, S.; Ventriglia, M.; Simonelli, I.; Bucossi, S.; Siotto, M.; Donno, S.; Vernieri, F.; Squitti, R. Association between Sex, Systemic Iron Variation and Probability of Parkinson’s Disease. Int. J. Neurosci. 2016, 126, 354–360. [Google Scholar] [CrossRef]
- Chen, T.Y.; Lin, C.L.; Wang, L.F.; Tsai, K.L.; Lin, J.Y.; Hsu, C. Targeting GPER1 to Suppress Autophagy as a Male-Specific Therapeutic Strategy for Iron-Induced Striatal Injury. Sci. Rep. 2019, 9, 6661. [Google Scholar] [CrossRef]
- Xu, M.; Tan, X.; Li, N.; Wu, H.; Wang, Y.; Xie, J.; Wang, J. Differential Regulation of Estrogen in Iron Metabolism in Astrocytes and Neurons. J. Cell. Physiol. 2019, 234, 4232–4242. [Google Scholar] [CrossRef] [PubMed]
- Sääksjärvi, K.; Knekt, P.; Lundqvist, A.; Männistö, S.; Heliövaara, M.; Rissanen, H.; Järvinen, R. A Cohort Study on Diet and the Risk of Parkinson’s Disease: The Role of Food Groups and Diet Quality. Br. J. Nutr. 2013, 109, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Micha, R.; Wallace, S.K.; Mozaffarian, D. Red and Processed Meat Consumption and Risk of Incident Coronary Heart Disease, Stroke, and Diabetes: A Systematic Review and Meta-Analysis. Circulation 2010, 121, 2271–2283. [Google Scholar] [CrossRef] [PubMed]
- Rohrmann, S.; Overvad, K.; Bueno-de-Mesquita, H.B.; Jakobsen, M.U.; Egeberg, R.; Tjønneland, A.; Nailler, L.; Boutron-Ruault, M.C.; Clavel-Chapelon, F.; Krogh, V.; et al. Meat Consumption and Mortality—Results from the European Prospective Investigation into Cancer and Nutrition. BMC Med. 2013, 11, 63. [Google Scholar] [CrossRef]
- Hare, D.J.; Cardoso, B.R.; Raven, E.P.; Double, K.L.; Finkelstein, D.I.; Szymlek-Gay, E.A.; Biggs, B.A. Excessive Early-Life Dietary Exposure: A Potential Source of Elevated Brain Iron and a Risk Factor for Parkinson’s disease. NPJ Parkinsons Dis. 2017, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Logroscino, G.; Chen, H.; Wing, A.; Ascherio, A. Blood Donations Iron, Stores, and Risk of Parkinson’s Disease. Mov. Disord. 2006, 21, 835–838. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Tanaka, K.; Fukushima, W.; Sasaki, S.; Kiyohara, C.; Tsuboi, Y.; Yamada, T.; Oeda, T.; Miki, T.; Kawamura, N. Dietary Intake of Metals and Risk of Parkinson’s Disease: A Case-Control Study in Japan. J. Neurol. Sci. 2011, 306, 98–102. [Google Scholar] [CrossRef]
- Deng, Q.; Zhou, X.; Chen, J.; Pan, M.; Gao, H.; Zhou, J.; Wang, D.; Chen, Q.; Zhang, X.; Wang, Q. Lower Hemoglobin Levels in Patients with Parkinson’s Disease Are Associated with Disease Severity and Iron Metabolism. Brain Res. 2017, 1655, 145–151. [Google Scholar] [CrossRef]
- Hong, C.T.; Huang, Y.H.; Liu, H.Y.; Chiou, H.Y.; Chan, L.; Chien, L.N. Newly Diagnosed Anemia Increases Risk of Parkinson’s disease: A Population-Based Cohort Study. Sci. Rep. 2016, 6, 29651. [Google Scholar] [CrossRef]
- Santiago, J.A.; Bottero, V.; Potashkin, J.A. Biological and Clinical Implications of Comorbidities in Parkinson’s Disease. Front. Aging Neurosci. 2017, 9, 394. [Google Scholar] [CrossRef]
