Nutrients, Mitochondrial Function, and Perinatal Health

 , , and
, , and 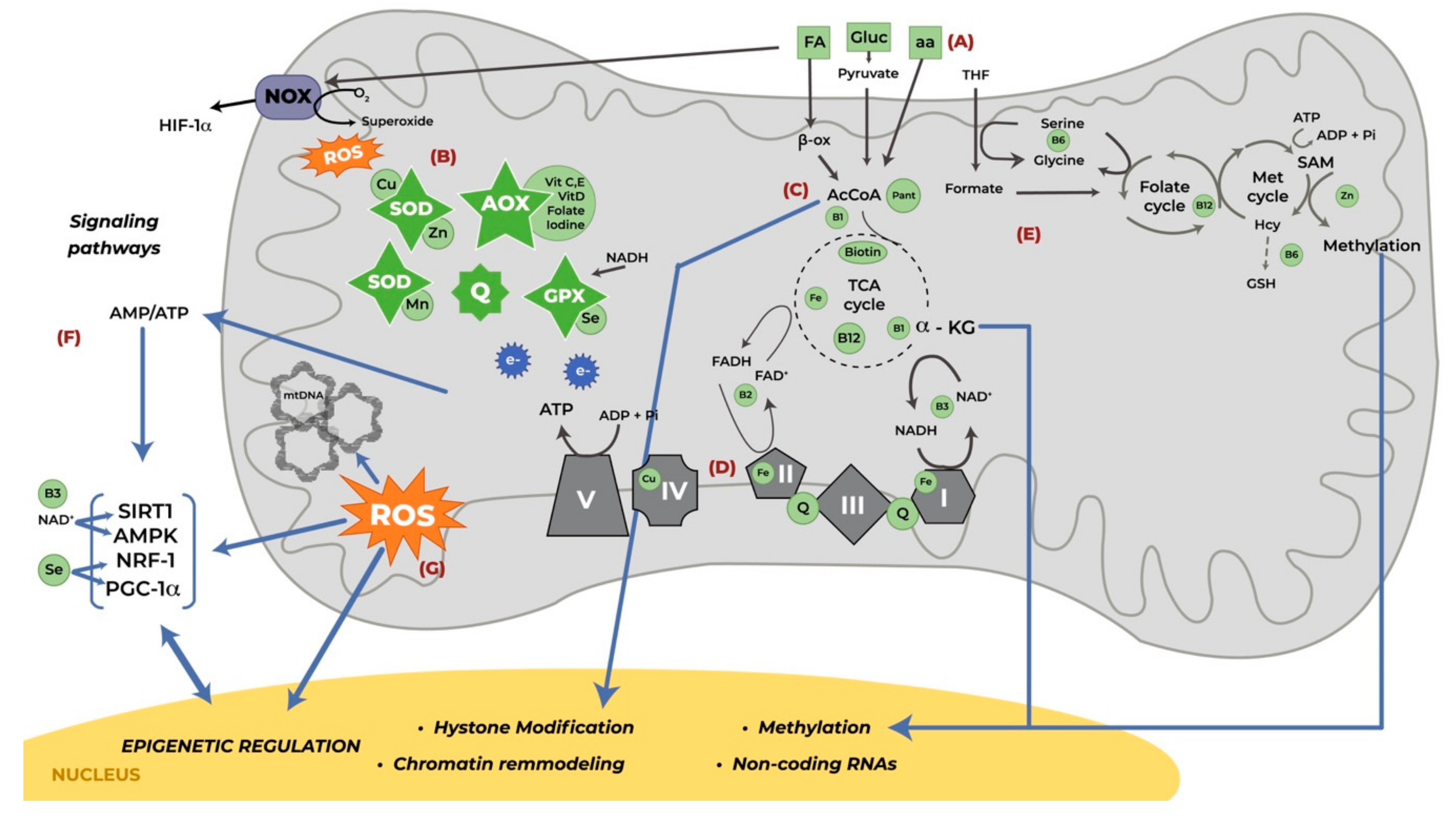{kind=link}
Abstract
1. Introduction
2. Mitochondrial Function
3. Mitochondrial Function in Pregnancy
4. Nutrients Involved in Mitochondrial Function During Pregnancy
4.1. Macronutrients
4.2. Micronutrients
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 1-C | One-carbon |
| α–KG | α-ketoglutarate |
| α-KGDH | α-KG dehydrogenase |
| AcCoA | Acetyl coenzyme A |
| AMP | Adenosine monophosphate |
| AMPK | Cyclic adenosine monophosphate activated protein kinase |
| ATP | Adenosine triphosphate |
| CoQ10 | Coenzyme Q10/Ubiquinone |
| ETC | Electron transport chain |
| FAD | Flavin adenine dinucleotide |
| GSH | Glutathione |
| GPX | Glutathione peroxidase |
| HIF-1α | Hypoxia-inducible factor 1α |
| IGF-2 | Insulin-like growth factor-2 |
| IL | Interleukin |
| IUGR | Intrauterine growth restriction |
| MAS | Malate aspartate transporter |
| mtDNA | Mitochondrial DNA |
| NAD/NADH | Oxidized/Reduced nicotinamide adenine dinucleotide |
| NCD | Non-communicable diseases |
| NF-κB | Nuclear Factor Kappa B |
| NOX | NADPH oxidase |
| NRF-1α | Nuclear Respiration Factor-1α |
| O2 | Oxygen |
| OXPHOS | Oxidative phosphorylation |
| PCR | C-Reactive protein |
| PDHc | Dehydrogenase complex |
| PE | Preeclampsia |
| PGC-1α | Peroxisome proliferator-activated receptor-gamma coactivator 1-α |
| PPAR | Peroxisome proliferator-activated receptors |
| ROS | Reactive oxygen species |
| SAM | S-adenosylmethionine |
| SIRT | Sirtuin |
| SOD | Superoxide dismutase |
| TCA | Tricarboxylic Acid cycle |
| TFAM | Mitochondrial Transcription Factor A |
| TNF-α | Tumor Necrosis Factor-α |
References
- Hsu, C.-N.; Tain, Y.-L. The Good, the Bad, and the Ugly of Pregnancy Nutrients and Developmental Programming of Adult Disease. Nutrients 2019, 11, 894. [Google Scholar] [CrossRef] [PubMed]
- Langley-Evans, S. Nutrition in early life and the programming of adult disease: A review. J. Hum. Nutr. Diet. 2015, 28 (Suppl. S1), 1–14. [Google Scholar] [CrossRef]
- Jousse, C.; Muranishi, Y.; Parry, L.; Montaurier, C.; Even, P.; Launay, J.M.; Carraro, V.; Maurin, A.C.; Averous, J.; Chaveroux, C.; et al. Perinatal protein malnutrition affects mitochondrial function in adult and results in a resistance to high fat diet-induced obesity. PLoS ONE 2014, 9, e104896. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, V.; Cardoso, R.C.; Puttabyatappa, M. Developmental Programming, a Pathway to Disease. Endocrinology 2016, 157, 1328–1340. [Google Scholar] [CrossRef] [PubMed]
- Colleoni, F.; Lattuada, D.; Garretto, A.; Massari, M.; Mandò, C.; Somigliana, E.; Cetin, I. Maternal blood mitochondrial DNA content during normal and intrauterine growth restricted (IUGR) pregnancy. Am. J. Obstet. Gynecol. 2010, 203, 365.e1–365.e6. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.J. Mitochondria in early development: Linking the microenvironment, metabolism and the epigenome. Reproduction 2019, 157, R159–R179. [Google Scholar] [CrossRef]
- Priliani, L.; Prado, E.L.; Restuadi, R.; Waturangi, D.E.; Shankar, A.H.; Malik, S.G. Maternal Multiple Micronutrient Supplementation Stabilizes Mitochondrial DNA Copy Number in Pregnant Women in Lombok, Indonesia. J. Nutr. 2019, 149, 1309–1316. [Google Scholar] [CrossRef]
- Al-Gubory, K.H.; Fowler, P.A.; Garrel, C. The roles of cellular reactive oxygen species, oxidative stress and antioxidants in pregnancy outcomes. Int. J. Biochem. Cell Biol. 2010, 42, 1634–1650. [Google Scholar] [CrossRef]
