The Impact of Dietary Glycemic Index and Glycemic Load on Postprandial Lipid Kinetics, Dyslipidemia and Cardiovascular Risk




Abstract
1. Introduction
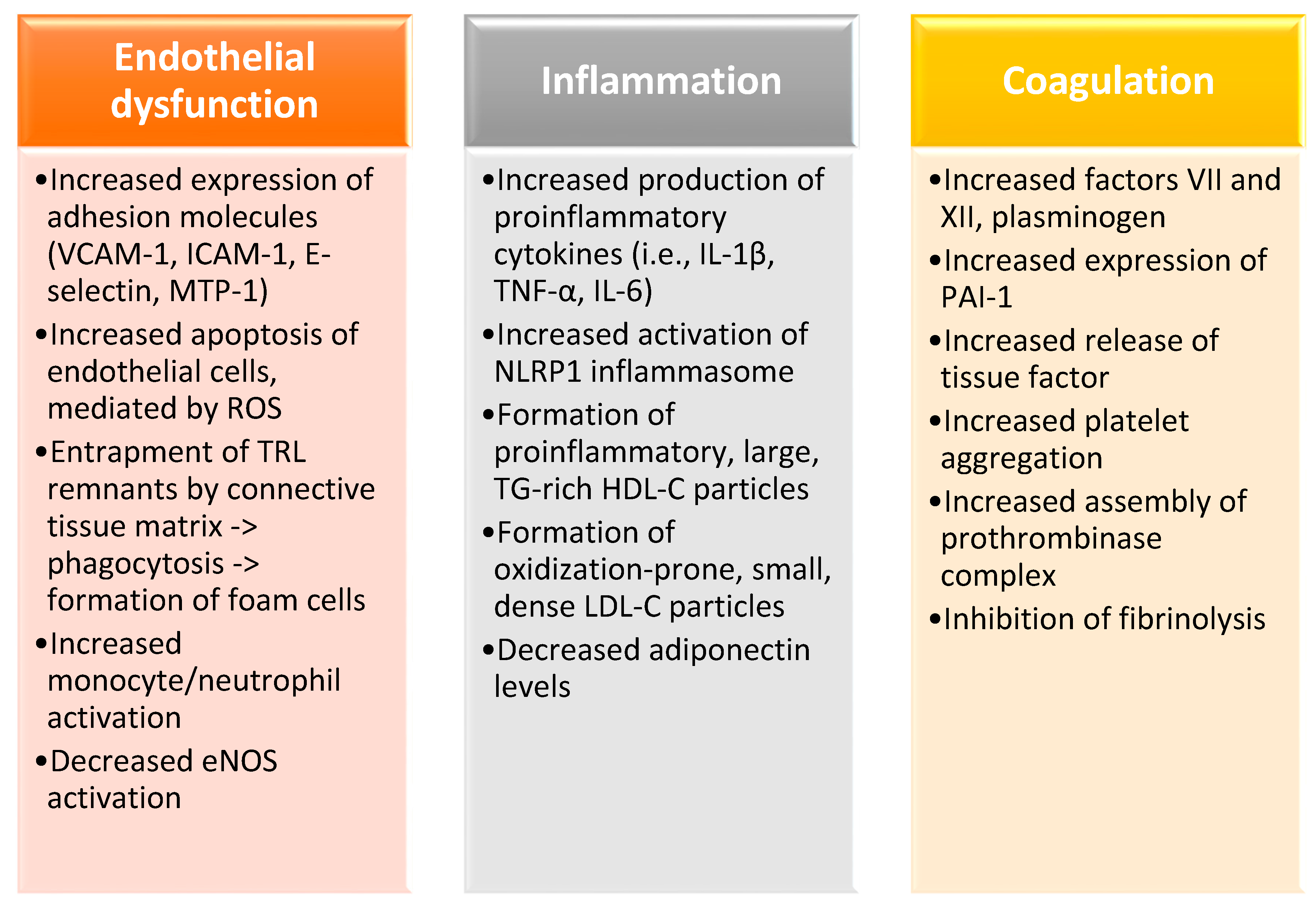
2. Postprandial Hypertriglyceridemia as a Risk Factor for Cardiovascular Disease
3. GI/GL: Definition, Measurement and Clinical Significance
3.1. Definition, Measurement and Limitations
3.2. GI/GL, Metabolic Health and Cardiovascular Risk
3.2.1. GI/GL and Satiety
3.2.2. GI/GL and Body Weight
3.2.3. GI/GL and Glucose Homeostasis
3.2.4. GI/GL and Cardiovascular Events
3.2.5. GI/GL and Blood Lipids
4. Postprandial Lipemia and Carbohydrates: Pathophysiology and Available Clinical Data
4.1. Regulation of Postprandial Lipemia and Association with Dietary Carbohydrates
4.2. GI/GL and Postprandial Hypertriglyceridemia: A Well-Established Association(?)
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABCA1 | ATP-binding cassette transporter |
| AD | atherogenic dyslipidemia |
| ApoB | apolipoprotein B |
| AUC | area-under-the-curve |
| BMI | body mass index |
| CETP | cholesteryl ester transfer protein |
| CHD | coronary heart disease |
| ChREBP | carbohydrate response element-binding protein |
| CMs | chylomicrons |
| CVD | cardiovascular disease |
| DM | diabetes mellitus |
| DNL | de novo lipogenesis |
| FFAs | free fatty acids |
| GI | glycemic index |
| GL | glycemic load |
| GLP-1 | glucagon-like peptide 1 |
| HDL-C | high-density lipoprotein cholesterol |
| HF | heart failure |
| HOMA-IR | Homeostatic Model Assessment for Insulin Resistance |
| IGF-1 | insulin-like growth factor 1 |
| IR | insulin resistance |
| LDL-C | low-density lipoprotein cholesterol |
| LPL | lipoprotein lipase |
| mRNA | messenger ribonucleic acid |
| MTP | microsomal triglyceride transfer protein |
| MUFAs | monounsaturated fatty acids |
| NAFLD | non-alcoholic fatty liver disease |
| PKC | protein kinase C |
| PUFAs | polyunsaturated fatty acids |
| PYY | peptide tyrosine-tyrosine |
| SFAs | saturated fatty acid |
| SREBP1c | sterol regulatory element-binding protein 1c |
| T2DM | type 2 diabetes mellitus |
| TAGs | triacylglycerols |
| TGs | triglycerides |
| TRL | triglyceride-rich lipoprotein |
| VLDLs | very low-density lipoproteins |
References
- Cardiovascular Diseases (CVDs). Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 28 April 2019).
