Impacts of Habitual Diets Intake on Gut Microbial Counts in Healthy Japanese Adults

Abstract
1. Introduction
2. Materials and Methods
2.1. Cohorts
2.2. Collection of Dietary Data
2.3. Collection and Preparation of Stool Samples and DNA Extraction
2.4. Quantification of Total Bacteria by Quantitative PCR
2.5. Amplification of the 16S rRNA Gene Region and Next-Generation Sequencing
2.6. Processing of 16S rRNA Gene Sequence Data
2.7. Estimation of the Number of Gut Bacteria
2.8. Stastical Analysis
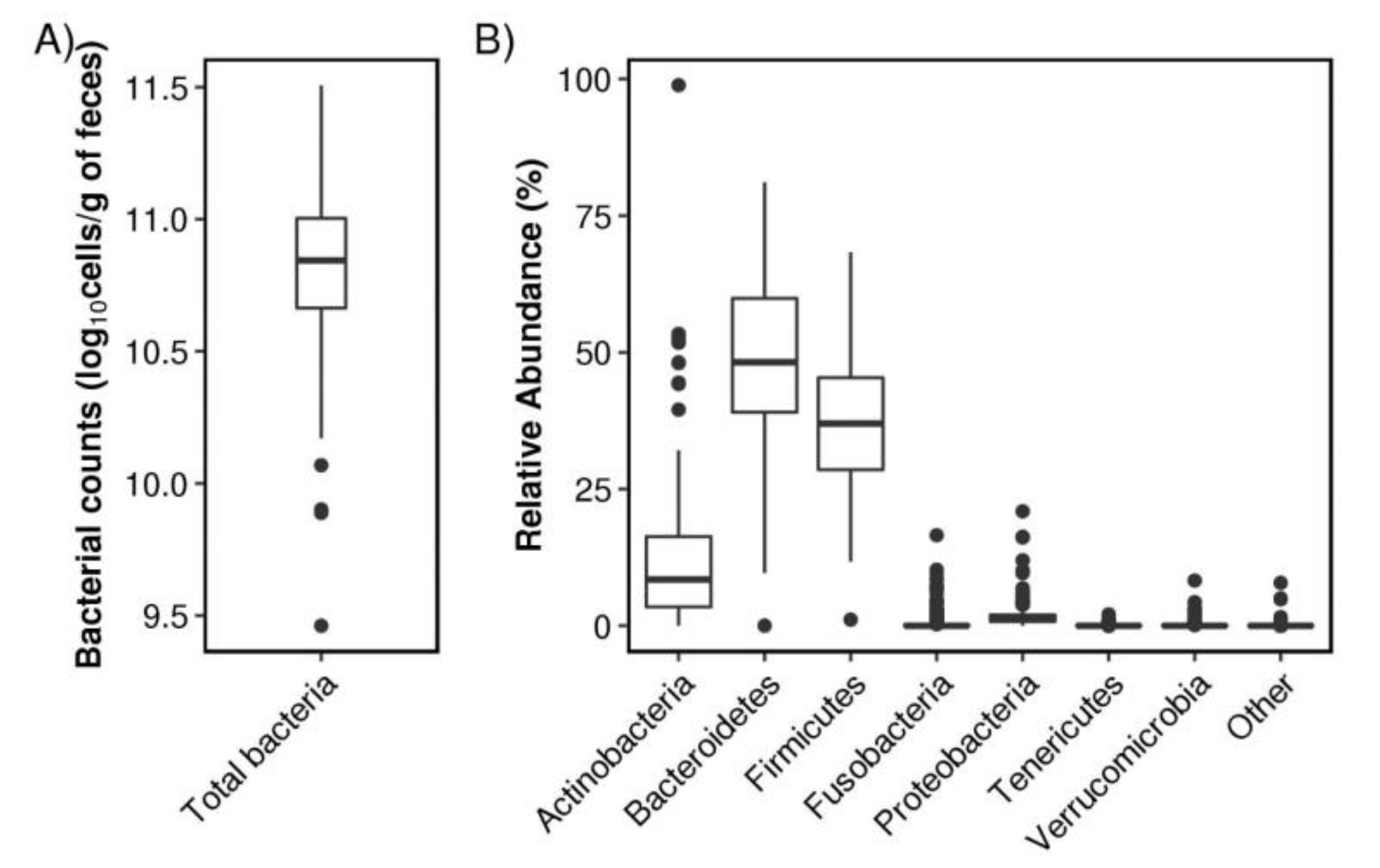
3. Results
3.1. Characteristics of Subjects
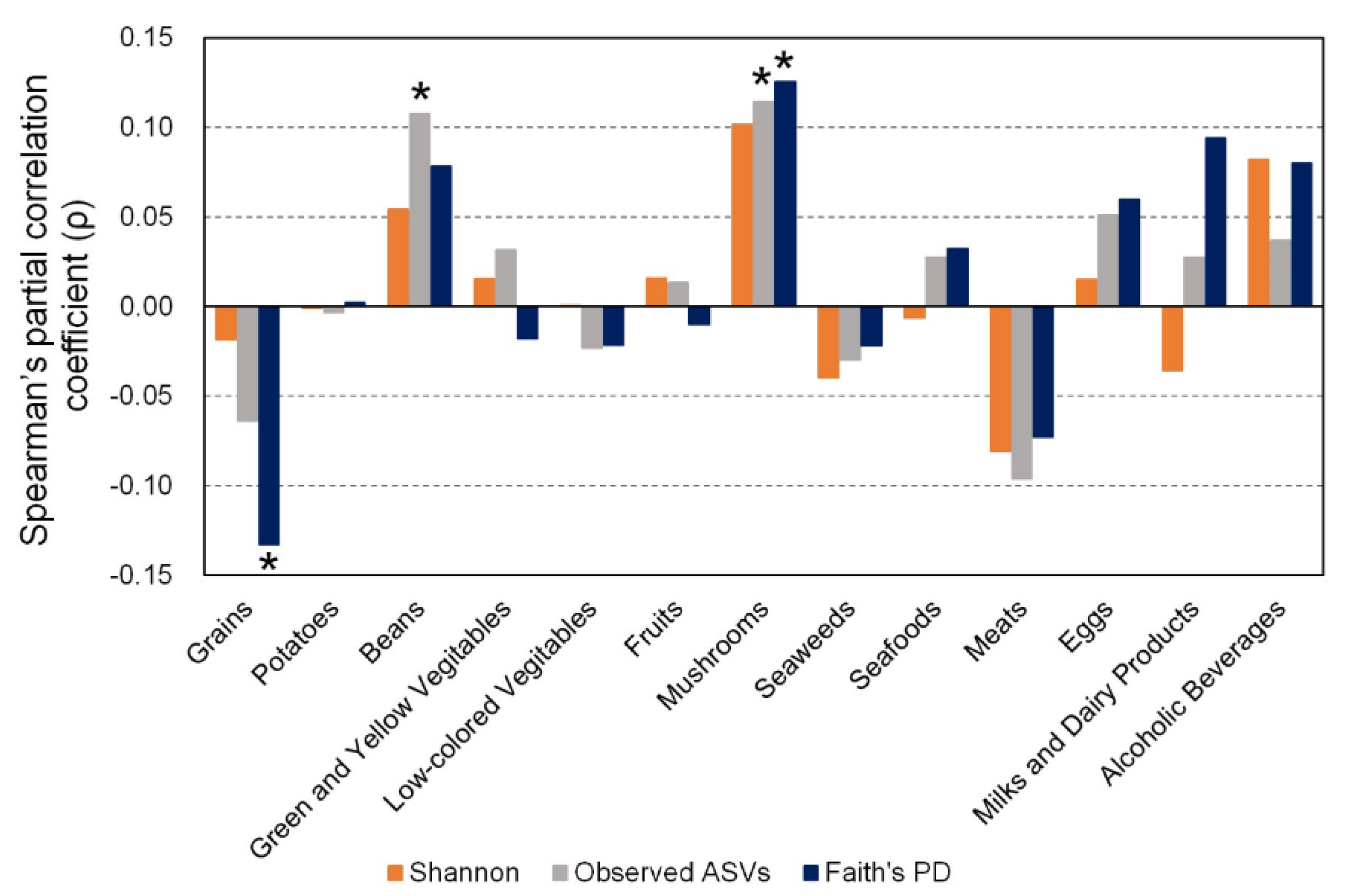
3.2. Association between α-Diversity of Gut Microbiota and Dietary Food Intake
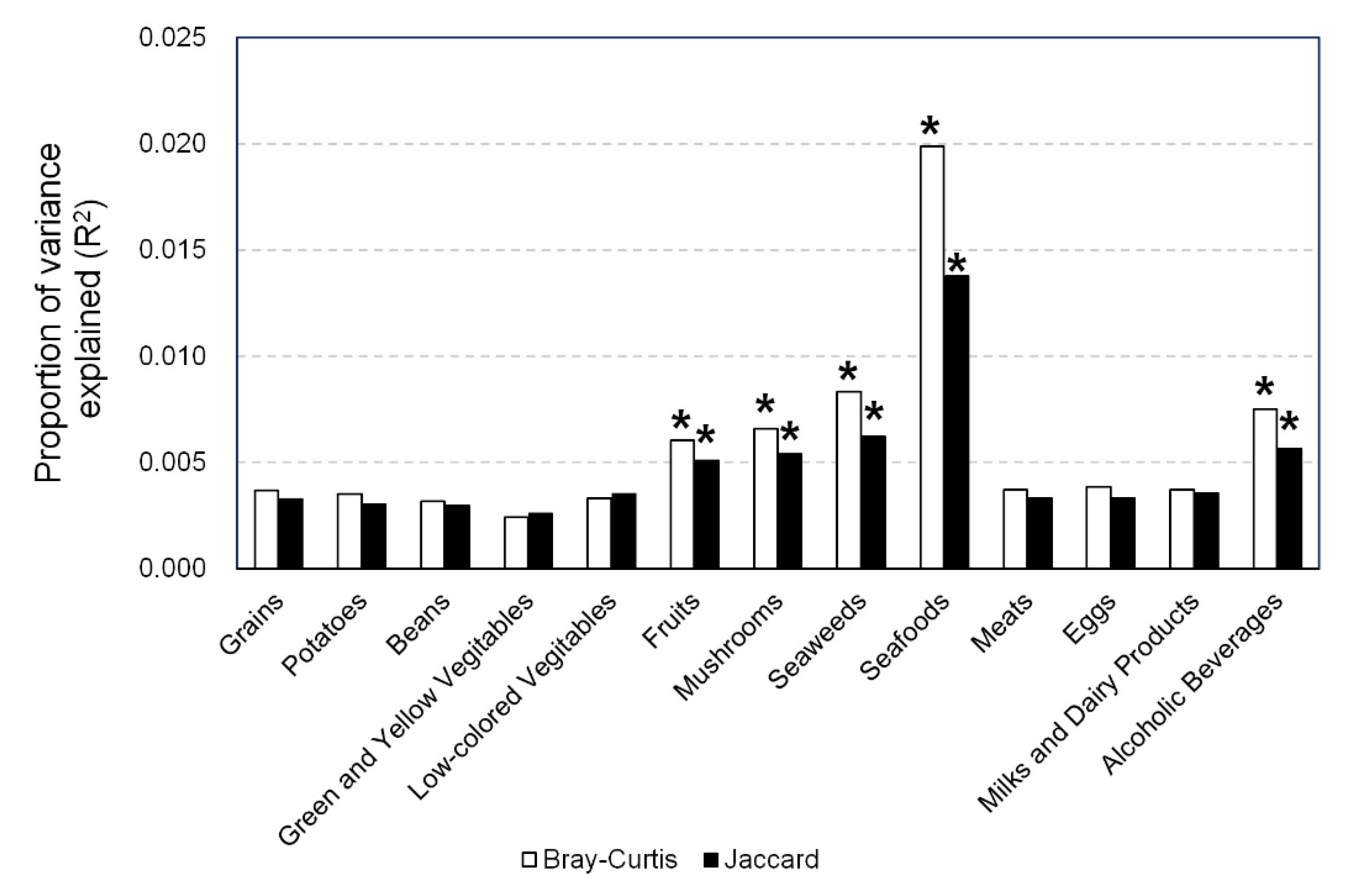
3.3. Association between β-Diversity of Gut Microbiota and Dietary Food Intake
3.4. Associations between Number of Gut Bacteria and Food Intake Related to α-Diversity of Gut Microbiota
3.5. Associations between Number of Gut Bacteria and Food Intake Related to β-Diversity of Gut Microbiota
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Lu, L.; Sun, J.; Petrof, E.O.; Claud, E.C. Preterm infant gut microbiota affects intestinal epithelial development in a humanized microbiome gnotobiotic mouse model. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G521–G532. [Google Scholar] [CrossRef] [PubMed]
- Takiishi, T.; Fenero, C.I.M.; Câmara, N.O.S. Intestinal barrier and gut microbiota: Shaping our immune responses throughout life. Tissue Barriers 2017, 5, e1373208. [Google Scholar] [CrossRef] [PubMed]