- Bhopal, R.S.; Rafnsson, S.B. Could mitochondrial efficiency explain the susceptibility to adiposity, metabolic syndrome, diabetes and cardiovascular diseases in South Asian populations? Int. J. Epidemiol. 2009, 38, 1072–1081. [Google Scholar] [CrossRef]
- Eisner, V.; Picard, M.; Hajnóczky, G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat. Cell Biol. 2018, 20, 755–765. [Google Scholar] [CrossRef]
- Pinto, M.; Moraes, C.T. Mechanisms linking mtDNA damage and aging. Free Radic. Biol. Med. 2015, 85, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [PubMed]
- Boenzi, S.; Diodato, D. Biomarkers for mitochondrial energy metabolism diseases. Essays Biochem. 2018, 62, 443–454. [Google Scholar]
- Mansouri, A.; Gattolliat, C.H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef] [PubMed]
- Shaughnessy, D.; McAllister, K.; Worth, L.; Haugen, A.; Meyer, J.; Domann, F.; Van Houten, B.; Mostoslavsky, R.; Bultman, S.; Baccarelli, A.; et al. Mitochondria, Energetics, Epigenetics, and Cellular Responses to Stress. Environ. Health Perspect. 2014, 122, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.N.; Czajka, A. Is mitochondrial DNA content a potential biomarker of mitochondrial dysfunction? Mitochondrion 2013, 13, 481–492. [Google Scholar] [CrossRef]
- Tan, B.L.; Norhaizan, M.E.; Liew, W.-P.-P. Review Article Nutrients and Oxidative Stress: Friend or Foe? Oxidative Med. Cell. Longev. 2018, 2018, 9719584. [Google Scholar] [CrossRef]
- Pereira, A.C.; Martel, F. Oxidative stress in pregnancy and fertility pathologies. Cell Biol. Toxicol. 2014, 30, 301–312. [Google Scholar] [CrossRef]
- Görlach, A.; Dimova, E.Y.; Petry, A.; Martínez-Ruiz, A.; Hernansanz-Agustín, P.; Rolo, A.P.; Palmeira, C.M.; Kietzmann, T. Reactive oxygen species, nutrition, hypoxia and diseases: Problems solved? Redox Biol. 2015, 6, 372–385. [Google Scholar] [CrossRef]
- Wesselink, E.; Koekkoek, W.A.C.; Grefte, S.; Witkamp, R.F.; van Zanten, A.R.H. Feeding mitochondria: Potential role of nutritional components to improve critical illness convalescence. Clin. Nutr. 2018, 31, 1–14. [Google Scholar] [CrossRef]
- Díaz, M.; Aragonés, G.; Sánchez-Infantes, D.; Bassols, J.; Pérez-Cruz, M.; De Zegher, F.; Lopez-Bermejo, A.; Ibáñez, L. Mitochondrial DNA in placenta: Associations with fetal growth and superoxide dismutase activity. Horm. Res. Paediatr. 2014, 82, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity: Implications for metabolic syndrome, diabetes, hypertension, dyslipidemia, atherosclerosis, and cancer. Obes. Res. Clin. Pract. 2013, 7, e330–e341. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Scheibye-Knudsen, M.; Chua, K.F.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. Nuclear DNA damage signalling to mitochondria in ageing. Nat. Rev. Mol. Cell Biol. 2016, 17, 308–321. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef]
- Chiaratti, M.R.; Malik, S.; Diot, A.; Rapa, E.; Macleod, L.; Morten, K.; Vatish, M.; Boyd, R.; Poulton, J. Is Placental Mitochondrial Function a Regulator that Matches Fetal and Placental Growth to Maternal Nutrient Intake in the Mouse? PLoS ONE 2017, 10, e0130631. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Jauniaux, E. Pathophysiology of placental-derived fetal growth restriction. Am. J. Obstet. Gynecol. 2018, 218, S745–S761. [Google Scholar] [CrossRef] [PubMed]
- Krishna, U.; Bhalerao, S. Placental insufficiency and fetal growth restriction. J. Obstet. Gynaecol. India 2011, 61, 505–511. [Google Scholar] [CrossRef]