- Mariani, S.; Ventriglia, M.; Simonelli, I.; Donno, S.; Bucossi, S.; Vernieri, F.; Melgari, J.M.; Pasqualetti, P.; Rossini, P.M.; Squitti, R. Fe and Cu do not Differ in Parkinson’s Disease: A Replication Study Plus Meta-Analysis. Neurobiol. Aging 2013, 34, 632–633. [Google Scholar] [CrossRef] [PubMed]
- Sian-Hülsmann, J.; Mandel, S.; Youdim, M.B.H.; Riederer, P. The Relevance of Iron in the Pathogenesis of Parkinson’s Disease. J. Neurochem. 2011, 118, 939–957. [Google Scholar] [CrossRef] [PubMed]
- Savica, R.; Grossardt, B.R.; Bower, J.H.; Ahlskog, J.E.; Rocca, W.A. Risk Factors for Parkinson’s Disease May Differ in Men and Women: An Exploratory Study. Horm. Behav. 2013, 63, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Seidl, S.E.; Santiago, J.A.; Bilyk, H.; Potashkin, J.A. The Emerging Role of Nutrition in Parkinson’s Disease. Front. Aging Neurosci. 2014, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Wang, X.; Geng, M. Alzheimer’s Disease Hypothesis and Related Therapies. Transl. Neurodegener. 2018, 7, 1–7. [Google Scholar] [CrossRef]
- Chen, M.; Zheng, J.; Liu, G.; Zeng, C.; Xu, E.; Zhu, W.; Anderson, G.J.; Chen, H. High Dietary Iron Disrupts Iron Homeostasis and Induces Amyloid-β and Phospho-τ Expression in the Hippocampus of Adult Wild-Type and APP/PS1 Transgenic Mice. J. Nutr. 2019, 149, 2247–2254. [Google Scholar] [CrossRef]
- Perry, G.; Cash, A.D.; Smith, M.A. Alzheimer Disease and Oxidative Stress. J. Biomed. Biotechnol. 2002, 2, 120–123. [Google Scholar] [CrossRef]
- Xu, J.; Jia, Z.; Knutson, M.D.; Leeuwenburgh, C. Impaired Iron Status in Aging Research. Int. J. Mol. Sci. 2012, 13, 2368–2386. [Google Scholar] [CrossRef]
- Ayton, S.; Faux, N.G.; Bush, A.I. Association of cerebrospinal fluid ferritin level with preclinical cognitive decline in APOE-epsilon4 carriers. JAMA Neurol. 2017, 74, 122–125. [Google Scholar] [CrossRef]
- Ayton, S.; Diouf, I.; Bush, A.I. Alzheimer’s Disease Neuroimaging, Evidence that Iron Accelerates Alzheimer’s Pathology: A CSF Biomarker Study. J. Neurol. Neurosurg. Psychiatry 2018, 89, 456–460. [Google Scholar] [CrossRef]
- Bartzokis, G.; Lu, P.H.; Tishler, T.A.; Peters, D.G.; Kosenko, A.; Barrall, K.A.; Finn, J.P.; Villablanca, P.; Laub, G.; Altshuler, L.L. Prevalent Iron Metabolism Gene Variants Associated with Increased Brain Ferritin Iron in Healthy Older Men. J. Alzheimers Dis. 2010, 20, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Burger, A.; Kotze, M.J.; Stein, D.J.; van Rensburg, S.J.; Howells, F.M. The Relationship between Measurement of vice versa Brain Glutamate and Markers of Iron Metabolism: A Proton Magnetic Resonance Spectroscopy Study in Healthy Adults. Eur. J. Neurosci. 2020, 51, 984–990. [Google Scholar] [CrossRef] [PubMed]
- Simpkins, J.W.; Perez, E.; Wang, X.; Yang, S.; Wen, Y.; Singh, M. The Potential for Estrogens in Preventing Alzheimer’s Disease and Vascular Dementia. Ther. Adv. Neurol. Disord. 2009, 2, 31–49. [Google Scholar] [CrossRef] [PubMed]
- Wahjoepramono, E.J.; Asih, P.R.; Aniwiyanti, V.; Taddei, K.; Dhaliwal, S.S.; Fuller, S.J.; Foster, J.; Carruthers, M.; Verdile, G.; Sohrabi, H.R.; et al. The Effects of Testosterone Supplementation on Cognitive Functioning in Older Men. CNS Neurol. Disord. Drug Targets 2016, 15, 337–343. [Google Scholar] [CrossRef]