- Natividad, J.M.; Verdu, E.F. Modulation of intestinal barrier by intestinal microbiota: Pathological and therapeutic implications. Pharm. Res. 2017, 69, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Baumler, A.J.; Sperandio, V. Interactions between the microbiota and pathogenic bacteria in the gut. Nature 2016, 535, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef]
- Rinninella, E.; Cintoni, M.; Raoul, P.; Lopetuso, L.R.; Scaldaferri, F.; Pulcini, G.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. Food Components and Dietary Habits: Keys for a Healthy Gut Microbiota Composition. Nutrients 2019, 11, 2393. [Google Scholar] [CrossRef]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef]
- So, D.; Whelan, K.; Rossi, M.; Morrison, M.; Holtmann, G.; Kelly, J.T.; Shanahan, E.R.; Staudacher, H.M.; Campbell, K.L. Dietary fiber intervention on gut microbiota composition in healthy adults: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2018, 107, 965–983. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Wang, F.; Yuan, J.; Li, J.; Jiang, D.; Zhang, J.; Li, H.; Wang, R.; Tang, J.; Huang, T.; et al. Effects of dietary fat on gut microbiota and faecal metabolites, and their relationship with cardiometabolic risk factors: A 6-month randomised controlled-feeding trial. Gut 2019, 68, 1417–1429. [Google Scholar] [CrossRef] [PubMed]
- Partula, V.; Mondot, S.; Torres, M.J.; Kesse-Guyot, E.; Deschasaux, M.; Assmann, K.; Latino-Martel, P.; Buscail, C.; Julia, C.; Galan, P.; et al. Milieu Intérieur Consortium. Associations between usual diet and gut microbiota composition: Results from the Milieu Interieur cross-sectional study. Am. J. Clin. Nutr. 2019, 109, 1472–1483. [Google Scholar] [CrossRef] [PubMed]