- Ference, B.A.; Graham, I.; Tokgozoglu, L.; Catapano, A.L. Impact of Lipids on Cardiovascular Health: JACC Health Promotion Series. J. Am. Coll. Cardiol. 2018, 72, 1141–1156. [Google Scholar] [CrossRef]
- Ferrari, R.; Aguiar, C.; Alegria, E.; Bonadonna, R.C.; Cosentino, F.; Elisaf, M.; Farnier, M.; Ferrières, J.; Filardi, P.P.; Hancu, N.; et al. Current practice in identifying and treating cardiovascular risk, with a focus on residual risk associated with atherogenic dyslipidaemia. Eur. Heart J. Suppl. J. Eur. Soc. Cardiol. 2016, 18, C2–C12. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Nakano, T.; Tokita, Y.; Nagamine, T.; Inazu, A.; Kobayashi, J.; Mabuchi, H.; Stanhope, K.L.; Havel, P.J.; Okazaki, M.; et al. Postprandial lipoprotein metabolism: VLDL vs chylomicrons. Clin. Chim. Acta 2011, 412, 1306–1318. [Google Scholar] [CrossRef]
- Pirillo, A.; Norata, G.D.; Catapano, A.L. Postprandial lipemia as a cardiometabolic risk factor. Curr. Med. Res. Opin. 2014, 30, 1489–1503. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, D.J.; Wolever, T.M.; Taylor, R.H.; Barker, H.; Fielden, H.; Baldwin, J.M.; Bowling, A.C.; Newman, H.C.; Jenkins, A.L.; Goff, D.V. Glycemic index of foods: A physiological basis for carbohydrate exchange. Am. J. Clin. Nutr. 1981, 34, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Schwingshackl, L.; Hoffmann, G. Long-term effects of low glycemic index/load vs. high glycemic index/load diets on parameters of obesity and obesity-associated risks: A systematic review and meta-analysis. Nutrition, metabolism, and cardiovascular diseases. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Desmarchelier, C.; Borel, P.; Lairon, D.; Maraninchi, M.; Valéro, R. Effect of Nutrient and Micronutrient Intake on Chylomicron Production and Postprandial Lipemia. Nutrients 2019, 11, 1299. [Google Scholar] [CrossRef]
- Watts, G.F.; Karpe, F. Republished review: Triglycerides and atherogenic dyslipidaemia: Extending treatment beyond statins in the high-risk cardiovascular patient. Postgrad. Med. J. 2011, 87, 776–782. [Google Scholar] [CrossRef]
- Dimitriadis, G.; Boutati, E.; Lambadiari, V.; Mitrou, P.; Maratou, E.; Brunel, P.; Raptis, S.A. Restoration of early insulin secretion after a meal in type 2 diabetes: Effects on lipid and glucose metabolism. Eur. J. Clin. Investig. 2004, 34, 490–497. [Google Scholar] [CrossRef]
- Lambadiari, V.; Mitrou, P.; Maratou, E.; Raptis, A.; Raptis, S.A.; Dimitriadis, G. Increases in muscle blood flow after a mixed meal are impaired at all stages of type 2 diabetes. Clin. Endocrinol. 2012, 76, 825–830. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Benn, M.; Schnohr, P.; Tybjaerg-Hansen, A. Nonfasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. JAMA 2007, 298, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Triglyceride Coronary Disease Genetics Consortium; Emerging Risk Factors Collaboration; Sarwar, N.; Sandhu, M.S.; Ricketts, S.L.; Butterworth, A.S.; Di Angelantonio, E.; Boekholdt, S.M.; Ouwehand, W.; Kastelein, J.J.; et al. Triglyceride-mediated pathways and coronary disease: Collaborative analysis of 101 studies. Lancet 2010, 375, 1634–1639, Erratum in 2010, 376, 90. [Google Scholar] [CrossRef]
- Tabas, I.; Williams, K.J.; Borén, J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: Update and therapeutic implications. Circulation 2007, 116, 1832–1844. [Google Scholar] [CrossRef] [PubMed]
- Borén, J.; Williams, K.J. The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: A triumph of simplicity. Curr. Opin. Lipidol. 2016, 27, 473–483. [Google Scholar] [CrossRef] [PubMed]
- ACCORD Study Group; Ginsberg, H.N.; Elam, M.B.; Lovato, L.C.; Crouse, J.R., 3rd; Leiter, L.A.; Linz, P.; Friedewald, W.T.; Buse, J.B.; Gerstein, H.C.; et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N. Engl. J. Med. 2010, 362, 1563–1574, Erratum in 2010, 362, 1748. [Google Scholar]
- Maki, K.C.; Dicklin, M.R. Do triglyceride-lowering drugs decrease risk of cardiovascular disease? Curr. Opin. Lipidol. 2017, 28, 374–379. [Google Scholar] [CrossRef]
- Scott, R.; O’Brien, R.; Fulcher, G.; Pardy, C.; D’Emden, M.; Tse, D.; Taskinen, M.R.; Ehnholm, C.; Keech, A.; The FIELD Study Investigators. Effects of fenofibrate treatment on cardiovascular disease risk in 9795 individuals with type 2 diabetes and various components of the metabolic syndrome: The Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study. Diabetes Care 2009, 32, 493–498. [Google Scholar] [CrossRef]
- Toth, P.P.; Fazio, S.; Wong, N.D.; Hull, M.; Nichols, G.A. Risk of cardiovascular events in patients with hypertriglyceridaemia: A review of real-world evidence. Diabetes Obes. Metab. 2020, 22, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, D.L.; Steg, P.G.; Miller, M.; Brinton, E.A.; Jacobson, T.A.; Ketchum, S.B.; Doyle, R.T., Jr.; Juliano, R.A.; Jiao, L.; Granowitz, C.; et al. Cardiovascular Risk Reduction with Icosapent Ethyl for Hypertriglyceridemia. N. Engl. J. Med. 2019, 380, 11–22. [Google Scholar] [CrossRef]
- Emerging Risk Factors Collaboration; Di Angelantonio, E.; Gao, P.; Pennells, L.; Kaptoge, S.; Caslake, M.; Thompson, A.; Butterworth, A.S.; Sarwar, N.; Wormser, D.; et al. Lipid-related markers and cardiovascular disease prediction. JAMA 2012, 307, 2499–2506. [Google Scholar]
- Ference, B.A.; Kastelein, J.J.P.; Ray, K.K.; Ginsberg, H.N.; Chapman, M.J.; Packard, C.J.; Laufs, U.; Oliver-Williams, C.; Wood, A.M.; Butterworth, A.S.; et al. Association of Triglyceride-Lowering LPL Variants and LDL-C-Lowering LDLR Variants with Risk of Coronary Heart Disease. JAMA 2019, 321, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Frikke-Schmidt, R.; Nordestgaard, B.G.; Stene, M.C.; Sethi, A.A.; Remaley, A.T.; Schnohr, P.; Grande, P.; Tybjærg-Hansen, A. Association of loss-of-function mutations in the ABCA1 gene with high-density lipoprotein cholesterol levels and risk of ischemic heart disease. JAMA 2008, 299, 2524–2532. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Olsson, A.G.; Abt, M.; Ballantyne, C.M.; Barter, P.J.; Brumm, J.; Chaitman, B.R.; Holme, I.M.; Kallend, D.; Leiter, L.A.; et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N. Engl. J. Med. 2012, 367, 2089–2099. [Google Scholar] [CrossRef]
