Does Calorie Restriction Modulate Inflammaging via FoxO Transcription Factors?


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
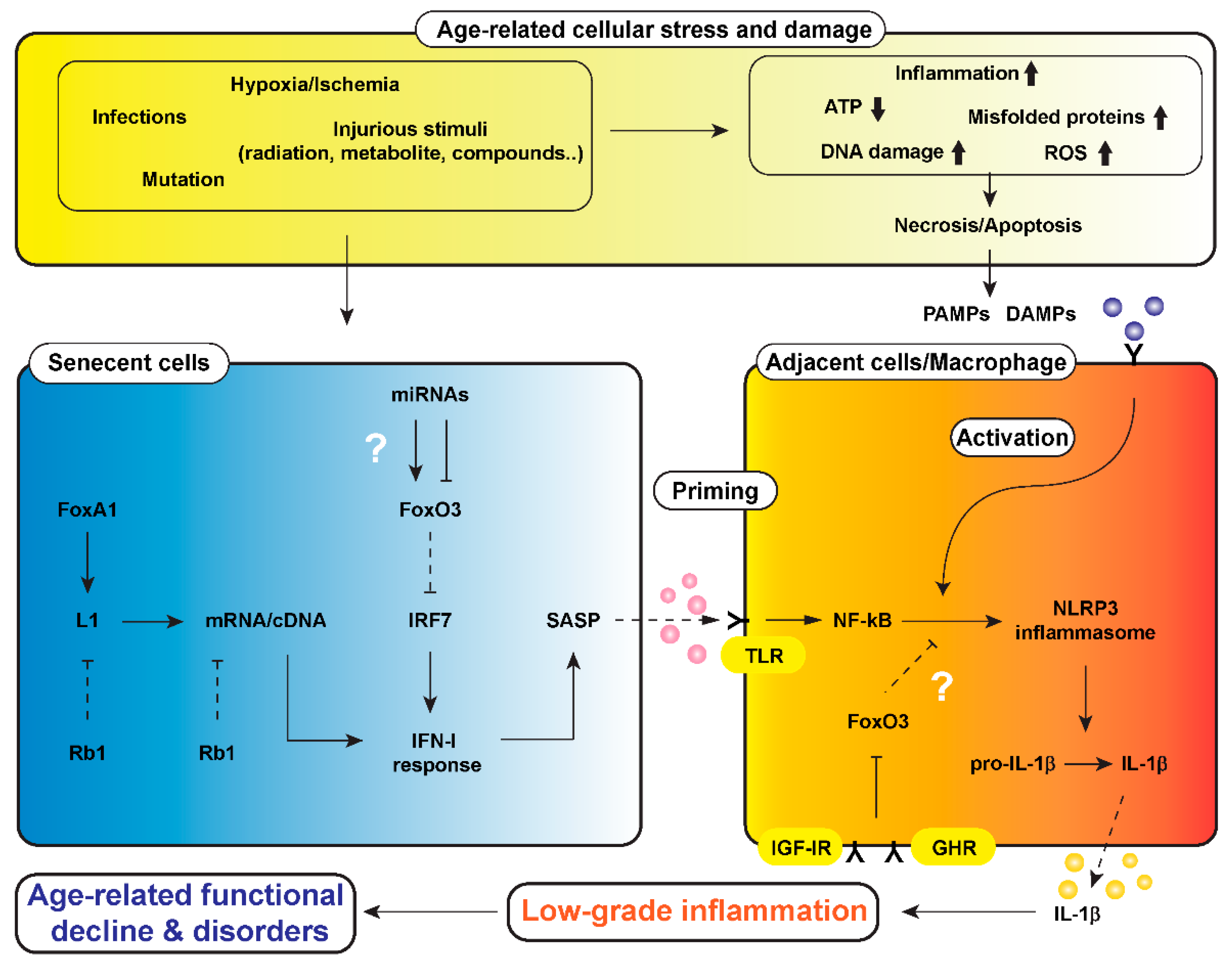
2. Chronic Low-Level Inflammation and Aging