- Seura, T.; Yoshino, Y.; Fukuwatari, T. The Relationship between Habitual Dietary Intake and Gut Microbiota in Young Japanese Women. J. Nutr. Sci. Vitam. (Tokyo) 2017, 63, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Shima, T.; Amamoto, R.; Kaga, C.; Kado, Y.; Sasai, T.; Watanabe, O.; Shiinoki, J.; Iwazaki, K.; Shigemura, H.; Tsuji, H. Association of life habits and fermented milk intake with stool frequency, defecatory symptoms and intestinal microbiota in healthy Japanese adults. Benef. Microbes 2019, 10, 841–854. [Google Scholar] [CrossRef] [PubMed]
- Tsubono, Y.; Takamori, S.; Kobayashi, M.; Takahashi, T.; Iwase, Y.; Iitoi, Y.; Akabane, M.; Yamaguchi, M.; Tsugane, S. A data-based approach for designing a semiquantitative food frequency questionnaire for a population-based prospective study in Japan. J. Epidemiol. 1996, 6, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Kubota, H.; Tsuji, H.; Matsuda, K.; Kurakawa, T.; Asahara, T.; Nomoto, K. Detection of human intestinal catalase-negative, Gram-positive cocci by rRNA-targeted reverse transcription-PCR. Appl. Environ. Microbiol. 2010, 76, 5440–5451. [Google Scholar] [CrossRef] [PubMed]
- Matsuki, T.; Watanabe, K.; Fujimoto, J.; Takada, T.; Tanaka, R. Use of 16S rRNA gene-targeted group-specific primers for real-time PCR analysis of predominant bacteria in human feces. Appl. Environ. Microbiol. 2004, 70, 7220–7228. [Google Scholar] [CrossRef]