- Voight, B.F.; Peloso, G.M.; Orho-Melander, M.; Frikke-Schmidt, R.; Barbalic, M.; Jensen, M.K.; Hindy, G.; Hólm, H.; Ding, E.L.; Johnson, T.; et al. Plasma HDL cholesterol and risk of myocardial infarction: A mendelian randomisation study. Lancet 2012, 380, 572–580, Erratum in 2012, 380, 564. [Google Scholar] [CrossRef]
- AIM-HIGH Investigators; Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2012, 365, 2255–2267, Erratum in 2012, 367, 189. [Google Scholar]
- HPS3/TIMI55–REVEAL Collaborative Group; Bowman, L.; Hopewell, J.C.; Chen, F.; Wallendszus, K.; Stevens, W.; Collins, R.; Wiviott, S.D.; Cannon, C.P.; Braunwald, E.; et al. Effects of Anacetrapib in Patients with Atherosclerotic Vascular Disease. N. Engl. J. Med. 2017, 377, 1217–1227. [Google Scholar]
- Santos, F.L.; Esteves, S.S.; Da Costa Pereira, A.; Yancy, W.S., Jr.; Nunes, J.P. Systematic review and meta-analysis of clinical trials of the effects of low carbohydrate diets on cardiovascular risk factors. Obes. Rev. 2012, 13, 1048–1066. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Lopez-Miranda, J.; Williams, C.; Lairon, D. Dietary, physiological, genetic and pathological influences on postprandial lipid metabolism. Br. J. Nutr. 2007, 98, 458–473. [Google Scholar] [CrossRef]
- Vogel, R.A.; Corretti, M.C.; Plotnick, G.D. Effect of a single high-fat meal on endothelial function in healthy subjects. Am. J. Cardiol. 1997, 79, 350–354. [Google Scholar] [CrossRef]
- Dimitriadis, G.; Lambadiari, V.; Mitrou, P.; Maratou, E.; Boutati, E.; Panagiotakos, D.B.; Economopoulos, T.; Raptis, S.A. Impaired postprandial blood flow in adipose tissue may be an early marker of insulin resistance in type 2 diabetes. Diabetes Care 2007, 30, 3128–3130. [Google Scholar] [CrossRef]
- Miller, G.J. Postprandial lipaemia and haemostatic factors. Atherosclerosis 1998, 141, S47–S51. [Google Scholar] [CrossRef]
- Sanders, T.A.; Oakley, F.R.; Cooper, J.A.; Miller, G.J. Influence of a stearic acid-rich structured triacylglycerol on postprandial lipemia, factor VII concentrations, and fibrinolytic activity in healthy subjects. Am. J. Clin. Nutr. 2001, 73, 715–721. [Google Scholar] [CrossRef]
- Nordoy, A.; Strom, E.; Gjesdal, K. The effect of alimentary hyperlipaemia and primary hypertriglyceridaemia on platelets in man. Scand. J. Haematol. 1974, 12, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Venn, B.J.; Green, T.J. Glycemic index and glycemic load: Measurement issues and their effect on diet-disease relationships. Eur. J. Clin. Nutr. 2007, 61, S122–S131. [Google Scholar] [CrossRef]
- Brand-Miller, J.C.; Thomas, M.; Swan, V.; Ahmad, Z.I.; Petocz, P.; Colagiuri, S. Physiological validation of the concept of glycemic load in lean young adults. J. Nutr. 2003, 133, 2728–2732. [Google Scholar] [CrossRef]
- Glade, M.J.; Smith, K. A glance at… glycemic index. Nutrition 2015, 31, 539–541. [Google Scholar] [CrossRef]
- Brand, J.C.; Nicholson, P.L.; Thorburn, A.W.; Truswell, A.S. Food processing and the glycemic index. Am. J. Clin. Nutr. 1985, 42, 1192–1196. [Google Scholar] [CrossRef]
- Dias, C.B.; Moughan, P.J.; Wood, L.G.; Singh, H.; Garg, M.L. Postprandial lipemia: Factoring in lipemic response for ranking foods for their healthiness. Lipids Health Dis. 2017, 16, 178. [Google Scholar] [CrossRef]
- Flint, A.; Møller, B.K.; Raben, A.; Pedersen, D.; Tetens, I.; Holst, J.J.; Astrup, A. The use of glycaemic index tables to predict glycaemic index of composite breakfast meals. Br. J. Nutr. 2004, 91, 979–989. [Google Scholar] [CrossRef]
- Wolever, T.M.; Yang, M.; Zeng, X.Y.; Atkinson, F.; Brand-Miller, J.C. Food glycemic index, as given in glycemic index tables, is a significant determinant of glycemic responses elicited by composite breakfast meals. Am. J. Clin. Nutr. 2006, 83, 1306–1312. [Google Scholar] [CrossRef] [PubMed]
- Hardy, D.S.; Hoelscher, D.M.; Aragaki, C.; Stevens, J.; Steffen, L.M.; Pankow, J.S.; Boerwinkle, E. Association of glycemic index and glycemic load with risk of incident coronary heart disease among Whites and African Americans with and without type 2 diabetes: The Atherosclerosis Risk in Communities study. Ann. Epidemiol. 2010, 20, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Barclay, A.W.; Petocz, P.; McMillan-Price, J.; Flood, V.M.; Prvan, T.; Mitchell, P.; Brand-Miller, J.C. Glycemic index, glycemic load, and chronic disease risk—A meta-analysis of observational studies. Am. J. Clin. Nutr. 2008, 87, 627–637. [Google Scholar] [CrossRef]
- Fan, J.; Song, Y.; Wang, Y.; Hui, R.; Zhang, W. Dietary glycemic index, glycemic load, and risk of coronary heart disease, stroke, and stroke mortality: A systematic review with meta-analysis. PLoS ONE 2012, 7, e52182. [Google Scholar] [CrossRef]
- Ma, X.Y.; Liu, J.P.; Song, Z.Y. Glycemic load, glycemic index and risk of cardiovascular diseases: Meta-analyses of prospective studies. Atherosclerosis 2012, 223, 491–496. [Google Scholar] [CrossRef]
- Simila, M.E.; Kontto, J.P.; Valsta, L.M.; Mannisto, S.; Albanes, D.; Virtamo, J. Carbohydrate substitution for fat or protein and risk of type 2 diabetes in male smokers. Eur. J. Clin. Nutr. 2012, 66, 716–721. [Google Scholar] [CrossRef]
- Sluijs, I.; Beulens, J.W.; Van Der Schouw, Y.T.; Van Der, A.D.; Buckland, G.; Kuijsten, A.; Schulze, M.B.; Amiano, P.; Ardanaz, E.; Balkau, B.; et al. Dietary glycemic index, glycemic load, and digestible carbohydrate intake are not associated with risk of type 2 diabetes in eight European countries. J. Nutr. 2013, 143, 93–99. [Google Scholar]
- VanWoudenbergh, G.J.; Kuijsten, A.; Sijbrands, E.J.; Hofman, A.; Witteman, J.C.; Feskens, E.J. Glycemic index and glycemic load and their association with C-reactive protein and incident type 2 diabetes. J. Nutr. Metab. 2011, 2011, 623076. [Google Scholar]
- Sahyoun, N.R.; Anderson, A.L.; Tylavsky, F.A.; Lee, J.S.; Sellmeyer, D.E.; Harris, T.B. Dietary glycemic index and glycemic load and the risk of type 2 diabetes in older adults. Am. J. Clin. Nutr. 2008, 87, 126–131. [Google Scholar] [CrossRef]
