Mechanism of Action of Ketogenic Diet Treatment: Impact of Decanoic Acid and Beta—Hydroxybutyrate on Sirtuins and Energy Metabolism in Hippocampal Murine Neurons


Abstract
1. Introduction
2. Materials and Methods
3. Results
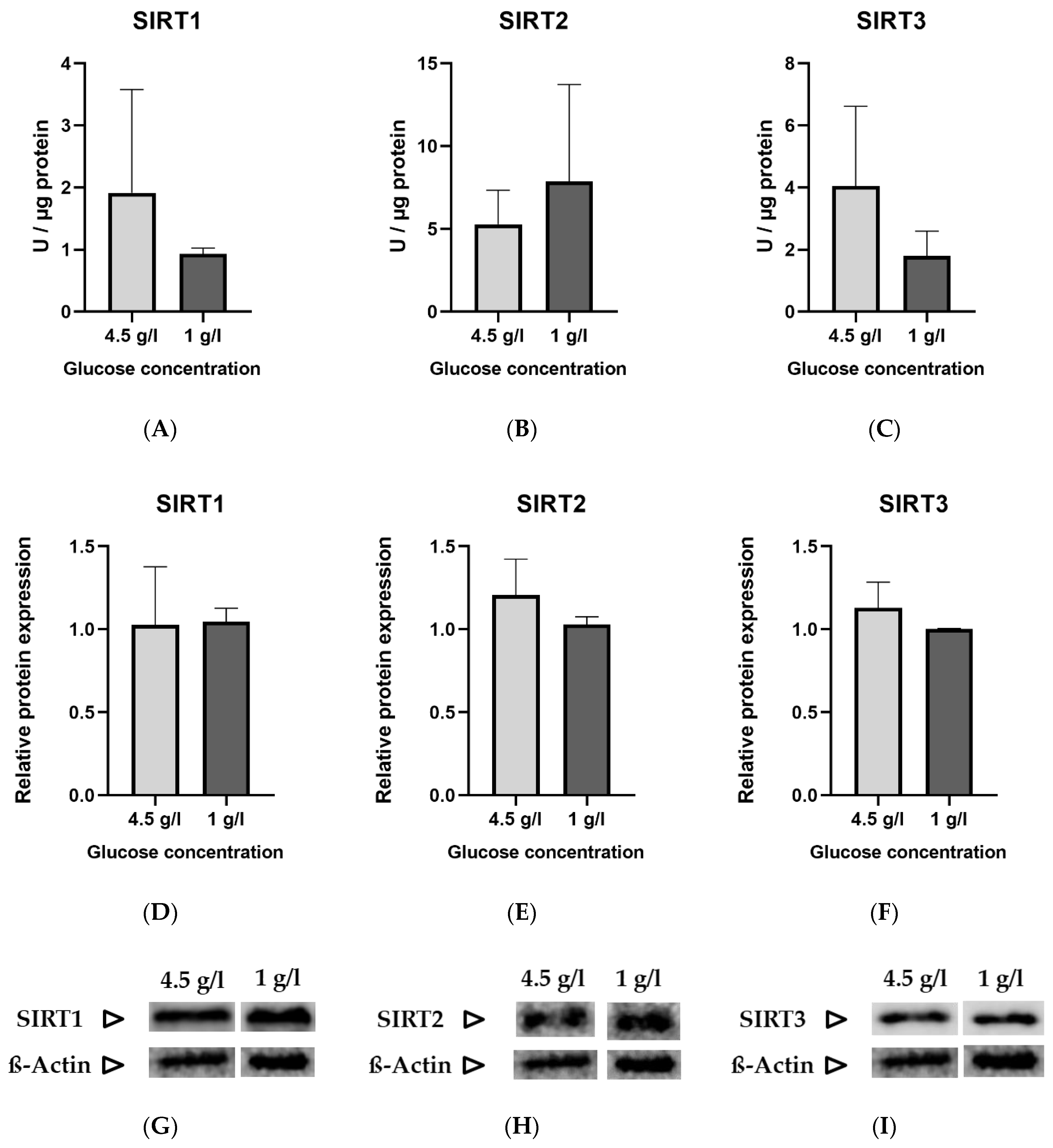
3.1. Measurement of Sirtuins under Standard and Low Glucose Concentrations
3.2. Incubation with the Predominant Ketone Body-BETA-Hydroxybutyrate (ßHB)
3.2.1. Sirtuin Enzyme Activity, Protein Expression and Gene Expression
3.2.2. Mitochondrial Respiratory Chain (MRC) Complexes and Citrate Synthase (CS)
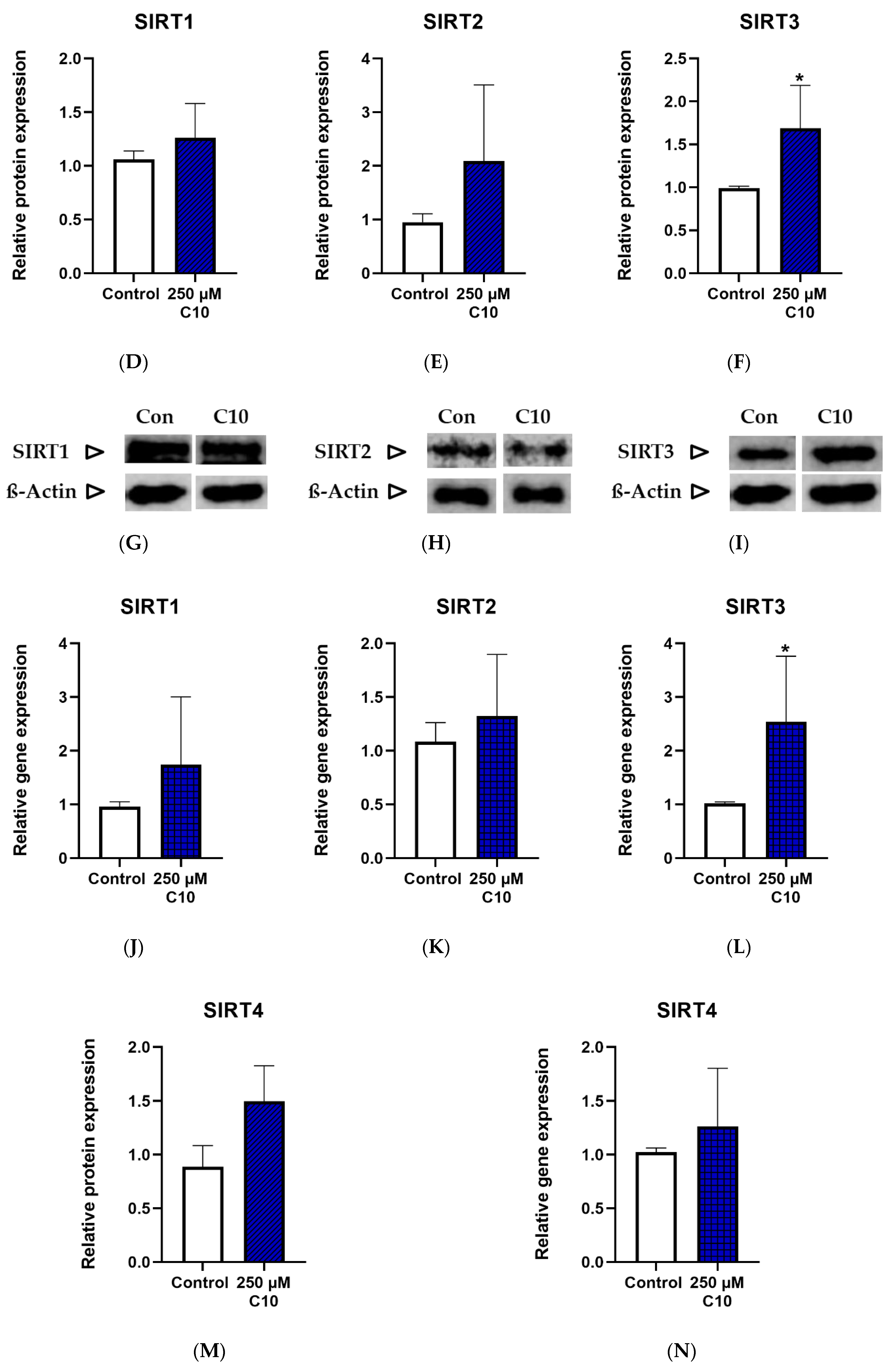
3.3. Incubation with the Medium Chain Fatty Acid Decanoic Acid (C10)
3.3.1. Sirtuin Enzyme Activity, Protein Expression and Gene Expression
3.3.2. Mitochondrial Respiratory Chain (MRC) Complexes and Citrate Synthase (CS)
3.4. Monocarboxylate Transporters (MCTr) 1 and 2
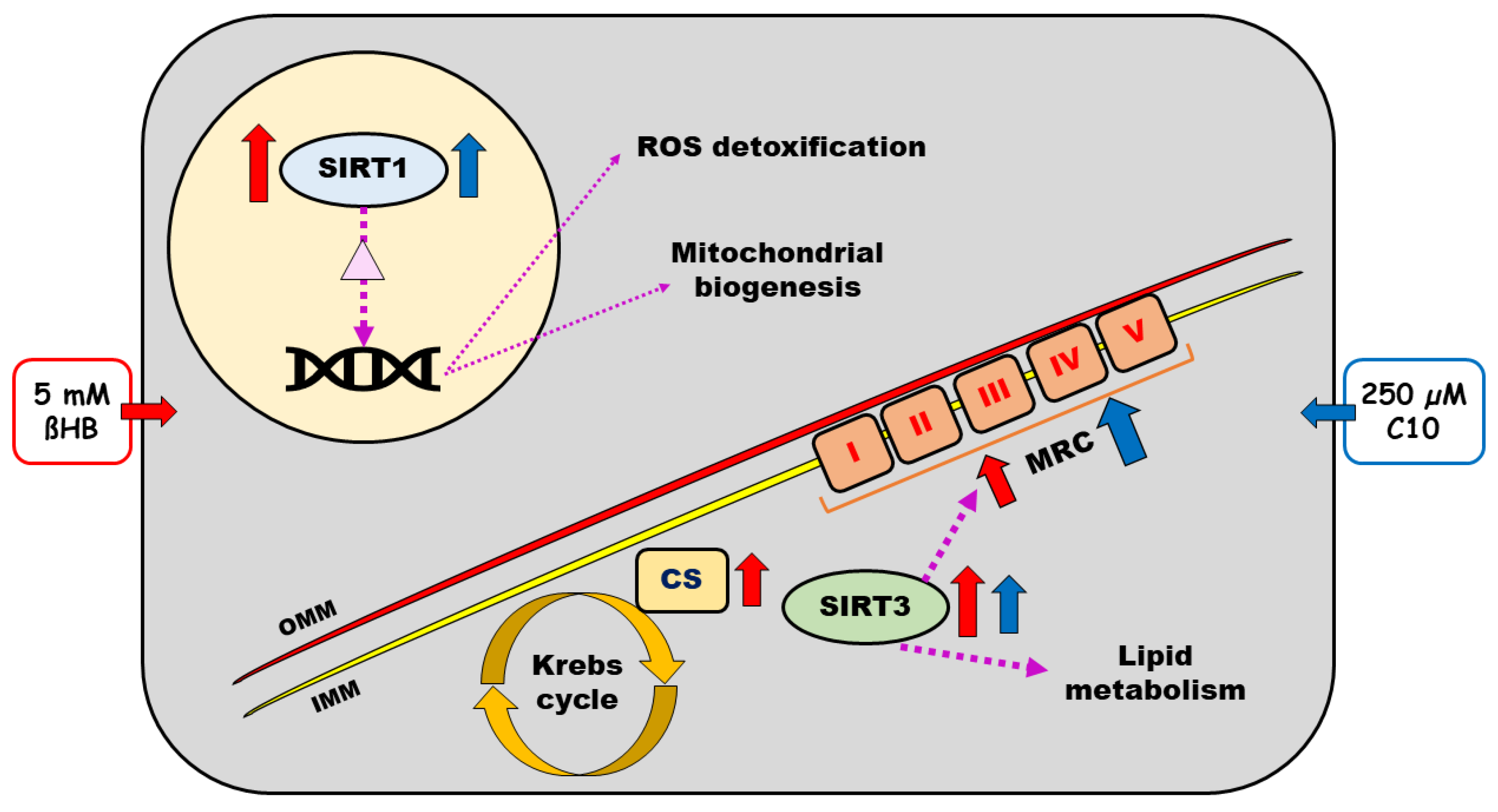
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wheless, J.W. History of the ketogenic diet. Epilepsia 2008, 49, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Huttenlocher, P.R. Ketonemia and Seizures: Metabolic and Anticonvulsant Effects of Two Ketogenic Diets in Childhood Epilepsy. Pediatr. Res. 1976, 10, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Huttenlocher, P.R.; Wilbourn, A.J.; Signore, J.M. Medium-chain triglycerides as a therapy for intractable childhood epilepsy. Neurology 1971, 21, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Hartman, A.L.; Vining, E.P. Clinical aspects of the ketogenic diet. Epilepsia 2007, 48, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Gano, L.B.; Patel, M.; Rho, J.M. Ketogenic diets, mitochondria, and neurological diseases. J. Lipid Res. 2014, 55, 2211–2228. [Google Scholar] [CrossRef] [PubMed]
- Kossoff, E.H.; Hartman, A.L. Ketogenic diets: New advances for metabolism-based therapies. Curr. Opin. Neurol. 2012, 25, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Kanabus, M.; Fassone, E.; Hughes, S.D.; Bilooei, S.F.; Rutherford, T.; Donnell, M.O.; Heales, S.J.R.; Rahman, S. The pleiotropic effects of decanoic acid treatment on mitochondrial function in fibroblasts from patients with complex I deficient Leigh syndrome. J. Inherit. Metab. Dis. 2016, 39, 415–426. [Google Scholar] [CrossRef]
- Mayorandan, S.; Meyer, U.; Hartmann, H.; Das, A.M. Glycogen storage disease type III: Modified Atkins diet improves myopathy. Orphanet J. Rare Dis. 2014, 9, 196. [Google Scholar] [CrossRef]