- Fuller, Z.; Louis, P.; Mihajlovski, A.; Rungapamestry, V.; Ratcliffe, B.; Duncan, A.J. Influence of cabbage processing methods and prebiotic manipulation of colonic microflora on glucosinolate breakdown in man. Br. J. Nutr. 2007, 98, 364–372. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA. 2011, 108 (Suppl. S1), 4516–4522. [Google Scholar] [CrossRef] [PubMed]
- Nagara, Y.; Takada, T.; Nagata, Y.; Kado, S.; Kushiro, A. Microscale spatial analysis provides evidence for adhesive monopolization of dietary nutrients by specific intestinal bacteria. PLoS ONE 2017, 12, e0175497. [Google Scholar] [CrossRef] [PubMed]
- Matsuki, T.; Yahagi, K.; Mori, H.; Matsumoto, H.; Hara, T.; Tajima, S.; Ogawa, E.; Kodama, H.; Yamamoto, K.; Yamada, T.; et al. A key genetic factor for fucosyllactose utilization affects infant gut microbiota development. Nat. Commun. 2016, 7, 11939. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5073. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximumlikelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Revell, L.J. phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 2011, 3, 217–223. [Google Scholar] [CrossRef]
- Vieira-Silva, S.; Sabino, J.; Valles-Colomer, M.; Falony, G.; Kathagen, G.; Caenepeel, C.; Cleynen, I.; van der Merwe, S.; Vermeire, S.; Raes, J. Quantitative microbiome profiling disentangles inflammation- and bile duct obstruction-associated microbiota alterations across PSC/IBD diagnoses. Nat. Microbiol. 2019, 4, 1826–1831. [Google Scholar] [CrossRef]