- Shahdadian, F.; Saneei, P.; Milajerdi, A.; Esmaillzadeh, A. Dietary glycemic index, glycemic load, and risk of mortality from all causes and cardiovascular diseases: A systematic review and dose-response meta-analysis of prospective cohort studies. Am. J. Clin. Nutr. 2019, 110, 921–937. [Google Scholar] [CrossRef]
- Choudhury, S.M.; Tan, T.M.; Bloom, S.R. Gastrointestinal hormones and their role in obesity. Curr. Opin. Endocrinol. Diabetes Obes. 2016, 23, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Vega-López, S.; Venn, B.J.; Slavin, J.L. Relevance of the Glycemic Index and Glycemic Load for Body Weight, Diabetes, and Cardiovascular Disease. Nutrients 2018, 10, 1361. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Lim, S.; Egger, G. The effect of a low glycaemic index breakfast on blood glucose, insulin, lipid profiles, blood pressure, body weight, body composition and satiety in obese and overweight individuals: A pilot study. J. Am. Coll. Nutr. 2008, 27, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.T.; Lampe, J.W.; Schwarz, Y.; Breymeyer, K.L.; Noar, K.A.; Song, X.; Neuhouser, M.L. Low glycemic load experimental diet more satiating than high glycemic load diet. Nutr. Cancer 2012, 64, 666–673. [Google Scholar] [CrossRef]
- Das, S.K.; Gilhooly, C.H.; Golden, J.K.; Pittas, A.G.; Fuss, P.J.; Cheatham, R.A.; Tyler, S.; Tsay, M.; McCrory, M.A.; Lichtenstein, A.H.; et al. Long-term effects of 2 energy-restricted diets differing in glycemic load on dietary adherence, body composition, and metabolism in calerie: A 1-y randomized controlled trial. Am. J. Clin. Nutr. 2007, 85, 1023–1030. [Google Scholar] [CrossRef]
- Juanola-Falgarona, M.; Salas-Salvado, J.; Ibarrola-Jurado, N.; Rabassa-Soler, A.; Diaz-Lopez, A.; Guasch-Ferre, M.; Hernandez-Alonso, P.; Balanza, R.; Bullo, M. Effect of the glycemic index of the diet on weight loss, modulation of satiety, inflammation, and other metabolic risk factors: A randomized controlled trial. Am. J. Clin. Nutr. 2014, 100, 27–35. [Google Scholar] [CrossRef]
- Abete, I.; Parra, D.; Martinez, J.A. Energy-restricted diets based on a distinct food selection affecting the glycemic index induce different weight loss and oxidative response. Clin. Nutr. 2008, 27, 545–551. [Google Scholar] [CrossRef]
- Karl, J.P.; Cheatham, R.A.; Das, S.K.; Hyatt, R.R.; Gilhooly, C.H.; Pittas, A.G.; Lieberman, H.R.; Lerner, D.; Roberts, S.B.; Saltzman, E. Effect of glycemic load on eating behavior self-efficacy during weight loss. Appetite 2014, 80, 204–211. [Google Scholar] [CrossRef]
- Buscemi, S.; Cosentino, L.; Rosafio, G.; Morgana, M.; Mattina, A.; Sprini, D.; Verga, S.; Rini, G.B. Effects of hypocaloric diets with different glycemic indexes on endothelial function and glycemic variability in overweight and in obese adult patients at increased cardiovascular risk. Clin. Nutr. 2013, 32, 346–352. [Google Scholar] [CrossRef]
- Sichieri, R.; Moura, A.S.; Genelhu, V.; Hu, F.; Willett, W.C. An 18-mo randomized trial of a low-glycemic-index diet and weight change in Brazilian women. Am. J. Clin. Nutr. 2007, 86, 707–713. [Google Scholar] [CrossRef]
- Zafar, M.I.; Mills, K.E.; Zheng, J.; Peng, M.M.; Ye, X.; Chen, L.L. Low glycaemic index diets as an intervention for obesity: A systematic review and meta-analysis. Obes. Rev. 2019, 20, 290–315. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.L.; Gellar, L.; Nathanson, B.H.; Pbert, L.; Ma, Y.; Ockene, I.; Rosal, M.C. Decrease in glycemic index associated with improved glycemic control among latinos with type 2 diabetes. J. Acad. Nutr. Diet. 2015, 115, 898–906. [Google Scholar] [CrossRef] [PubMed]
- Farvid, M.S.; Homayouni, F.; Shokoohi, M.; Fallah, A.; Farvid, M.S. Glycemic index, glycemic load and their association with glycemic control among patients with type 2 diabetes. Eur. J. Clin. Nutr. 2014, 68, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Mayer-Davis, E.J.; Dhawan, A.; Liese, A.D.; Teff, K.; Schulz, M. Towards understanding of glycaemic index and glycaemic load in habitual diet: Associations with measures of glycaemia in the insulin resistance atherosclerosis study. Br. J. Nutr. 2006, 95, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Hodge, A.M.; English, D.R.; O’Dea, K.; Giles, G.G. Glycemic index and dietary fiber and the risk of type 2 diabetes. Diabetes Care 2004, 27, 2701–2706. [Google Scholar] [CrossRef]
- Salmerón, J.; Ascherio, A.; Rimm, E.B.; Colditz, G.A.; Spiegelman, D.; Jenkins, D.J.A.; Stampfer, M.J.; Wing, A.L.; Willett, W.C. Dietary fiber, glycemic load, and risk of NIDDM in men. Diabetes Care 1997, 20, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Runchey, S.S.; Pollak, M.N.; Valsta, L.M.; Coronado, G.D.; Schwarz, Y.; Breymeyer, K.L.; Wang, C.; Wang, C.Y.; Lampe, J.W.; Neuhouser, M.L. Glycemic load effect on fasting and post-prandial serum glucose, insulin, IGF-1 and IGFBP-3 in a randomized, controlled feeding study. Eur. J. Clin. Nutr. 2012, 66, 1146–1152. [Google Scholar] [CrossRef]
- Ojo, O.; Ojo, O.O.; Adebowale, F.; Wang, X.H. The effect of dietary glycaemic index on glycaemia in patients with type 2 diabetes: A systematic review and meta-analysis of randomized controlled trials. Nutrients 2018, 10, 373. [Google Scholar] [CrossRef]
- Bhupathiraju, S.N.; Tobias, D.K.; Malik, V.S.; Pan, A.; Hruby, A.; Manson, J.E.; Willett, W.C.; Hu, F.B. Glycemic index, glycemic load, and risk of type 2 diabetes: Results from 3 large US cohorts and an updated meta-analysis. Am. J. Clin. Nutr. 2014, 100, 218–232. [Google Scholar] [CrossRef]
- Dong, J.Y.; Zhang, L.; Zhang, Y.H.; Qin, L.Q. Dietary glycaemic index and glycaemic load in relation to the risk of type 2 diabetes: A meta-analysis of prospective cohort studies. Br. J. Nutr. 2011, 106, 1649–1654. [Google Scholar] [CrossRef]