- Wang, S.; Lin, C.; Shi, H.; Xie, M.; Zhang, W.; Lv, J. Correlation of the Mitochondrial Activity of Two-Cell Embryos Produced In Vitro and the Two-Cell Block In Kunming and B6C3F1 Mice. Anat. Rec. 2009, 292, 661–669. [Google Scholar] [CrossRef]
- Yang, S.-G.; Park, H.-J.; Kim, J.-W.; Jung, J.-M.; Kim, M.-J.; Jegal, H.-G.; Kim, I.-S.; Kang, M.-J.; Wee, G.; Yang, H.-Y.; et al. Mito-TEMPO improves development competence by reducing superoxide in preimplantation porcine embryos. Sci. Rep. 2018, 8, 10130. [Google Scholar] [CrossRef]
- Gardner, D.K.; Harvey, A.J. Blastocyst metabolism. Reprod. Fertil. Dev. 2015, 27, 638–654. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.-H.; Lo, L.-M.; Chiu, T.-H.; Li, M.-J.; Yeh, Y.-L.; Chen, S.-F.; Hsieh, T.-T. A Longitudinal Study of Oxidative Stress and Antioxidant Status in Women With Uncomplicated Pregnancies Throughout Gestation. Reprod. Sci. 2010, 17, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Toescu, V.; Nuttall, S.L.; Martin, U.; Kendall, M.J.; Dunne, F. Oxidative stress and normal pregnancy. Clin. Endocrinol. (Oxf.) 2002, 57, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Mistry, H.D.; Williams, P.J. The Importance of Antioxidant Micronutrients in Pregnancy. Oxidative Med. Cell. Longev. 2011, 2011, 841749. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Hevner, K.; Abetew, D.; Sedensky, M.; Morgan, P.; Enquobahrie, D.A.; Williams, M.A. Mitochondrial DNA copy number and oxidative DNA damage in placental tissues from gestational diabetes and control pregnancies: A pilot study. Clin. Lab. 2013, 59, 655–660. [Google Scholar] [CrossRef]
- Mele, J.; Muralimanoharan, S.; Maloyan, A.; Myatt, L. Impaired mitochondrial function in human placenta with increased maternal adiposity. Am. J. Physiol. Metab. 2014, 307, E419–E425. [Google Scholar] [CrossRef]
- Myatt, L.; Maloyan, A. Obesity and Placental Function. Semin. Reprod. Med. 2016, 34, 42–49. [Google Scholar] [CrossRef]
- Holland, O.J.; Cuffe, J.S.M.; Dekker Nitert, M.; Callaway, L.; Kwan Cheung, K.A.; Radenkovic, F.; Perkins, A.V. Placental mitochondrial adaptations in preeclampsia associated with progression to term delivery. Cell Death Dis. 2018, 9, 1150. [Google Scholar] [CrossRef]
- Mandò, C.; Anelli, G.M.; Novielli, C.; Panina-Bordignon, P.; Massari, M.; Mazzocco, M.I.; Cetin, I. Impact of Obesity and Hyperglycemia on Placental Mitochondria. Oxidative Med. Cell. Longev. 2018, 2018, 2378189. [Google Scholar] [CrossRef]
- Novielli, C.; Mandò, C.; Tabano, S.; Anelli, G.M.; Fontana, L.; Antonazzo, P.; Miozzo, M.; Cetin, I. Mitochondrial DNA content and methylation in fetal cord blood of pregnancies with placental insufficiency. Placenta 2017, 55, 63–70. [Google Scholar] [CrossRef]
- Zhou, X.; Han, T.-L.; Chen, H.; Baker, P.N.; Qi, H.; Zhang, H. Impaired mitochondrial fusion, autophagy, biogenesis and dysregulated lipid metabolism is associated with preeclampsia. Exp. Cell Res. 2017, 359, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Vishnyakova, P.A.; Volodina, M.A.; Tarasova, N.V.; Marey, M.V.; Tsvirkun, D.V.; Vavina, O.V.; Khodzhaeva, Z.S.; Kan, N.E.; Menon, R.; Vysokikh, M.Y.; et al. Mitochondrial role in adaptive response to stress conditions in preeclampsia. Sci. Rep. 2016, 6, 32410. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.J.; Bartho, L.A.; Perkins, A.V.; Holland, O.J. Placental mitochondria and reactive oxygen species in the physiology and pathophysiology of pregnancy. Clin. Exp. Pharmacol. Physiol. 2020, 47, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Ausman, J.; Abbade, J.; Ermini, L.; Farrell, A.; Tagliaferro, A.; Post, M.; Caniggia, I. Ceramide-induced BOK promotes mitochondrial fission in preeclampsia. Cell Death Dis. 2018, 9, 298. [Google Scholar] [CrossRef] [PubMed]