- Vandeputte, D.; Kathagen, G.; D’hoe, K.; Vieira-Silva, S.; Valles-Colomer, M.; Sabino, J.; Wang, J.; Tito, R.Y.; De Commer, L.; Darzi, Y.; et al. Quantitative microbiome profiling links gut community variation to microbial load. Nature 2017, 551, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Falony, G.; Joossens, M.; Vieira-Silva, S.; Wang, J.; Darzi, Y.; Faust, K.; Kurilshikov, A.; Bonder, M.J.; Valles-Colomer, M.; Vandeputte, D.; et al. Population-level analysis of gut microbiome variation. Science 2016, 352, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Ministry of Health, Labor and Welfare. The National Health Nutrition Survey in Japan. 2018. Available online: https://www.nibiohn.go.jp/eiken/kenkounippon21/en/eiyouchousa/koumoku_syokuhin_chousa.html (accessed on 22 April 2020).
- Varshney, J.; Ooi, J.H.; Jayarao, B.M.; Albert, I.; Fisher, J.; Smith, R.L.; Patterson, A.D.; Cantorna, M.T. White button mushrooms increase microbial diversity and accelerate the resolution of Citrobacter rodentium infection in mice. J. Nutr. 2013, 143, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Zhernakova, A.; Kurilshikov, A.; Bonder, M.J.; Tigchelaar, E.F.; Schirmer, M.; Vatanen, T.; Mujagic, Z.; Vila, A.V.; Falony, G.; Vieira-Silva, S.; et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 2016, 352, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Zava, T.T.; Zava, D.T. Assessment of Japanese iodine intake based on seaweed consumption in Japan: A literature-based analysis. Thyroid Res. 2011, 4, 14. [Google Scholar] [CrossRef]
- Ministry of Agriculture, Forestry and Fisheries. White Paper on Fisheries. 2018. Available online: https://www.jfa.maff.go.jp/j/kikaku/wpaper/ (accessed on 22 April 2020).