- Livesey, G.; Taylor, R.; Livesey, H.; Liu, S. Is there a dose-response relation of dietary glycemic load to risk of type 2 diabetes? Meta-analysis of prospective cohort studies. Am. J. Clin. Nutr. 2013, 97, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Shu, X.O.; Li, H.; Xiang, Y.B.; Yang, G.; Gao, Y.T.; Zheng, W.; Zhang, X. Dietary carbohydrates, refined grains, glycemic load, and risk of coronary heart disease in Chinese adults. Am. J. Epidemiol. 2013, 178, 1542–1549. [Google Scholar] [CrossRef] [PubMed]
- Burger, K.N.; Beulens, J.W.; Boer, J.M.; Spijkerman, A.M.; Van Der, A.D. Dietary glycemic load and glycemic index and risk of coronary heart disease and stroke in Dutch men and women: The EPIC-MORGEN study. PLoS ONE 2011, 6, e25955. [Google Scholar] [CrossRef] [PubMed]
- Sieri, S.; Krogh, V.; Berrino, F.; Evangelista, A.; Agnoli, C.; Brighenti, F.; Pellegrini, N.; Palli, D.; Masala, G.; Sacerdote, C.; et al. Dietary glycemic load and index and risk of coronary heart disease in a large Italian cohort: The EPICOR study. Arch. Intern. Med. 2010, 170, 640–647. [Google Scholar] [CrossRef]
- Levitan, E.B.; Mittleman, M.A.; Hakansson, N.; Wolk, A. Dietary glycemic index, dietary glycemic load, and cardiovascular disease in middle-aged and older Swedish men. Am. J. Clin. Nutr. 2007, 85, 1521–1526. [Google Scholar] [CrossRef]
- Burger, K.N.; Beulens, J.W.; Van Der Schouw, Y.T.; Sluijs, I.; Spijkerman, A.M.; Sluik, D.; Boeing, H.; Kaaks, R.; Teucher, B.; Dethlefsen, C.; et al. Dietary fiber, carbohydrate quality and quantity, and mortality risk of individuals with diabetes mellitus. PLoS ONE 2012, 7, e43127. [Google Scholar] [CrossRef]
- Levitan, E.B.; Mittleman, M.A.; Wolk, A. Dietary glycemic index, dietary glycemic load, and incidence of heart failure events: A prospective study of middle-aged and elderly women. J. Am. Coll. Nutr. 2010, 29, 65–71. [Google Scholar] [CrossRef]
- Levitan, E.B.; Cook, N.R.; Stampfer, M.J.; Ridker, P.M.; Rexrode, K.M.; Buring, J.E.; Manson, J.E.; Liu, S. Dietary glycemic index, dietary glycemic load, blood lipids, and c-reactive protein. Metabolism 2008, 57, 437–443. [Google Scholar] [CrossRef]
- Liese, A.D.; Gilliard, T.; Schulz, M.; D’Agostino, R.B., Jr.; Wolever, T.M. Carbohydrate nutrition, glycaemic load, and plasma lipids: The insulin resistance atherosclerosis study. Eur. Heart J. 2007, 28, 80–87. [Google Scholar] [CrossRef]
- Milton, J.E.; Briche, B.; Brown, I.J.; Hickson, M.; Robertson, C.E.; Frost, G.S. Relationship of glycaemic index with cardiovascular risk factors: Analysis of the national diet and nutrition survey for people aged 65 and older. Public Health Nutr. 2007, 10, 1321–1335. [Google Scholar] [CrossRef]
- Castro-Quezada, I.; Artacho, R.; Molina-Montes, E.; Serrano, F.A.; Ruiz-Lopez, M.D. Dietary glycaemic index and glycaemic load in a rural elderly population (60–74 years of age) and their relationship with cardiovascular risk factors. Eur. J. Nutr. 2015, 54, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Sasaki, S.; Takahashi, Y.; Okubo, H.; Hosoi, Y.; Horiguchi, H.; Oguma, E.; Kayama, F. Dietary glycemic index and load in relation to metabolic risk factors in Japanese female farmers with traditional dietary habits. Am. J. Clin. Nutr. 2006, 83, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Shikany, J.M.; Tinker, L.F.; Neuhouser, M.L.; Ma, Y.; Patterson, R.E.; Phillips, L.S.; Liu, S.; Redden, D.T. Association of glycemic load with cardiovascular disease risk factors: The women’s health initiative observational study. Nutrition 2010, 26, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Hosseinpour-Niazi, S.; Sohrab, G.; Asghari, G.; Mirmiran, P.; Moslehi, N.; Azizi, F. Dietary glycemic index, glycemic load, and cardiovascular disease risk factors: Tehran lipid and glucose study. Arch. Iran. Med. 2013, 16, 401–407. [Google Scholar] [PubMed]
- McKeown, N.M.; Meigs, J.B.; Liu, S.; Rogers, G.; Yoshida, M.; Saltzman, E.; Jacques, P.F. Dietary carbohydrates and cardiovascular disease risk factors in the framingham offspring cohort. J. Am. Coll. Nutr. 2009, 28, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Masson, C.J.; Mensink, R.P. Exchanging saturated fatty acids for (n-6) polyunsaturated fatty acids in a mixed meal may decrease postprandial lipemia and markers of inflammation and endothelial activity in overweight men. J. Nutr. 2011, 141, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Mekki, N.; Charbonnier, M.; Borel, P.; Leonardi, J.; Juhel, C.; Portugal, H.; Lairon, D. Butter differs from olive oil and sunflower oil in its effects on postprandial Lipemia and Triacylglycerol-rich lipoproteins after single mixed meals in healthy young men. J. Nutr. 2002, 132, 3642–3649. [Google Scholar] [CrossRef]
- Peairs, A.D.; Rankin, J.W.; Lee, Y.W. Effects of acute ingestion of different fats on oxidative stress and inflammation in overweight and obese adults. Nutr. J. 2011, 10, 122. [Google Scholar] [CrossRef]
- Tulk, H.M.F.; Robinson, L.E. Modifying the n-6/n-3 polyunsaturated fatty acid ratio of a high–saturated fat challenge does not acutely attenuate postprandial changes in inflammatory markers in men with metabolic syndrome. Metab. Clin. Exp. 2009, 58, 1709–1716. [Google Scholar] [CrossRef]
- Westphal, S.; Leodolter, A.; Kahl, S.; Dierkes, J.; Malfertheiner, P.; Luley, C. Addition of glucose to a fatty meal delays chylomicron and suppresses VLDL in healthy subjects. Eur. J. Clin. Investig. 2002, 32, 322–327. [Google Scholar] [CrossRef]
- Westphal, S.; Kastner, S.; Taneva, E.; Leodolter, A.; Dierkes, J.; Luley, C. Postprandial lipid and carbohydrate responses after the ingestion of a casein-enriched mixed meal. Am. J. Clin. Nutr. 2004, 80, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Brannon, P.M. Adaptation of the exocrine pancreas to diet. Annu. Rev. Nutr. 1990, 10, 85–105. [Google Scholar] [CrossRef] [PubMed]
- Armand, M.; Hamosh, M.; DiPalma, J.S.; Gallagher, J.; Benjamin, S.B.; Philpott, J.R.; Lairon, D.; Hamosh, P. Dietary fat modulates gastric lipase activity in healthy humans. Am. J. Clin. Nutr. 1995, 62, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, B.; Armand, M.; Guillon, F.; Castelain, C.; Borel, P.; Barry, J.L.; Pieroni, G.; Lairon, D. Viscous soluble dietary fibers alter emulsification and lipolysis of triacylglycerols in duodenal medium in vitro. J. Nutr. Biochem. 1996, 7, 293–302. [Google Scholar] [CrossRef]