- Paoli, A.; Bianco, A.; Damiani, E.; Bosco, G. Ketogenic diet in neuromuscular and neurodegenerative diseases. Biomed. Res. Int. 2014, 2014, 474296. [Google Scholar] [CrossRef]
- Klement, R.J. Beneficial effects of ketogenic diets for cancer patients: A realist review with focus on evidence and confirmation. Med. Oncol. 2017, 34, 132. [Google Scholar] [CrossRef]
- Rogawski, M.A.; Loscher, W.; Rho, J.M. Mechanisms of Action of Antiseizure Drugs and the Ketogenic Diet. Cold Spring Harb. Perspect. Med. 2016, 6, a022780. [Google Scholar] [CrossRef] [PubMed]
- Masino, S.A.; Rho, J.M. Mechanisms of Ketogenic Diet Action. In Jasper’s Basic Mechanisms of the Epilepsies [Internet], 4th ed.; Noebels, J.L., Avoli, M., Rogawski, M.A., Delgado-Escueta, A.V., Olsen, R.W., Eds.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2012. [Google Scholar]
- Simeone, T.A.; Simeone, K.A.; Stafstrom, C.E.; Rho, J.M. Do ketone bodies mediate the anti-seizure effects of the ketogenic diet? Neuropharmacology 2018, 133, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Haidukewych, D.; Forsythe, W.I.; Sills, M. Monitoring Octanoic and Decanoic Acids in Plasma From Children With Intractable Epilepsy Treated With Medium-Chain Triglyceride Diet. Clin. Chem. 1982, 28, 642–645. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.D.; Kanabus, M.; Anderson, G.; Hargreaves, I.P.; Rutherford, T.; O’Donnell, M.; Cross, J.H.; Rahman, S.; Eaton, S.; Heales, S.J. The ketogenic diet component decanoic acid increases mitochondrial citrate synthase and complex I activity in neuronal cells. J. Neurochem. 2014, 129, 426–433. [Google Scholar] [CrossRef]
- Bough, K.J.; Wetherington, J.; Hassel, B.; Pare, J.F.; Gawryluk, J.W.; Greene, J.G.; Shaw, R.; Smith, Y.; Geiger, J.D.; Dingledine, R.J. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann. Neurol. 2006, 60, 223–235. [Google Scholar] [CrossRef]
- Hasan-Olive, M.M.; Lauritzen, K.H.; Ali, M.; Rasmussen, L.J.; Storm-Mathisen, J.; Bergersen, L.H. A Ketogenic Diet Improves Mitochondrial Biogenesis and Bioenergetics via the PGC1alpha-SIRT3-UCP2 Axis. Neurochem. Res. 2019, 44, 22–37. [Google Scholar] [CrossRef]
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999, 13, 2570–2580. [Google Scholar] [CrossRef]
- Haigis, M.C.; Guarente, L.P. Mammalian sirtuins--emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006, 20, 2913–2921. [Google Scholar] [CrossRef]
- Feldman, J.L.; Dittenhafer-Reed, K.E.; Denu, J.M. Sirtuin catalysis and regulation. J. Biol. Chem. 2012, 287, 42419–42427. [Google Scholar] [CrossRef]