- Pejznochová, M.; Tesarová, M.; Honzík, T.; Hansiková, H.; Magner, M.; Zeman, J. The developmental changes in mitochondrial DNA content per cell in human cord blood leukocytes during gestation. Physiol. Res. 2008, 57, 1–20. [Google Scholar]
- Lattuada, D.; Colleoni, F.; Martinelli, A.; Garretto, A.; Magni, R.; Radaelli, T.; Cetin, I. Higher Mitochondrial DNA Content in Human IUGR Placenta. Placenta 2008, 29, 1029–1033. [Google Scholar] [CrossRef]
- Sharma, N.; Pasala, M.S.; Prakash, A. Mitochondrial DNA: Epigenetics and environment. Environ. Mol. Mutagen. 2019, 60, 668–682. [Google Scholar] [CrossRef]
- Aon, M.A.; Cortassa, S.; Juhaszova, M.; Sollott, S.J. Mitochondrial health, the epigenome and healthspan. Clin. Sci. 2016, 130, 1285–1305. [Google Scholar] [CrossRef]
- Burgueño, A.L.; Cabrerizo, R.; Gonzales Mansilla, N.; Sookoian, S.; Pirola, C.J. Maternal high-fat intake during pregnancy programs metabolic-syndrome-related phenotypes through liver mitochondrial DNA copy number and transcriptional activity of liver PPARGC1A. J. Nutr. Biochem. 2013, 24, 6–13. [Google Scholar] [CrossRef]
- Jorgensen, W.; Gam, C.; Andersen, J.L.; Schjerling, P.; Scheibye-Knudsen, M.; Mortensen, O.H.; Grunnet, N.; Nielsen, M.O.; Quistorff, B. Changed mitochondrial function by pre- and/or postpartum diet alterations in sheep. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1349–E1357. [Google Scholar] [CrossRef]
- Zou, T.; Yu, B.; Yu, J.; Mao, X.; Zheng, P.; He, J.; Huang, Z.; Liu, Y.; Chen, D. Moderately decreased maternal dietary energy intake during pregnancy reduces fetal skeletal muscle mitochondrial biogenesis in the pigs. Genes Nutr. 2016, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Tian, F.-J.; Lin, Y. Oxidative Stress in Placenta: Health and Diseases. Biomed Res. Int. 2015, 2015, 293271. [Google Scholar] [CrossRef] [PubMed]
- Rani, V.; Deep, G.; Singh, R.K.; Palle, K.; Yadav, U.C.S. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016, 148, 183–193. [Google Scholar] [CrossRef]
- Muñoz, A.; Costa, M. Nutritionally mediated oxidative stress and inflammation. Oxidative Med. Cell. Longev. 2013, 2013, 610950. [Google Scholar] [CrossRef] [PubMed]
- Stowe, D.F.; Camara, A.K.S. Mitochondrial Reactive Oxygen Species Production in Excitable Cells: Modulators of Mitochondrial and Cell Function. Antioxid. Redox Signal. 2009, 11, 1373–1414. [Google Scholar] [CrossRef]
- Dan Dunn, J.; Alvarez, L.A.J.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef]
- Hernansanz-Agustín, P.; Izquierdo-Álvarez, A.; Sánchez-Gómez, F.J.; Ramos, E.; Villa-Piña, T.; Lamas, S.; Bogdanova, A.; Martínez-Ruiz, A. Acute hypoxia produces a superoxide burst in cells. Free Radic. Biol. Med. 2014, 71, 146–156. [Google Scholar] [CrossRef]
- Rudich, A.; Kanety, H.; Bashan, N. Adipose stress-sensing kinases: Linking obesity to malfunction. Trends Endocrinol. Metab. 2007, 18, 291–299. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Latouche, C.; Heywood, S.E.; Henry, S.L.; Ziemann, M.; Lazarus, R.; El-Osta, A.; Armitage, J.A.; Kingwell, B.A. Maternal Overnutrition Programs Changes in the Expression of Skeletal Muscle Genes That Are Associated with Insulin Resistance and Defects of Oxidative Phosphorylation in Adult Male Rat Offspring. J. Nutr. 2014, 144, 237–244. [Google Scholar] [CrossRef]