- Ausar, S.F.; Landa, C.A.; Bianco, I.D.; Castagna, L.F.; Beltramo, D.M. Hydrolysis of a chitosan-induced milk aggregate by pepsin, trypsin and pancreatic lipase. Biosci. Biotechnol. Biochem. 2001, 65, 2412–2418. [Google Scholar] [CrossRef]
- D’Souza, V.M.; Shertzer, H.G.; Menon, A.G.; Pauletti, G.M. High glucose concentration in isotonic media alters caco-2 cell permeability. AAPS PharmSci 2003, 5, E24. [Google Scholar]
- D’Souza, V.M.; Buckley, D.J.; Buckley, A.R.; Pauletti, G.M. Extracellular glucose concentration alters functional activity of the intestinal oligopeptide transporter (PepT-1) in Caco-2 cells. J. Pharm. Sci. 2003, 92, 594–603. [Google Scholar] [CrossRef]
- Yamamoto, M.; Acevedo-Duncan, M.; Chalfant, C.E.; Patel, N.A.; Watson, J.E.; Cooper, D.R. Acute glucose-induced downregulation of PKCbetaII accelerates cultured VSMC proliferation. Am. J. Physiol. Cell Physiol. 2000, 279, 587–595. [Google Scholar] [CrossRef]
- Abe-Dohmae, S.; Ikeda, Y.; Matsuo, M.; Hayashi, M.; Okuhira, K.; Ueda, K.; Yokoyama, S. Human ABCA7 supports apolipoprotein-mediated release of cellular cholesterol and phospholipid to generate high density lipoprotein. J. Biol. Chem. 2004, 279, 604–611. [Google Scholar] [CrossRef]
- Kobayashi, T.; Ogawa, Y.; Watanabe, Y.; Furuya, M.; Kataoka, S.; Garcia del Saz, E.; Tsunawaki, S.; Dinauer, M.C.; Seguchi, H. Mitochondrial transmembrane potential is diminished in phorbol myristate acetate-stimulated peritoneal resident macrophages isolated from wild-type mice, but not in those from gp91-phoxdeficient mice. Histochem. Cell Biol. 2004, 122, 323–332. [Google Scholar] [CrossRef]
- Iizuka, K.; Bruick, R.K.; Liang, G.; Horton, J.D.; Uyeda, K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. USA 2004, 101, 7281–7286. [Google Scholar] [CrossRef] [PubMed]
- Aragno, M.; Tomasinelli, C.E.; Vercellinatto, I.; Catalano, M.G.; Collino, M.; Fantozzi, R.; Danni, O.; Boccuzzi, G. SREBP-1c in nonalcoholic fatty liver disease induced by western-type high-fat diet plus fructose in rats. Free Radic. Biol. Med. 2009, 47, 1067–1074. [Google Scholar] [CrossRef]
- Bidwell, A.J. Chronic Fructose Ingestion as a Major Health Concern: Is a Sedentary Lifestyle Making It Worse? A Review. Nutrients 2017, 9, 549. [Google Scholar] [CrossRef] [PubMed]
- Tappy, L.; Randin, J.P.; Felber, J.P.; Chiolero, R.; Simonson, D.C.; Jequier, E.; DeFronzo, R.A. Comparison of thermogenic effect of fructose and glucose in normal humans. Am. J. Physiol. 1986, 250, E718–E724. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L.; Havel, P.J. Fructose consumption: Potential mechanisms for its effects to increase visceral adiposity and induce dyslipidemia and insulin resistance. Curr. Opin. Lipidol. 2008, 19, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.Z.; Empie, M.W. Fructose metabolism in humans—What isotopic tracer studies tell us. Nutr. Metab. 2012, 9, 89. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology 2012, 142, 1592–1609. [Google Scholar] [CrossRef]
- Mayes, P.A. Intermediary metabolism of fructose. Am. J. Clin. Nutr. 1993, 58, 754S–765S. [Google Scholar] [CrossRef]
- Ma, J.; Fox, C.; Speliotes, E.; Hoffmann, U.; Smith, C.; Saltzman, E.; Jacques, P.; McKeown, N. Sugar-sweetened beverage intake is associated with fatty liver in the Framingham offspring study. J. Hepatol. 2015, 63, 462–469. [Google Scholar] [CrossRef]
- Ouyang, X.; Cirillo, P.; Sautin, Y.; McCall, S.; Bruchette, J.L.; Diehl, A.M.; Johnson, R.J.; Abdelmalek, M.F. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J. Hepatol. 2008, 48, 993–999. [Google Scholar] [CrossRef]
- Kanerva, N.; Sandboge, S.; Kaartinen, N.E.; Mannisto, S.; Eriksson, J.G. Higher fructose intake is inversely associated with risk of nonalcoholic fatty liver disease in older Finnish adults. Am. J. Clin. Nutr. 2014, 100, 1133–1138. [Google Scholar] [CrossRef] [PubMed]
- Abdelmalek, M.F.; Suzuki, A.; Guy, C.; Unalp-Arida, A.; Colvin, R.; Johnson, R.J.; Diehl, A.M. Increased fructose consumption is associated with fibrosis severity in patients with NAFLD. Hepatology 2010, 51, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.; Ma, J.; Patel, K.; Berger, S.; Lau, J.; Lichtenstein, A.H. Fructose, high-fructose corn syrup, sucrose, and nonalcoholic fatty liver disease or indexes of liver health: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2014, 100, 833–849. [Google Scholar] [CrossRef] [PubMed]
- Chiu, S.; Sievenpiper, J.L.; De Souza, R.J.; Cozma, A.I.; Mirrahimi, A.; Carleton, A.J.; Ha, V.; Di Buono, M.; Jenkins, A.L.; Leiter, L.A.; et al. Effect of fructose on markers of non-alcoholic fatty liver disease (NAFLD): A systematic review and meta-analysis of controlled feeding trials. Eur. J. Clin. Nutr. 2014, 68, 416–423. [Google Scholar] [CrossRef]
- Ter Horst, K.W.; Serlie, M.J. Fructose Consumption, Lipogenesis, and Non-Alcoholic Fatty Liver Disease. Nutrients 2017, 9, 981. [Google Scholar] [CrossRef]
- Matikainen, N.; Söderlund, S.; Björnson, E.; Bogl, L.H.; Pietiläinen, K.H.; Hakkarainen, A.; Lundbom, N.; Eliasson, B.; Räsänen, S.M.; Rivellese, A.; et al. Fructose intervention for 12 weeks does not impair glycemic control or incretin hormone responses during oral glucose or mixed meal tests in obese men. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Swarbrick, M.M.; Stanhope, K.L.; Elliott, S.S.; Graham, J.L.; Krauss, R.M.; Christiansen, M.P.; Griffen, S.C.; Keim, N.L.; Havel, P.J. Consumption of fructose-sweetened beverages for 10 weeks increases postprandial triacylglycerol and apolipoprotein-B concentrations in overweight and obese women. Br. J. Nutr. 2008, 100, 947–952. [Google Scholar] [CrossRef]
- Stanhope, K.L.; Griffen, S.C.; Bremer, A.A.; Vink, R.G.; Schaefer, E.J.; Nakajima, K.; Schwarz, J.-M.; Beysen, C.; Berglund, L.; Keim, N.L.; et al. Metabolic responses to prolonged consumption of glucose- and fructose-sweetened beverages are not associated with postprandial or 24-h glucose and insulin excursions. Am. J. Clin. Nutr. 2011, 94, 112–119. [Google Scholar] [CrossRef]