- Korpela, K. Diet, Microbiota, and Metabolic Health: Trade-Off Between Saccharolytic and Proteolytic Fermentation. Annu. Rev. Food Sci. Technol. 2018, 9, 65–84. [Google Scholar] [CrossRef]
- Hjorth, M.F.; Roager, H.M.; Larsen, T.M.; Poulsen, S.K.; Licht, T.R.; Bahl, M.I.; Zohar, Y.; Astrup, A. Pre-treatment microbial Prevotella-to-Bacteroides ratio, determines body fat loss success during a 6-month randomized controlled diet intervention. Int. J. Obes. (Lond.) 2018, 42, 580–583. [Google Scholar] [CrossRef]
- Costabile, A.; Klinder, A.; Fava, F.; Napolitano, A.; Fogliano, V.; Leonard, C.; Gibson, G.R.; Tuohy, K.M. Whole-grain wheat breakfast cereal has a prebiotic effect on the human gut microbiota: A double-blind, placebo-controlled, crossover study. Br. J. Nutr. 2008, 99, 110–120. [Google Scholar] [CrossRef]
- Martinez, I.; Kim, J.; Duffy, P.R.; Schlegel, V.L.; Walter, J. Resistant starches types 2 and 4 have differential effects on the composition of the fecal microbiota in human subjects. PLoS ONE 2010, 5, e15046. [Google Scholar] [CrossRef]
- Venkataraman, A.; Sieber, J.R.; Schmidt, A.W.; Waldron, C.; Theis, K.R.; Schmidt, T.M. Variable responses of human microbiomes to dietary supplementation with resistant starch. Microbiome 2016, 4, 33. [Google Scholar] [CrossRef]
- Tomova, A.; Bukovsky, I.; Rembert, E.; Yonas, W.; Alwarith, J.; Barnard, N.D.; Kahleova, H. The Effects of Vegetarian and Vegan Diets on Gut Microbiota. Front. Nutr. 2019, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Halmos, E.P.; Christophersen, C.T.; Bird, A.R.; Shepherd, S.J.; Gibson, P.R.; Muir, J.G. Diets that differ in their FODMAP content alter the colonic luminal microenvironment. Gut 2015, 64, 93–100. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, M.; Montemurno, E.; Vannini, L.; Cosola, C.; Cavallo, N.; Gozzi, G.; Maranzano, V.; Di Cagno, R.; Gobbetti, M.; Gesualdo, L. Effect of Whole-Grain Barley on the Human Fecal Microbiota and Metabolome. Appl. Environ. Microbiol. 2015, 81, 7945–7956. [Google Scholar] [CrossRef] [PubMed]
- Hess, J.; Wang, Q.; Gould, T.; Slavin, J. Impact of Agaricus bisporus Mushroom Consumption on Gut Health Markers in Healthy Adults. Nutrients 2018, 10, 1402. [Google Scholar] [CrossRef] [PubMed]
- Henning, S.M.; Yang, J.; Shao, P.; Lee, R.P.; Huang, J.; Ly, A.; Hsu, M.; Lu, Q.Y.; Thames, G.; Heber, D.; et al. Health benefit of vegetable/fruit juice-based diet: Role of microbiome. Sci. Rep. 2017, 7, 2167. [Google Scholar] [CrossRef]
- Cherry, P.; Yadav, S.; Strain, C.R.; Allsopp, P.J.; McSorley, E.M.; Ross, R.P.; Stanton, C. Prebiotics from Seaweeds: An Ocean of Opportunity? Mar. Drugs 2019, 17, 327. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kwon, Y.; Kim, I.S.; Kim, J.A.; Yu, D.Y.; Adhikari, B.; Lee, S.S.; Choi, I.S.; Cho, K.K. Effects of the Brown Seaweed Laminaria japonica Supplementation on Serum Concentrations of IgG, Triglycerides, and Cholesterol, and Intestinal Microbiota Composition in Rats. Front. Nutr. 2018, 5, 23. [Google Scholar] [CrossRef]
- Holmstrom, K.; Collins, M.D.; Moller, T.; Falsen, E.; Lawson, P.A. Subdoligranulum variabile gen. nov., sp. nov. from human feces. Anaerobe 2004, 10, 197–203. [Google Scholar] [CrossRef]