- Taylor, P.D.; McConnell, J.; Khan, I.Y.; Holemans, K.; Lawrence, K.M.; Asare-Anane, H.; Persaud, S.J.; Jones, P.M.; Petrie, L.; Hanson, M.A.; et al. Impaired glucose homeostasis and mitochondrial abnormalities in offspring of rats fed a fat-rich diet in pregnancy. AJP Regul. Integr. Comp. Physiol. 2005, 288, R134–R139. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Li, R.; Cong, R.; Yang, X.; Sun, Q.; Parvizi, N.; Zhao, R. Maternal Low-Protein Diet Affects Epigenetic Regulation of Hepatic Mitochondrial DNA Transcription in a Sex-Specific Manner in Newborn Piglets Associated with GR Binding to Its Promoter. PLoS ONE 2013, 8, e63855. [Google Scholar] [CrossRef] [PubMed]
- Gould, R.L.; Pazdro, R. Impact of Supplementary Amino Acids, Micronutrients, and Overall Diet on Glutathione Homeostasis. Nutrients 2019, 11, 1056. [Google Scholar] [CrossRef]
- Taravati, A.; Tohidi, F. Comprehensive analysis of oxidative stress markers and antioxidants status in preeclampsia. Taiwan. J. Obstet. Gynecol. 2018, 57, 779–790. [Google Scholar] [CrossRef] [PubMed]
- Hawk, S.N.; Lanoue, L.; Keen, C.L.; Kwik-Uribe, C.L.; Rucker, R.B.; Uriu-Adams, J.Y. Copper-Deficient Rat Embryos Are Characterized by Low Superoxide Dismutase Activity and Elevated Superoxide Anions1. Biol. Reprod. 2003, 68, 896–903. [Google Scholar] [CrossRef]
- Picco, S.J.; De Luca, J.C.; Mattioli, G.; Dulout, F.N. DNA damage induced by copper deficiency in cattle assessed by the Comet assay. Mutat. Res. Toxicol. Environ. Mutagen. 2001, 498, 1–6. [Google Scholar] [CrossRef]
- Lynch, S.M.; Balz, F.; Morrow, J.D.; Roberts, L.J.; Xu, A.; Jackson, T.; Reyna, R.; Klevay, L.M.; Vita, J.A.; Keaney, J.F. Vascular Superoxide Dismutase Deficiency Impairs Endothelial Vasodilator Function Through Direct Inactivation of Nitric Oxide and Increased Lipid Peroxidation. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2975–2981. [Google Scholar] [CrossRef]
- Lewandowska, M.; Sajdak, S.; Marciniak, W.; Lubiński, J. First Trimester Serum Copper or Zinc Levels, and Risk of Pregnancy-Induced Hypertension. Nutrients 2019, 11, 2479. [Google Scholar] [CrossRef]
- Özdemir, Y.; Börekci, B.; Levet, A.; Kurudirek, M. Assessment of trace element concentration distribution in human placenta by wavelength dispersive X-ray fluorescence: Effect of neonate weight and maternal age. Appl. Radiat. Isot. 2009, 67, 1790–1795. [Google Scholar] [CrossRef]
- Ota, E.; Mori, R.; Middleton, P.; Tobe-gai, R.; Mahomed, K.; Miyazaki, C.; Bhutta, Z.A. Zinc supplementation for improving pregnancy and infant outcome. Cochrane Database Syst. Rev. 2015, CD000230. [Google Scholar] [CrossRef]
- Xie, K.; Sheppard, A. Dietary Micronutrients Promote Neuronal Differentiation by Modulating the Mitochondrial-Nuclear Dialogue. BioEssays 2018, 40, 1800051. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.; Dong, L.; Holland, O.; Vanderlelie, J.; Pasdar, E.A.; Neuzil, J.; Perkins, A. V Selenium supplementation induces mitochondrial biogenesis in trophoblasts. Placenta 2015, 36, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Guo, D.; Gu, H.; Zhang, L.; Lv, S. Selenium and Preeclampsia: A Systematic Review and Meta-analysis. Biol. Trace Elem. Res. 2016, 171, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Vanderlelie, J.; Perkins, A.V.A. Selenium and preeclampsia: A global perspective. Pregnancy Hypertens. Int. J. Women’s Cardiovasc. Health 2011, 1, 213–224. [Google Scholar] [CrossRef]