- Chuang, Y.C.; Chen, S.D.; Jou, S.B.; Lin, T.K.; Chen, S.F.; Chen, N.C.; Hsu, C.Y. Sirtuin 1 Regulates Mitochondrial Biogenesis and Provides an Endogenous Neuroprotective Mechanism Against Seizure-Induced Neuronal Cell Death in the Hippocampus Following Status Epilepticus. Int. J. Mol. Sci. 2019, 20, 3588. [Google Scholar] [CrossRef]
- Olmos, Y.; Sanchez-Gomez, F.J.; Wild, B.; Garcia-Quintans, N.; Cabezudo, S.; Lamas, S.; Monsalve, M. SirT1 regulation of antioxidant genes is dependent on the formation of a FoxO3a/PGC-1alpha complex. Antioxid. Redox Signal. 2013, 19, 1507–1521. [Google Scholar] [CrossRef] [PubMed]
- Singh, C.K.; Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; Mack, N.J.; Ahmad, N. The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid. Redox Signal. 2018, 28, 643–661. [Google Scholar] [CrossRef] [PubMed]
- Pacifici, F.; Di Cola, D.; Pastore, D.; Abete, P.; Guadagni, F.; Donadel, G.; Bellia, A.; Esposito, E.; Salimei, C.; Sinibaldi Salimei, P.; et al. Proposed Tandem Effect of Physical Activity and Sirtuin 1 and 3 Activation in Regulating Glucose Homeostasis. Int. J. Mol. Sci. 2019, 20, 4748. [Google Scholar] [CrossRef] [PubMed]
- Zang, Y.; Fan, L.; Chen, J.; Huang, R.; Qin, H. Improvement of Lipid and Glucose Metabolism by Capsiate in Palmitic Acid-Treated HepG2 Cells via Activation of the AMPK/SIRT1 Signaling Pathway. J. Agric. Food Chem. 2018, 66, 6772–6781. [Google Scholar] [CrossRef]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef]
- Verdin, E.; Hirschey, M.D.; Finley, L.W.; Haigis, M.C. Sirtuin regulation of mitochondria: Energy production, apoptosis, and signaling. Trends Biochem. Sci. 2010, 35, 669–675. [Google Scholar] [CrossRef]
- Laurent, G.; de Boer, V.C.; Finley, L.W.; Sweeney, M.; Lu, H.; Schug, T.T.; Cen, Y.; Jeong, S.M.; Li, X.; Sauve, A.A.; et al. SIRT4 represses peroxisome proliferator-activated receptor alpha activity to suppress hepatic fat oxidation. Mol. Cell. Biol. 2013, 33, 4552–4561. [Google Scholar] [CrossRef]
- Laurent, G.; German, N.J.; Saha, A.K.; de Boer, V.C.; Davies, M.; Koves, T.R.; Dephoure, N.; Fischer, F.; Boanca, G.; Vaitheesvaran, B.; et al. SIRT4 coordinates the balance between lipid synthesis and catabolism by repressing malonyl CoA decarboxylase. Mol. Cell 2013, 50, 686–698. [Google Scholar] [CrossRef]
- Nasrin, N.; Wu, X.; Fortier, E.; Feng, Y.; Bare, O.C.; Chen, S.; Ren, X.; Wu, Z.; Streeper, R.S.; Bordone, L. SIRT4 regulates fatty acid oxidation and mitochondrial gene expression in liver and muscle cells. J. Biol. Chem. 2010, 285, 31995–32002. [Google Scholar] [CrossRef]