- Chong, M.F.; Fielding, B.A.; Frayn, K.N. Mechanisms for the acute effect of fructose on postprandial lipemia. Am. J. Clin. Nutr. 2007, 85, 1511–1520. [Google Scholar] [CrossRef]
- Bantle, J.P.; Raatz, S.K.; Thomas, W.; Georgopoulos, A. Effects of dietary fructose on plasma lipids in healthy subjects. Am. J. Clin. Nutr. 2000, 72, 1128–1134. [Google Scholar] [CrossRef]
- Stanhope, K.L.; Schwarz, J.M.; Keim, N.L.; Griffen, S.C.; Bremer, A.A.; Graham, J.L.; Hatcher, B.; Cox, C.L.; Dyachenko, A.; Zhang, W.; et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J. Clin. Investig. 2009, 119, 1322–1334. [Google Scholar] [CrossRef]
- Harbis, A.; Perdreau, S.; Vincent-Baudry, S.; Charbonnier, M.; Bernard, M.C.; Raccah, D.; Senft, M.; Lorec, A.M.; Defoort, C.; Portugal, H.; et al. Glycemic and insulinemic meal responses modulate postprandial hepatic and intestinal lipoprotein accumulation in obese, insulin-resistant subjects. Am. J. Clin. Nutr. 2004, 80, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Harbis, A.; Defoort, C.; Narbonne, H.; Juhel, C.; Senft, M.; Latgé, C.; Delenne, B.; Portugal, H.; Atlan-Gepner, C.; Vialettes, B.; et al. Acute hyperinsulinism modulates plasma apolipoprotein B-48 triglyceride-rich lipoproteins in healthy subjects during the postprandial period. Diabetes 2001, 50, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Bouché, C.; Rizkalla, S.W.; Luo, J.; Vidal, H.; Veronese, A.; Pacher, N.; Fouquet, C.; Lang, V.; Slama, G. Five-week, low-glycemic index diet decreases total fat mass and improves plasma lipid profile in moderately overweight nondiabetic men. Diabetes Care 2002, 25, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Bukkapatnam, R.N.; Berglund, L.; Anuurad, E.; Devaraj, S.; Hyson, D.; Rafii, F.; Malmstein, C.; Villablanca, A.C. Postprandial metabolic responses to dietary glycemic index in hypercholesterolemic postmenopausal women. Prev. Cardiol. 2010, 13, 29–35. [Google Scholar] [CrossRef]
- Sun, L.; Tan, K.W.J.; Lim, J.Z.; Magkos, F.; Henry, C.J. Dietary fat and carbohydrate quality have independent effects on postprandial glucose and lipid responses. Eur. J. Nutr. 2018, 57, 243–250. [Google Scholar] [CrossRef]
- Despland, C.; Walther, B.; Kast, C.; Campos, V.; Rey, V.; Stefanoni, N.; Tappy, L. A randomized-controlled clinical trial of high fructose diets from either Robinia honey or free fructose and glucose in healthy normal weight males. Clin. Nutr. ESPEN 2017, 19, 16–22. [Google Scholar] [CrossRef][Green Version]
- Campos, V.; Despland, C.; Brandejsky, V.; Kreis, R.; Schneiter, P.; Boesch, C.; Tappy, L. Metabolic Effects of Replacing Sugar-Sweetened Beverages with Artificially-Sweetened Beverages in Overweight Subjects with or without Hepatic Steatosis: A Randomized Control Clinical Trial. Nutrients 2017, 9, 202. [Google Scholar] [CrossRef]
- Livesey, G.; Taylor, R. Fructose consumption and consequences for glycation, plasma triacylglycerol, and body weight: Meta-analyses and meta-regression models of intervention studies. Am. J. Clin. Nutr. 2008, 88, 1419–1437. [Google Scholar]
- David Wang, D.; Sievenpiper, J.L.; De Souza, R.J.; Cozma, A.I.; Chiavaroli, L.; Ha, V.; Mirrahimi, A.; Carleton, A.J.; Di Buono, M.; Jenkins, A.L.; et al. Effect of fructose on postprandial triglycerides: A systematic review and meta-analysis of controlled feeding trials. Atherosclerosis 2014, 232, 125–133. [Google Scholar] [CrossRef]


{kind=link}
| Study | Type of Study | Sample | Results |
|---|---|---|---|
| Wang et al. [63] | Cross-sectional | 238 low income Latino adults w/T2DM, 45–67 years, 33–36 kg/m2 | Positive association between GI and HbA1c (but not GL) |
| Farvid et al. [64] | Cross-sectional | 640 adults w/T2DM, 28–75 years | Positive association between GL and FSG, HbA1c No association of GI and HbA1c or FSG |
| Mayer-Davis et al. [65] | Cross-sectional | 1255 adults with/without IR or T2DM, 55.3 years, 29.1 kg/m2 | No association of GI/GL with glucose homeostasis |
| Hodge et al. [66] | Prospective | 36,787 men and women aged 40–69 years without diabetes | Low GI and high CHO intake—> decreased risk of DM |
| Salmeron et al. [67] | Prospective | 42,759 men without DM or cardiovascular disease, 40–75 years, 6 years of follow-up | Positive association between high GI and incidence of DM (RR: 1.37; 95% CI, 1.02–1.83). Positive association of high GL/low cereal fiber intake and incidence of DM (RR: 2.17, 95% CI; 1.04–4.54) |
| Hardy et al. [43] | Sub-analysis | 13,051 individuals aged 45–64 years from the Atherosclerosis Risk in Communities (ARIC) study | High GI—> increased risk of CHD in African Americans High GL—> increased risk of CHD in Whites (without DM) |
| Simila et al. [47] | Prospective | 25,943 male smokers, 50–69 years | No association of GI/GL with glucose homeostasis |
| Sluijs et al. [48] | Prospective | 37,843 Netherlands adults, 21–70 years | No association of GI/GL with glucose homeostasis |
| Van Woudenbergh et al. [49] | Prospective | 4366 Netherlands adults, ≥55 years | No association of GI/GL with glucose homeostasis |
| Sahyoun et al. [50] | Prospective | 1898 adults, 70–79 years | No association of GI/GL with glucose homeostasis |
| Bhupathiraju et al. [70] | Meta-analysis | 74,248 women from the Nurses’ Health Study, 90,411 women from the Nurses’ Health Study II, and 40,498 men from the Health Professionals Follow-Up Study | Positive association of high GI (RR: 1.19; 95% CI: 1.14–1.24) and GL (RR: 1.13; 95% CI: 1.08–1.17) with T2DM |
| Barclay et al. [44] | Meta-analysis | 37 prospective cohort studies | Positive association of high GI (RR: 1.40; 95% CI: 1.23–1.59) and GL (RR: 1.27; 95% CI: 1.12–1.45) with T2DM |
| Dong et al. [71] | Meta-analysis | 13 prospective cohort studies | Positive association of high GI (RR: 1.16; 95% CI: 1.06–1.26) and GL (RR: 1.20; 95% CI: 1.11–1.30) with T2DM |
| Livesey et al. [72] | Meta-analysis | 24 prospective cohort studies | Positive association of GL with T2D (RR: 1.45 for a 100 g increment in GL; 95% CI: 1.31–1.61) |