- Lewandowska, M.; Więckowska, B.; Sajdak, S.; Lubiński, J. First Trimester Microelements and their Relationships with Pregnancy Outcomes and Complications. Nutrients 2020, 12, 1108. [Google Scholar] [CrossRef] [PubMed]
- Lewandowska, M.; Sajdak, S.; Lubiński, J. The Role of Early Pregnancy Maternal Selenium Levels on the Risk for Small-for-Gestational Age Newborns. Nutrients 2019, 11, 2298. [Google Scholar] [CrossRef] [PubMed]
- Cuellar-Rufino, S.; Navarro-Meza, M.; García-Solís, P.; Xochihua-Rosas, I.; Arroyo-Helguera, O. Iodine levels are associated with oxidative stress and antioxidant status in pregnant women with hypertensive disease. Nutr. Hosp. 2017, 34, 661–666. [Google Scholar] [CrossRef]
- Pearce, E.N.; Andersson, M.; Zimmermann, M.B. Global Iodine Nutrition: Where Do We Stand in 2013? Thyroid 2013, 23, 523–528. [Google Scholar] [CrossRef]
- Vidal, Z.E.O.; Rufino, S.C.; Tlaxcalteco, E.H.; Trejo, C.H.; Campos, R.M.; Meza, M.N.; Rodríguez, R.C.; Arroyo-Helguera, O. Oxidative Stress Increased in Pregnant Women with Iodine Deficiency. Biol. Trace Elem. Res. 2014, 157, 211–217. [Google Scholar] [CrossRef]
- Xiao, Y.; Sun, H.; Li, C.; Li, Y.; Peng, S.; Fan, C.; Teng, W.; Shan, Z. Effect of Iodine Nutrition on Pregnancy Outcomes in an Iodine-Sufficient Area in China. Biol. Trace Elem. Res. 2018, 182, 231–237. [Google Scholar] [CrossRef]
- Rumbold, A.; Ota, E.; Nagata, C.; Shahrook, S.; Crowther, C.A. Vitamin C supplementation in pregnancy. Cochrane Database Syst. Rev. 2015, CD004072. [Google Scholar] [CrossRef]
- Poston, L.; Igosheva, N.; Mistry, H.D.; Seed, P.T.; Shennan, A.H.; Rana, S.; Karumanchi, S.A.; Chappell, L.C. Role of oxidative stress and antioxidant supplementation in pregnancy disorders. Am. J. Clin. Nutr. 2011, 94, 1980S–1985S. [Google Scholar] [CrossRef] [PubMed]
- Rumbold, A.; Ota, E.; Hori, H.; Miyazaki, C.; Crowther, C.A. Vitamin E supplementation in pregnancy. Cochrane Database Syst. Rev. 2015, CD004069. [Google Scholar] [CrossRef]
- Asemi, Z.; Samimi, M.; Tabassi, Z.; Shakeri, H.; Esmaillzadeh, A. Vitamin D Supplementation Affects Serum High-Sensitivity C-Reactive Protein, Insulin Resistance, and Biomarkers of Oxidative Stress in Pregnant Women. J. Nutr. 2013, 143, 1432–1438. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.M.; Gillespie, S.L.; Thiele, D.K.; Ralph, J.L.; Ohm, J.E. Effects of Maternal Vitamin D Supplementation on the Maternal and Infant Epigenome. Breastfeed. Med. Off. J. Acad. Breastfeed. Med. 2018, 13, 371–380. [Google Scholar] [CrossRef]
- Liu, J.; Yu, B.; Mao, X.; He, J.; Yu, J.; Zheng, P.; Huang, Z.; Chen, D. Effects of intrauterine growth retardation and maternal folic acid supplementation on hepatic mitochondrial function and gene expression in piglets. Arch. Anim. Nutr. 2012, 66, 357–371. [Google Scholar] [CrossRef]
- Kalhan, S.C. One carbon metabolism in pregnancy: Impact on maternal, fetal and neonatal health. Mol. Cell. Endocrinol. 2016, 435, 48–60. [Google Scholar] [CrossRef]
- McKee, S.E.; Reyes, T.M. Effect of supplementation with methyl-donor nutrients on neurodevelopment and cognition: Considerations for future research. Nutr. Rev. 2018, 76, 497–511. [Google Scholar] [CrossRef]
- Friso, S.; Udali, S.; De Santis, D.; Choi, S.-W. One-carbon metabolism and epigenetics. Mol. Aspects Med. 2017, 54, 28–36. [Google Scholar] [CrossRef]
- Brosnan, M.E.; Tingley, G.; MacMillan, L.; Harnett, B.; Pongnopparat, T.; Marshall, J.D.; Brosnan, J.T. Plasma Formate Is Greater in Fetal and Neonatal Rats Compared with Their Mothers. J. Nutr. 2020, 150, 1068–1075. [Google Scholar] [CrossRef]