- Ahn, B.H.; Kim, H.S.; Song, S.; Lee, I.H.; Liu, J.; Vassilopoulos, A.; Deng, C.X.; Finkel, T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 14447–14452. [Google Scholar] [CrossRef]
- Cimen, H.; Han, M.J.; Yang, Y.; Tong, Q.; Koc, H.; Koc, E.C. Regulation of succinate dehydrogenase activity by SIRT3 in mammalian mitochondria. Biochemistry 2010, 49, 304–311. [Google Scholar] [PubMed]
- Finley, L.W.; Haas, W.; Desquiret-Dumas, V.; Wallace, D.C.; Procaccio, V.; Gygi, S.P.; Haigis, M.C. Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PLoS ONE 2011, 6, e23295. [Google Scholar] [CrossRef] [PubMed]
- Finley, L.W.; Haigis, M.C. Metabolic regulation by SIRT3: Implications for tumorigenesis. Trends Mol. Med. 2012, 18, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Lu, H.F.; Alano, C.C. Neuronal Sirt3 protects against excitotoxic injury in mouse cortical neuron culture. PLoS ONE 2011, 6, e14731. [Google Scholar] [CrossRef] [PubMed]
- Kendrick, A.A.; Choudhury, M.; Rahman, S.M.; McCurdy, C.E.; Friederich, M.; Van Hove, J.L.; Watson, P.A.; Birdsey, N.; Bao, J.; Gius, D.; et al. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem. J. 2011, 433, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Potthast, A.B.; Heuer, T.; Warneke, S.J.; Das, A.M. Alterations of sirtuins in mitochondrial cytochrome c-oxidase deficiency. PLoS ONE 2017, 12, e0186517. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.T.; Lee, H.C.; Liao, C.C.; Wei, Y.H. Regulation of mitochondrial F(o)F(1)ATPase activity by Sirt3-catalyzed deacetylation and its deficiency in human cells harboring 4977bp deletion of mitochondrial DNA. Biochim. Biophys. Acta 2013, 1832, 216–227. [Google Scholar] [CrossRef]
- Tang, B.L. Sirt1 and the Mitochondria. Mol. Cells 2016, 39, 87–95. [Google Scholar]
- Wang, S.J.; Zhao, X.H.; Chen, W.; Bo, N.; Wang, X.J.; Chi, Z.F.; Wu, W. Sirtuin 1 activation enhances the PGC-1alpha/mitochondrial antioxidant system pathway in status epilepticus. Mol. Med. Rep. 2015, 11, 521–526. [Google Scholar] [CrossRef]
- Bernier, F.P.; Boneh, A.; Dennett, X.; Chow, C.W.; Cleary, M.A.; Thorburn, D.R. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology 2002, 59, 1406–1411. [Google Scholar] [CrossRef]
- Itkonen, O.; Suomalainen, A.; Turpeinen, U. Mitochondrial Coenzyme Q10 Determination by Isotope-Dilution Liquid Chromatography–Tandem Mass Spectrometry. Clin. Chem. 2013, 59, 1260–1267. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Selak, M.A.; de Chadarevian, J.P.; Melvin, J.J.; Grover, W.D.; Salganicoff, L.; Kaye, E.M. Mitochondrial activity in pompe’s disease. Pediatr. Neurol. 2000, 23, 54–57. [Google Scholar] [CrossRef]