| Study | Type of Study | Sample | Results |
|---|---|---|---|
| Yu et al. [73] | Prospective | 117,366 Chinese adults; 40–74 years; without history of diabetes, CHD, stroke or cancer; F/U of 9.8 years for women, 5.4 years for men | Positive association of GL and CHD |
| Burger et al. [74] | Prospective | 8855 men, 10,753 women, 21–64 years, F/U of 11.9 years | No association between GI/GL and CVD |
| Sieri et al. [75] | Prospective | 44,132 adults, F/U of 7.9 years | No association between GI/GL and CHD |
| Levitan et al. [76] | Prospective | 36,246 Swedish men, 45–79 years, F/U of 6 years | No association between GI/GL and CVD mortality |
| Burger et al. [77] | Prospective | 6192 adults with T2DM, F/U of 9.2 years | No association between GI/GL and CVD mortality |
| Levitan et al. [78] | Prospective | 36,019 women, 48–83 years, F/U of 9 years | No association between GI/GL and HF |
| Shahdadian et al. [51] | Meta-analysis | 18 cohort studies, 251,497 participants | No association between GI/GL and CVD mortality |
| Barclay et al. [44] | Meta-analysis | 37 prospective cohort studies | Positive association of GI with CHD (RR: 1.25; 95% CI: 1.00–1.56) |
| Fan et al. [45] | Meta-analysis | 15 prospective cohort studies, 438,073 participants | Positive association of GL with CHD (RR: 1.49; 95% CI: 1.27−1.73), only in women Positive association of GL with stroke (RR: 1.19; 95% CI: 1.00−1.43) |
| Ma et al. [46] | Meta-analysis | 14 prospective cohort studies, 229,213 participants | Positive association of GI (RR: 1.13; 95% CI: 1.04–1.22) and GL (RR: 1.23; 95% CI: 1.11–1.36) with CVD, both associations stronger for women |
| Study | Type of Study | Sample | Results |
|---|---|---|---|
| Matikainen et al. [117] | Prospective | 66 obese men consumed fructose-sweetened beverages containing 75 g fructose/day (high GL) for 12 weeks | Increased postprandial TGs |
| Swarbrick et al. [118] | Prospective | 7 overweight or obese postmenopausal women on high GL intervention diet, which included a fructose-sweetened beverage with each meal, for 10 weeks | 14 h postprandial TAG profiles were significantly increased (iAUC 141% higher) |
| Stanhope et al. [119] | Prospective | Overweight and obese subjects, 8-week consumption of fructose-sweetened beverages | Increased postprandial TGs |
| Chong et al. [120] | Crossover | 14 subjects, fructose or glucose test meal after an overnight fast | At 4 h postprandially, newly synthesized fatty acids from fructose = 0.4% of circulating VLDL-triacylglycerol, newly synthesized triacylglycerol-glycerol = 38%, newly synthesized fatty acids and triacylglycerol-glycerol from glucose = none of VLDL-triacylglycerol |
| Bantle et al. [121] | Prospective | 24 healthy adult volunteers, diet with 17% of energy as fructose or diet sweetened with glucose, for 6 weeks | Higher fasting, postprandial, and daylong plasma triacylglycerol concentrations with fructose |
| Stanhope et al. [122] | Prospective | Overweight and obese subjects consumed glucose- or fructose-sweetened beverages providing 25% of energy requirements for 10 weeks | Fructose increased postprandial TGs and DNL |
| Harbis et al. [123] | Crossover | 9 obese subjects with insulin resistance randomly ingested 2 test meals with different quantities of slowly available glucose | High GI meal increased accumulation of TRL-apoB-48 and TRL-apoB-100 at 4 and 2 h postprandially, respectively |
| Harbis et al. [124] | Crossover | 10 healthy men, 4 isolipidic meals with various GIs | Positive association of GI and apoB-48 plasma concentration at 6 h postprandially |
| Bouché et al. [125] | Prospective | 11 healthy men, 5-week low GI diet versus high GI diet | Low GI diet lowered plasma triacylglycerol excursion after lunch |
| Study | Type of Study | Sample | Results |
|---|---|---|---|
| Bukkapatnam et al. [126] | Crossover | 15 healthy postmenopausal women, low or high GI meal | Increased postprandial TGs with low GI meal |
| Sun et al. [127] | Crossover | 20 healthy Chinese men, isocaloric meals different in carbohydrate and fat quality, in random order | No association of GI with postprandial iAUC for TGs |
| Despland et al. [128] | Prospective | 8 healthy males, diet containing 25% energy as honey or pure fructose–glucose compared to an isocaloric starch diet, for 8 days | No difference in postprandial triglycerides regardless of GI |
| Campos et al. [129] | Prospective | 26 obese or overweight subjects, substitution of high sugar-sweetened beverages by artificially sweetened beverages for 12 weeks | No difference in postprandial triglycerides |
| Livesey et al. [130] | Meta-analysis | 42 reports | Significant effects on postprandial triacylglycerols with intakes of ≥50 g fructose/day Significant effects are seen on fasting triacylglycerol with intakes of ≥100 g fructose/day |
| Wang et al. [131] | Meta-analysis | 14 clinical trials | Fructose in isocaloric exchange for other carbohydrate does not increase postprandial TGs. Fructose providing excess energy increases postprandial TGs. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lambadiari, V.; Korakas, E.; Tsimihodimos, V. The Impact of Dietary Glycemic Index and Glycemic Load on Postprandial Lipid Kinetics, Dyslipidemia and Cardiovascular Risk. Nutrients 2020, 12, 2204. https://doi.org/10.3390/nu12082204
Lambadiari V, Korakas E, Tsimihodimos V. The Impact of Dietary Glycemic Index and Glycemic Load on Postprandial Lipid Kinetics, Dyslipidemia and Cardiovascular Risk. Nutrients. 2020; 12(8):2204. https://doi.org/10.3390/nu12082204
Chicago/Turabian StyleLambadiari, Vaia, Emmanouil Korakas, and Vasilios Tsimihodimos. 2020. "The Impact of Dietary Glycemic Index and Glycemic Load on Postprandial Lipid Kinetics, Dyslipidemia and Cardiovascular Risk" Nutrients 12, no. 8: 2204. https://doi.org/10.3390/nu12082204
APA StyleLambadiari, V., Korakas, E., & Tsimihodimos, V. (2020). The Impact of Dietary Glycemic Index and Glycemic Load on Postprandial Lipid Kinetics, Dyslipidemia and Cardiovascular Risk. Nutrients, 12(8), 2204. https://doi.org/10.3390/nu12082204