- Urwin, H.J.; Miles, E.A.; Noakes, P.S.; Kremmyda, L.S.; Vlachava, M.; Diaper, N.D.; Godfrey, K.M.; Calder, P.C.; Vulevic, J.; Yaqoob, P. Effect of salmon consumption during pregnancy on maternal and infant faecal microbiota, secretory IgA and calprotectin. Br. J. Nutr. 2014, 111, 773–784. [Google Scholar] [CrossRef]
- Fava, F.; Gitau, R.; Griffin, B.A.; Gibson, G.R.; Tuohy, K.M.; Lovegrove, J.A. The type and quantity of dietary fat and carbohydrate alter faecal microbiome and short-chain fatty acid excretion in a metabolic syndrome ’at-risk’ population. Int. J. Obes. (Lond.) 2013, 37, 216–223. [Google Scholar] [CrossRef]
- Engen, P.A.; Green, S.J.; Voigt, R.M.; Forsyth, C.B.; Keshavarzian, A. The Gastrointestinal Microbiome: Alcohol Effects on the Composition of Intestinal Microbiota. Alcohol Res. 2015, 37, 223–236. [Google Scholar] [PubMed]
- Mutlu, E.A.; Gillevet, P.M.; Rangwala, H.; Sikaroodi, M.; Naqvi, A.; Engen, P.A.; Kwasny, M.; Lau, C.K.; Keshavarzian, A. Colonic microbiome is altered in alcoholism. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G966–G978. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, F.; Lu, H.; Wang, B.; Chen, Y.; Lei, D.; Wang, Y.; Zhu, B.; Li, L. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology 2011, 54, 562–572. [Google Scholar] [CrossRef]
- Feng, Y.L.; Shu, L.; Zheng, P.F.; Zhang, X.Y.; Si, C.J.; Yu, X.L.; Gao, W.; Zhang, L. Dietary patterns and colorectal cancer risk: A meta-analysis. Eur. J. Cancer Prev. 2017, 26, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Myung, S.K.; Lee, J.H. Light Alcohol Drinking and Risk of Cancer: A Meta-Analysis of Cohort Studies. Cancer Res. Treat. 2018, 50, 474–487. [Google Scholar] [CrossRef]
- Mima, K.; Nishihara, R.; Qian, Z.R.; Cao, Y.; Sukawa, Y.; Nowak, J.A.; Yang, J.; Dou, R.; Masugi, Y.; Song, M.; et al. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut 2016, 65, 1973–1980. [Google Scholar] [CrossRef]
- Komiya, Y.; Shimomura, Y.; Higurashi, T.; Sugi, Y.; Arimoto, J.; Umezawa, S.; Uchiyama, S.; Matsumoto, M.; Nakajima, A. Patients with colorectal cancer have identical strains of Fusobacterium nucleatum in their colorectal cancer and oral cavity. Gut 2019, 68, 1335–1337. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Characteristic | Median (IQR) | n (%) | p Value |
|---|---|---|---|
| Sex | |||
| Female | 173 (48.9) | 0.60 | |
| Male | 181 (51.1) | ||
| Age (years) | 40 (29–50) | ||
| 20–29 | 89 (25.1) | 0.96 | |
| 30–39 | 85 (24.0) | ||
| 40–49 | 89 (25.1) | ||
| 50–59 | 91 (25.7) | ||
| BMI (kg/m2) | 21.9 (20.0–24.0) | ||
| <18.5 | 33 (9.3) | <0.001 | |
| 18.5–<25.0 | 258 (72.9) | ||
| ≥25.0 | 63 (17.8) | ||
| Smoking status | |||
| Never | 266 (75.1) | <0.001 | |
| Former | 34 (9.6) | ||
| Current | 54 (15.3) |
| Food Intake | |
|---|---|
| Total energy (kJ/day) | 8537.5 ± 1069.4 |
| Food group (g/day) | |
| Grains | 381.6 ± 95.6 |
| Potatoes | 41.2 ± 8.7 |
| Beans | 77.3 ± 46.3 |
| Green and yellow vegetables | 162.9 ± 50.2 |
| Light-colored vegetables | 181.0 ± 36.6 |
| Fruits | 148.1 ± 27.1 |
| Mushrooms | 16.5 ± 11.7 |
| Seaweeds | 10.8 ± 5.3 |
| Seafoods | 92.8 ± 3.1 |
| Meats | 76.7 ± 20.0 |
| Eggs | 35.4 ± 8.6 |
| Milks and dairy products | 192.5 ± 85.0 |
| Alcoholic beverage | 139.3 ± 195.3 |