- Parry, H.A.; Kephart, W.C.; Mumford, P.W.; Romero, M.A.; Mobley, C.B.; Zhang, Y.; Roberts, M.D.; Kavazis, A.N. Ketogenic diet increases mitochondria volume in the liver and skeletal muscle without altering oxidative stress markers in rats. Heliyon 2018, 4, e00975. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P. The monocarboxylate transporter family--Structure and functional characterization. IUBMB Life 2012, 64, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Price, N.T. The Proton-Linked Monocarboxylate Transporter (MCT) Family-Structure, Function and Regulation. Biochem. J. 1999, 343, 281–299. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Wilson, M.C. The monocarboxylate transporter family--role and regulation. IUBMB Life 2012, 64, 109–119. [Google Scholar] [CrossRef]
- Garcia, C.K.; Brown, M.S.; Pathak, R.K.; Goldstein, J.L. cDNA Cloning of MCT2, a Second Monocarboxylate Transporter Expressed in Different Cells Than MCT1. J. Biol. Chem. 1995, 270, 1843–1849. [Google Scholar] [CrossRef]
- Koehler-Stec, E.M.; Simpson, I.A.; Vannucci, S.J.; Landschulz, K.T.; Landschulz, W.H. Monocarboxylate transporter expression in mouse brain. Am. J. Physiol.-Endocrinol. Metab. 1998, 275, E516–E524. [Google Scholar] [CrossRef]
- Pellerin, L.; Pellegri, G.; Martin, J.-L.; Magistretti, P.J. Expression of monocarboxylate transporter mRNAs in mouse brain: Support for a distinct role of lactate as an energy substrate for the neonatal vs. adult brain. Proc. Natl. Acad. Sci. USA 1998, 95, 3990–3995. [Google Scholar] [CrossRef]
- Leino, R.L.; Gerhart, D.Z.; Duelli, R.; Enerson, B.E.; Drewes, L.R. Diet-induced ketosis increases monocarboxylate transporter (MCT1) levels in rat brain. Neurochem. Int. 2001, 38, 519–527. [Google Scholar] [CrossRef]
- Forero-Quintero, L.S.; Deitmer, J.W.; Becker, H.M. Reduction of epileptiform activity in ketogenic mice: The role of monocarboxylate transporters. Sci. Rep. 2017, 7, 4900. [Google Scholar] [CrossRef] [PubMed]
- Wlaź, P.; Socała, K.; Nieoczym, D.; Żarnowski, T.; Żarnowska, I.; Czuczwar, S.J.; Gasior, M. Acute anticonvulsant effects of capric acid in seizure tests in mice. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2015, 57, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, Q.; Zhou, J.; Wu, X.; Zhu, Q. β hydroxybutyrate levels in serum and cerebrospinal fluid under ketone body metabolism in rats. Exp. Anim. 2017, 66, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.; Nugent, S.; Tremblay-Mercier, J.; Tremblay, S.; Courchesne-Loyer, A.; Beaudoin, J.F.; Tremblay, L.; Descoteaux, M.; Lecomte, R.; Cunnane, S.C. The ketogenic diet increases brain glucose and ketone uptake in aged rats: A dual tracer PET and volumetric MRI study. Brain Res. 2012, 1488, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; Preter, K.D.; Pattyn, F.; Poppe, B.; Roy, N.V.; Paepe, A.D.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, research0034. [Google Scholar] [CrossRef]
- Das, A.M.; Byrd, D.J.; Brodehl, J. Regulation of the mitochondrial ATP-synthase in human fibroblasts. Clin. Chim. Acta 1994, 231, 61–68. [Google Scholar] [CrossRef]
- Lücke, T.; Höppner, W.; Schmidt, E.; Illsinger, S.; Das, A.M. Fabry disease: Reduced activities of respiratory chain enzymes with decreased levels of energy-rich phosphates in fibroblasts. Mol. Genet. Metab. 2004, 82, 93–97. [Google Scholar] [CrossRef]
- Fischer, J.C.; Ruitenbeek, W.; Trijbels, J.M.F.; Veerkamp, J.H.; Stadhouders, A.M.; Sengers, R.C.A.; Janssen, A.J.M. Estimation of NADH oxidation in human skeletal muscle mitochondria. Clin. Chim. Acta 1986, 155, 263–273. [Google Scholar] [CrossRef]
- Stumpf, D.A.; Parks, J.K. Human mitochondrial electron transport chain: Assay of succinate:Cytochrome c reductase in leukocytes, platelets and cultured fibroblasts. Biochem. Med. 1981, 25, 234–238. [Google Scholar] [CrossRef]
- Das, A.M.; Harris, D.A. Regulation of the mitochondrial ATP synthase in intact rat cardiomyocytes. Biochem. J. 1990, 266, 355–361. [Google Scholar] [CrossRef]
- Rosing, J.; Harris, D.A.; Kemp, A.; Slater, E.C. Nucleotide-binding properties of native and cold-treated mitochondrial ATPase. Biochim. Biophys. Acta (BBA)-Bioenerg. 1975, 376, 13–26. [Google Scholar] [CrossRef]
- Srere, P.A. [1] Citrate synthase: [EC 4.1.3.7. Citrate oxaloacetate-lyase (CoA-acetylating)]. Methods Enzymol. 1969, 13, 3–11. [Google Scholar]
- Bensadoun, A.; Weinstein, D. Assay of proteins in the presence of interfering materials. Anal. Biochem. 1976, 70, 241–250. [Google Scholar] [CrossRef]
- Augustin, K.; Khabbush, A.; Williams, S.; Eaton, S.; Orford, M.; Cross, J.H.; Heales, S.J.R.; Walker, M.C.; Williams, R.S.B. Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol. 2018, 17, 84–93. [Google Scholar] [CrossRef]