- Momb, J.; Lewandowski, J.P.; Bryant, J.D.; Fitch, R.; Surman, D.R.; Vokes, S.A.; Appling, D.R. Deletion of Mthfd1l causes embryonic lethality and neural tube and craniofacial defects in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Brosnan, M.E.; Brosnan, J.T. Formate: The Neglected Member of One-Carbon Metabolism. Annu. Rev. Nutr. 2016, 36, 369–388. [Google Scholar] [CrossRef]
- Taormina, G.; Russo, A.; Latteri, M.A.; Mirisola, M.G. Mitochondrion at the Crossroad Between Nutrients and Epigenome. Front. Endocrinol. (Lausanne) 2019, 10, 673. [Google Scholar] [CrossRef] [PubMed]
- Steegers-Theunissen, R.P.; Obermann-Borst, S.A.; Kremer, D.; Lindemans, J.; Siebel, C.; Steegers, E.A.; Slagboom, P.E.; Heijmans, B.T. Periconceptional Maternal Folic Acid Use of 400 µg per Day Is Related to Increased Methylation of the IGF2 Gene in the Very Young Child. PLoS ONE 2009, 4, e7845. [Google Scholar] [CrossRef]
- Joubert, B.R.; den Dekker, H.T.; Felix, J.F.; Bohlin, J.; Ligthart, S.; Beckett, E.; Tiemeier, H.; van Meurs, J.B.; Uitterlinden, A.G.; Hofman, A.; et al. Maternal plasma folate impacts differential DNA methylation in an epigenome-wide meta-analysis of newborns. Nat. Commun. 2016, 7, 10577. [Google Scholar] [CrossRef]
- Adaikalakoteswari, A.; Vatish, M.; Alam, M.T.; Ott, S.; Kumar, S.; Saravanan, P. Low Vitamin B12 in Pregnancy Is Associated With Adipose-Derived Circulating miRs Targeting PPARγ and Insulin Resistance. J. Clin. Endocrinol. Metab. 2017, 102, 4200–4209. [Google Scholar] [CrossRef]
- McCullough, L.E.; Miller, E.E.; Mendez, M.A.; Murtha, A.P.; Murphy, S.K.; Hoyo, C. Maternal B vitamins: Effects on offspring weight and DNA methylation at genomically imprinted domains. Clin. Epigenet. 2016, 8, 8. [Google Scholar] [CrossRef]
- Nye, M.D.; King, K.E.; Darrah, T.H.; Maguire, R.; Jima, D.D.; Huang, Z.; Mendez, M.A.; Fry, R.C.; Jirtle, R.L.; Murphy, S.K.; et al. Maternal blood lead concentrations, DNA methylation of MEG3 DMR regulating the DLK1/MEG3 imprinted domain and early growth in a multiethnic cohort. Environ. Epigenet. 2016, 2, dvv009. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Salas, P.; Moore, S.E.; Baker, M.S.; Bergen, A.W.; Cox, S.E.; Dyer, R.A.; Fulford, A.J.; Guan, Y.; Laritsky, E.; Silver, M.J.; et al. Maternal nutrition at conception modulates DNA methylation of human metastable epialleles. Nat. Commun. 2014, 5, 3746. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, S.; Duca, R.C.; Devlieger, R.; Freson, K.; Straetmans, D.; Van Herck, E.; Huybrechts, I.; Koppen, G.; Godderis, L. Maternal Methyl-Group Donor Intake and Global DNA (Hydroxy)Methylation before and during Pregnancy. Nutrients 2016, 8, 474. [Google Scholar] [CrossRef]
- Plumptre, L.; Tammen, S.A.; Sohn, K.-J.; Masih, S.P.; Visentin, C.E.; Aufreiter, S.; Malysheva, O.; Schroder, T.H.; Ly, A.; Berger, H.; et al. Maternal and Cord Blood Folate Concentrations Are Inversely Associated with Fetal DNA Hydroxymethylation, but Not DNA Methylation, in a Cohort of Pregnant Canadian Women. J. Nutr. 2019, 150, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-Y.; Liu, S.-M.; Zhang, Y.-Z. Maternal Folic Acid Supplementation Mediates Offspring Health via DNA Methylation. Reprod. Sci. 2020, 27, 963–976. [Google Scholar] [CrossRef] [PubMed]