| Food Group | Unstandardized Coefficients | Standardized Coefficients | p Value | ||
|---|---|---|---|---|---|
| B | Std. Error | β | |||
| Grains | Bacteroidetes | ||||
| Bacteroides | 0.000953 | 0.000401 | 0.190 | 0.018 | |
| Firmicutes | |||||
| Lactobacillus | −0.00410 | 0.00207 | −0.161 | 0.048 | |
| Lactococcus | −0.00414 | 0.00158 | −0.211 | 0.0093 | |
| Streptococcus | 0.00182 | 0.000846 | 0.173 | 0.032 | |
| Veillonella | 0.00482 | 0.00209 | 0.188 | 0.022 | |
| Beans | Bacteroidetes | ||||
| Prevotella | 0.00852 | 0.00401 | 0.120 | 0.034 | |
| Firmicutes | |||||
| Bacillus | 0.00991 | 0.00284 | 0.201 | <0.001 | |
| Clostridium | 0.00630 | 0.00270 | 0.133 | 0.020 | |
| Roseburia | 0.00670 | 0.00295 | 0.130 | 0.024 | |
| Faecalibacterium | 0.00529 | 0.00207 | 0.143 | 0.011 | |
| Ruminococcus | 0.00473 | 0.00197 | 0.136 | 0.017 | |
| Meganomas | 0.00716 | 0.00335 | 0.122 | 0.034 | |
| Eubacterium | −0.00757 | 0.00249 | −0.176 | 0.0025 | |
| Fusobacteria | |||||
| Fusobacterium | −0.00912 | 0.00313 | −0.162 | 0.0039 | |
| Mushrooms | Bacteroidetes | ||||
| Parabacteroides | −0.0323 | 0.00675 | −0.261 | <0.001 | |
| Food Group | Unstandardized Coefficients | Standardized Coefficients | p Value | ||
|---|---|---|---|---|---|
| B | Std. Error | β | |||
| Fruits | Bacteroidetes | ||||
| Alistipes | −0.0132 | 0.00528 | −0.163 | 0.013 | |
| Firmicutes | |||||
| Streptococcus | 0.00519 | 0.00245 | 0.139 | 0.035 | |
| Butyricicoccus | 0.00982 | 0.00492 | 0.134 | 0.047 | |
| Seaweeds | Firmicutes | ||||
| Streptococcus | −0.0440 | 0.0208 | −0.137 | 0.035 | |
| Subdoligranulum | 0.125 | 0.0545 | 0.151 | 0.022 | |
| Seafoods | Bacteroidetes | ||||
| Bacteroides | −0.0111 | 0.00355 | −0.309 | 0.0020 | |
| Alcoholic beverage | Actinobacteria | ||||
| Actinomyces | −0.00128 | 0.000606 | −0.121 | 0.036 | |
| Firmicutes | |||||
| Clostridium | −0.00145 | 0.000629 | −0.129 | 0.022 | |
| Fusobacteria | |||||
| Fusobacterium | 0.00170 | 0.000734 | 0.127 | 0.021 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sugimoto, T.; Shima, T.; Amamoto, R.; Kaga, C.; Kado, Y.; Watanabe, O.; Shiinoki, J.; Iwazaki, K.; Shigemura, H.; Tsuji, H.; et al. Impacts of Habitual Diets Intake on Gut Microbial Counts in Healthy Japanese Adults. Nutrients 2020, 12, 2414. https://doi.org/10.3390/nu12082414
Sugimoto T, Shima T, Amamoto R, Kaga C, Kado Y, Watanabe O, Shiinoki J, Iwazaki K, Shigemura H, Tsuji H, et al. Impacts of Habitual Diets Intake on Gut Microbial Counts in Healthy Japanese Adults. Nutrients. 2020; 12(8):2414. https://doi.org/10.3390/nu12082414
Chicago/Turabian StyleSugimoto, Takuya, Tatsuichiro Shima, Ryuta Amamoto, Chiaki Kaga, Yukiko Kado, Osamu Watanabe, Junko Shiinoki, Kaoru Iwazaki, Hiroko Shigemura, Hirokazu Tsuji, and et al. 2020. "Impacts of Habitual Diets Intake on Gut Microbial Counts in Healthy Japanese Adults" Nutrients 12, no. 8: 2414. https://doi.org/10.3390/nu12082414
APA StyleSugimoto, T., Shima, T., Amamoto, R., Kaga, C., Kado, Y., Watanabe, O., Shiinoki, J., Iwazaki, K., Shigemura, H., Tsuji, H., & Matsumoto, S. (2020). Impacts of Habitual Diets Intake on Gut Microbial Counts in Healthy Japanese Adults. Nutrients, 12(8), 2414. https://doi.org/10.3390/nu12082414