- Scholl-Burgi, S.; Holler, A.; Pichler, K.; Michel, M.; Haberlandt, E.; Karall, D. Ketogenic diets in patients with inherited metabolic disorders. J. Inherit. Metab. Dis. 2015, 38, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S. Pathophysiology of mitochondrial disease causing epilepsy and status epilepticus. Epilepsy Behav. 2015, 49, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Kann, O.; Kovacs, R.; Njunting, M.; Behrens, C.J.; Otahal, J.; Lehmann, T.N.; Gabriel, S.; Heinemann, U. Metabolic dysfunction during neuronal activation in the ex vivo hippocampus from chronic epileptic rats and humans. Brain 2005, 128, 2396–2407. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 2019, 49, 35–45. [Google Scholar] [CrossRef]
- Das, A.M. Regulation of the mitochondrial ATP-synthase in health and disease. Mol. Genet. Metab. 2003, 79, 71–82. [Google Scholar] [CrossRef]
- Das, A.M.; Steuerwald, U.; Illsinger, S. Inborn errors of energy metabolism associated with myopathies. J. Biomed. Biotechnol. 2010, 2010, 340849. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 602–614. [Google Scholar] [CrossRef] [PubMed]
- Boison, D. New insights into the mechanisms of the ketogenic diet. Curr. Opin. Neurol. 2017, 30, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Hebert, A.S.; Dittenhafer-Reed, K.E.; Yu, W.; Bailey, D.J.; Selen, E.S.; Boersma, M.D.; Carson, J.J.; Tonelli, M.; Balloon, A.J.; Higbee, A.J.; et al. Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol. Cell 2013, 49, 186–199. [Google Scholar] [CrossRef]
- Bao, J.; Lu, Z.; Joseph, J.J.; Carabenciov, D.; Dimond, C.C.; Pang, L.; Samsel, L.; McCoy, J.P., Jr.; Leclerc, J.; Nguyen, P.; et al. Characterization of the murine SIRT3 mitochondrial localization sequence and comparison of mitochondrial enrichment and deacetylase activity of long and short SIRT3 isoforms. J. Cell. Biochem. 2010, 110, 238–247. [Google Scholar] [CrossRef]
- Kincaid, B.; Bossy-Wetzel, E. Forever young: SIRT3 a shield against mitochondrial meltdown, aging and neurodegeneration. Front. Aging Neurosci. 2013, 5, 48. [Google Scholar] [CrossRef]
- Potthast, A.B.; Nebl, J.; Wasserfurth, P.; Haufe, S.; Eigendorf, J.; Hahn, A.; Das, A. Impact of Nutrition on Short-Term Exercise-Induced Sirtuin Regulation: Vegans Differ from Omnivores and Lacto-Ovo Vegetarians. Nutrients 2020, 12, 1004. [Google Scholar] [CrossRef]
- Xin, L.; Ipek, O.; Beaumont, M.; Shevlyakova, M.; Christinat, N.; Masoodi, M.; Greenberg, N.; Gruetter, R.; Cuenoud, B. Nutritional Ketosis Increases NAD(+)/NADH Ratio in Healthy Human Brain: An in Vivo Study by (31)P-MRS. Front. Nutr. 2018, 5, 62. [Google Scholar] [CrossRef]
- Flick, F.; Luscher, B. Regulation of sirtuin function by posttranslational modifications. Front. Pharmacol. 2012, 3, 29. [Google Scholar] [CrossRef]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha}. J. Biol. Chem. 2005, 280, 16456–16460. [Google Scholar] [CrossRef]