- Lei, Y.; Renyuan, Z. Effects of Androgens on the Amyloid-β Protein in Alzheimer’s Disease. Endocrinology 2018, 159, 3885–3894. [Google Scholar] [CrossRef]
- Ruiz-Larrea, B.; Leal, A.; Martín, C.; Martínez, R.; Lacort, M. Effects of Estrogens on the Redox Chemistry of Iron: A Possible Mechanism of the Antioxidant Action of Estrogens. Steroids 1995, 60, 780–783. [Google Scholar] [CrossRef]
- Requintina, P.J.; Oxenkrug, G.F. The in Vitro Effect of Estradiol and Testosterone on Iron-Induced Lipid Peroxidation in Rat Brain and Kidney Tissues. Ann. N. Y. Acad. Sci. 2005, 1053, 400–404. [Google Scholar] [CrossRef]
- Wolters, F.J.; HZonneveld, H.I.; Licher, S.; Cremers, L.G.M.; Heart Brain Connection Collaborative Research Group; Ikram, M.K.; Koudstaal, P.J.; Vernooij, M.W.; Ikram, M.A. Hemoglobin and Anemia in Relation to Dementia Risk and Accompanying Changes on Brain MRI. Neurology 2019, 93, e917–e926. [Google Scholar] [CrossRef]
- Atti, A.R.; Palmer, K.; Volpato, S.; Zuliani, G.; Winblad, B.; Fratiglioni, L. Anaemia Increases the Risk of Dementia in Cognitively Intact Elderly. Neurobiol. Aging 2006, 27, 278–284. [Google Scholar] [CrossRef]
- Shah, R.C.; Buchman, A.S.; Wilson, R.S.; Leurgans, S.E.; Bennett, D.A. Hemoglobin Level in Older Persons and Incident Alzheimer Disease: Prospective Cohort Analysis. Neurology 2011, 77, 219–226. [Google Scholar] [CrossRef]
- Carlson, E.S.; Magid, R.; Petryk, A.; Georgieff, M.K. Iron Deficiency Alters Expression of Genes Implicated in Alzheimer Disease Pathogenesis. Brain Res. 2008, 1237, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Falkingham, M.; Abdelhamid, A.; Curtis, P.; Fairweather-Tait, S.; Dye, L.; Hooper, L. The Effects of Oral Iron Supplementation on Cognition in Older Children and Adults: A Systematic Review and Meta-Analysis. Nutr. J. 2010, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Garry, P.J.; Hunt, W.C.; Baumgartner, R.N. Effects of Iron Intake on Iron Stores in Elderly Men and Women: Longitudinal and Cross-Sectional Results. J. Am. Coll. Nutr. 2000, 19, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Rutten, B.P.F.; Schmitz, C.; Gerlach, O.H.H.; Oyen, H.M.; de Mesquita, E.B.; Steinbusch, H.W.M.; Korr, H. The Aging Brain: Accumulation of DNA Damage or Neuron Loss? Neurobiol. Aging 2007, 28, 91–98. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grubić Kezele, T.; Ćurko-Cofek, B. Age-Related Changes and Sex-Related Differences in Brain Iron Metabolism. Nutrients 2020, 12, 2601. https://doi.org/10.3390/nu12092601
Grubić Kezele T, Ćurko-Cofek B. Age-Related Changes and Sex-Related Differences in Brain Iron Metabolism. Nutrients. 2020; 12(9):2601. https://doi.org/10.3390/nu12092601
Chicago/Turabian StyleGrubić Kezele, Tanja, and Božena Ćurko-Cofek. 2020. "Age-Related Changes and Sex-Related Differences in Brain Iron Metabolism" Nutrients 12, no. 9: 2601. https://doi.org/10.3390/nu12092601
APA StyleGrubić Kezele, T., & Ćurko-Cofek, B. (2020). Age-Related Changes and Sex-Related Differences in Brain Iron Metabolism. Nutrients, 12(9), 2601. https://doi.org/10.3390/nu12092601