- Boeke, C.E.; Gillman, M.W.; Hughes, M.D.; Rifas-Shiman, S.L.; Villamor, E.; Oken, E. Choline Intake During Pregnancy and Child Cognition at Age 7 Years. Am. J. Epidemiol. 2012, 177, 1338–1347. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.T.F.; Dyer, R.A.; King, D.J.; Richardson, K.J.; Innis, S.M. Early Second Trimester Maternal Plasma Choline and Betaine Are Related to Measures of Early Cognitive Development in Term Infants. PLoS ONE 2012, 7, e43448. [Google Scholar] [CrossRef] [PubMed]
- Kuszak, A.J.; Espey, M.G.; Falk, M.J.; Holmbeck, M.A.; Manfredi, G.; Shadel, G.S.; Vernon, H.J.; Zolkipli-Cunningham, Z. Nutritional Interventions for Mitochondrial OXPHOS Deficiencies: Mechanisms and Model Systems. Annu. Rev. Pathol. Mech. Dis. 2018, 13, 163–191. [Google Scholar] [CrossRef] [PubMed]
- Makarov, M.V.; Trammell, S.A.J.; Migaud, M.E. The chemistry of the vitamin B3 metabolome. Biochem. Soc. Trans. 2019, 47, 131–147. [Google Scholar] [CrossRef]
- Ames, B.N. Low micronutrient intake may accelerate the degenerative diseases of aging through allocation of scarce micronutrients by triage. Proc. Natl. Acad. Sci. USA 2006, 103, 17589–17594. [Google Scholar] [CrossRef]
- Zempleni, J.; Wijeratne, S.S.K.; Hassan, Y.I. Biotin. Biofactors 2009, 35, 36–46. [Google Scholar] [CrossRef]
- Mock, D.M.; Quirk, J.G.; Mock, N.I. Marginal biotin deficiency during normal pregnancy. Am. J. Clin. Nutr. 2002, 75, 295–299. [Google Scholar] [CrossRef]
- Mock, D.M. Marginal biotin deficiency is common in normal human pregnancy and is highly teratogenic in mice. J. Nutr. 2009, 139, 154–157. [Google Scholar] [CrossRef]
- Fujiwara, T.; Harigae, H. Biology of Heme in Mammalian Erythroid Cells and Related Disorders. Biomed Res. Int. 2015, 2015, 278536. [Google Scholar] [CrossRef] [PubMed]
- von Hardenberg, A.; Maack, C. Mitochondrial Therapies in Heart Failure. In Heart Failure; Bauersachs, J., Butler, J., Sandner, P., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 491–514. ISBN 978-3-319-59659-4. [Google Scholar]
- Teran, E.; Racines-Orbe, M.; Vivero, S.; Escudero, C.; Molina, G.; Calle, A. Preeclampsia is associated with a decrease in plasma coenzyme Q10 levels. Free Radic. Biol. Med. 2003, 35, 1453–1456. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Pan, J.-R.; Zhang, Y.-Z. CoQ10 alleviate preeclampsia symptoms by enhancing the function of mitochondria in the placenta of pregnant rats with preeclampsia. Hypertens. Pregnancy 2019, 38, 217–222. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Preventing and Controlling Micronutrient Deficiencies in Populations Affected by An Emergency; World Health Organization: Geneva, Switzerland, 2007. [Google Scholar]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Cano, A.M.; Calzada-Mendoza, C.C.; Estrada-Gutierrez, G.; Mendoza-Ortega, J.A.; Perichart-Perera, O. Nutrients, Mitochondrial Function, and Perinatal Health. Nutrients 2020, 12, 2166. https://doi.org/10.3390/nu12072166
Rodríguez-Cano AM, Calzada-Mendoza CC, Estrada-Gutierrez G, Mendoza-Ortega JA, Perichart-Perera O. Nutrients, Mitochondrial Function, and Perinatal Health. Nutrients. 2020; 12(7):2166. https://doi.org/10.3390/nu12072166
Chicago/Turabian StyleRodríguez-Cano, Ameyalli M, Claudia C Calzada-Mendoza, Guadalupe Estrada-Gutierrez, Jonatan A Mendoza-Ortega, and Otilia Perichart-Perera. 2020. "Nutrients, Mitochondrial Function, and Perinatal Health" Nutrients 12, no. 7: 2166. https://doi.org/10.3390/nu12072166
APA StyleRodríguez-Cano, A. M., Calzada-Mendoza, C. C., Estrada-Gutierrez, G., Mendoza-Ortega, J. A., & Perichart-Perera, O. (2020). Nutrients, Mitochondrial Function, and Perinatal Health. Nutrients, 12(7), 2166. https://doi.org/10.3390/nu12072166