- Bae, Y.S.; Oh, H.; Rhee, S.G.; Yoo, Y.D. Regulation of reactive oxygen species generation in cell signaling. Mol. Cells 2011, 32, 491–509. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Zhou, Z. Post-translational modifications of nuclear sirtuins. Genome Instab. Dis. 2019, 1, 34–45. [Google Scholar] [CrossRef]
- Dabek, A.; Wojtala, M.; Pirola, L.; Balcerczyk, A. Modulation of Cellular Biochemistry, Epigenetics and Metabolomics by Ketone Bodies. Implications of the Ketogenic Diet in the Physiology of the Organism and Pathological States. Nutrients 2020, 12, 788. [Google Scholar] [CrossRef] [PubMed]
- Hartman, A.L.; Gasior, M.; Vining, E.P.; Rogawski, M.A. The neuropharmacology of the ketogenic diet. Pediatr. Neurol. 2007, 36, 281–292. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sirtuin | Localization and Enzymatic Activity | Relevant Physiological Processes and Targets * | Reference |
|---|---|---|---|
| SIRT1 | Nucleus Deacetylation | Mitochondrial biogenesis
| [21,22,23,24,25] |
ROS detoxification
| |||
Glucose metabolism
| |||
| SIRT2 | Cytoplasm Deacetylation | ROS detoxification
| [23] |
| SIRT3 | Mitochondria Deacetylation | Energy metabolism (MRC) (Discussed in detail in the next sub-section) | [26,27] |
Fatty acid oxidation
| |||
| SIRT4 | Mitochondria ADP-ribosylation (predominant) Deacetylation (weak) | Fatty acid oxidation
| [28,29,30] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dabke, P.; Das, A.M. Mechanism of Action of Ketogenic Diet Treatment: Impact of Decanoic Acid and Beta—Hydroxybutyrate on Sirtuins and Energy Metabolism in Hippocampal Murine Neurons. Nutrients 2020, 12, 2379. https://doi.org/10.3390/nu12082379
Dabke P, Das AM. Mechanism of Action of Ketogenic Diet Treatment: Impact of Decanoic Acid and Beta—Hydroxybutyrate on Sirtuins and Energy Metabolism in Hippocampal Murine Neurons. Nutrients. 2020; 12(8):2379. https://doi.org/10.3390/nu12082379
Chicago/Turabian StyleDabke, Partha, and Anibh M. Das. 2020. "Mechanism of Action of Ketogenic Diet Treatment: Impact of Decanoic Acid and Beta—Hydroxybutyrate on Sirtuins and Energy Metabolism in Hippocampal Murine Neurons" Nutrients 12, no. 8: 2379. https://doi.org/10.3390/nu12082379
APA StyleDabke, P., & Das, A. M. (2020). Mechanism of Action of Ketogenic Diet Treatment: Impact of Decanoic Acid and Beta—Hydroxybutyrate on Sirtuins and Energy Metabolism in Hippocampal Murine Neurons. Nutrients, 12(8), 2379. https://doi.org/10.3390/nu12082379