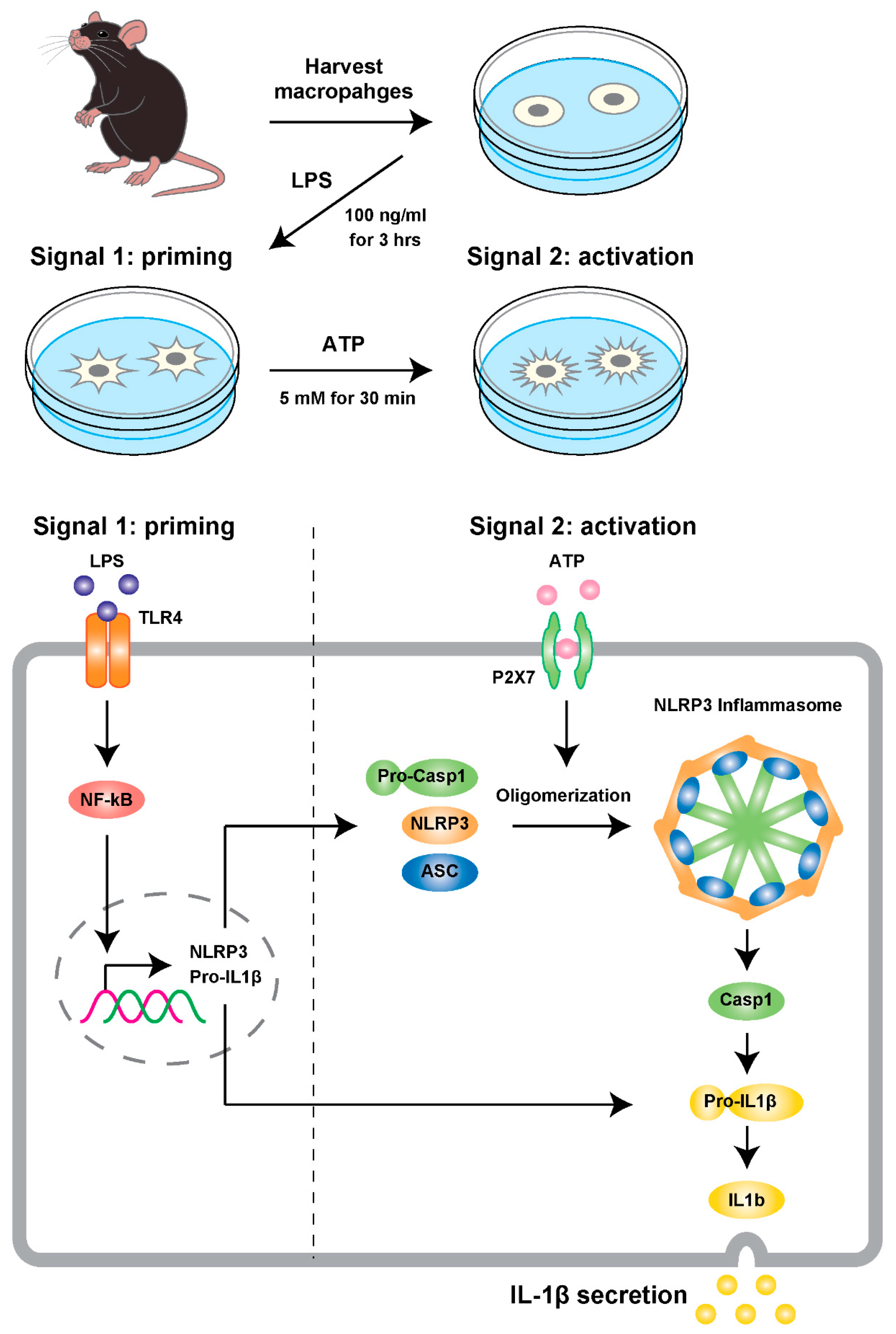
3. Activation of Inflammasome in the Aging Process
4. Inflammaging in the Brain
4.1. Inflammaging in Neural Stem/Progenitor Cells
4.2. Effects of CR on Adult Neurogenesis: An Implication of FoxO3
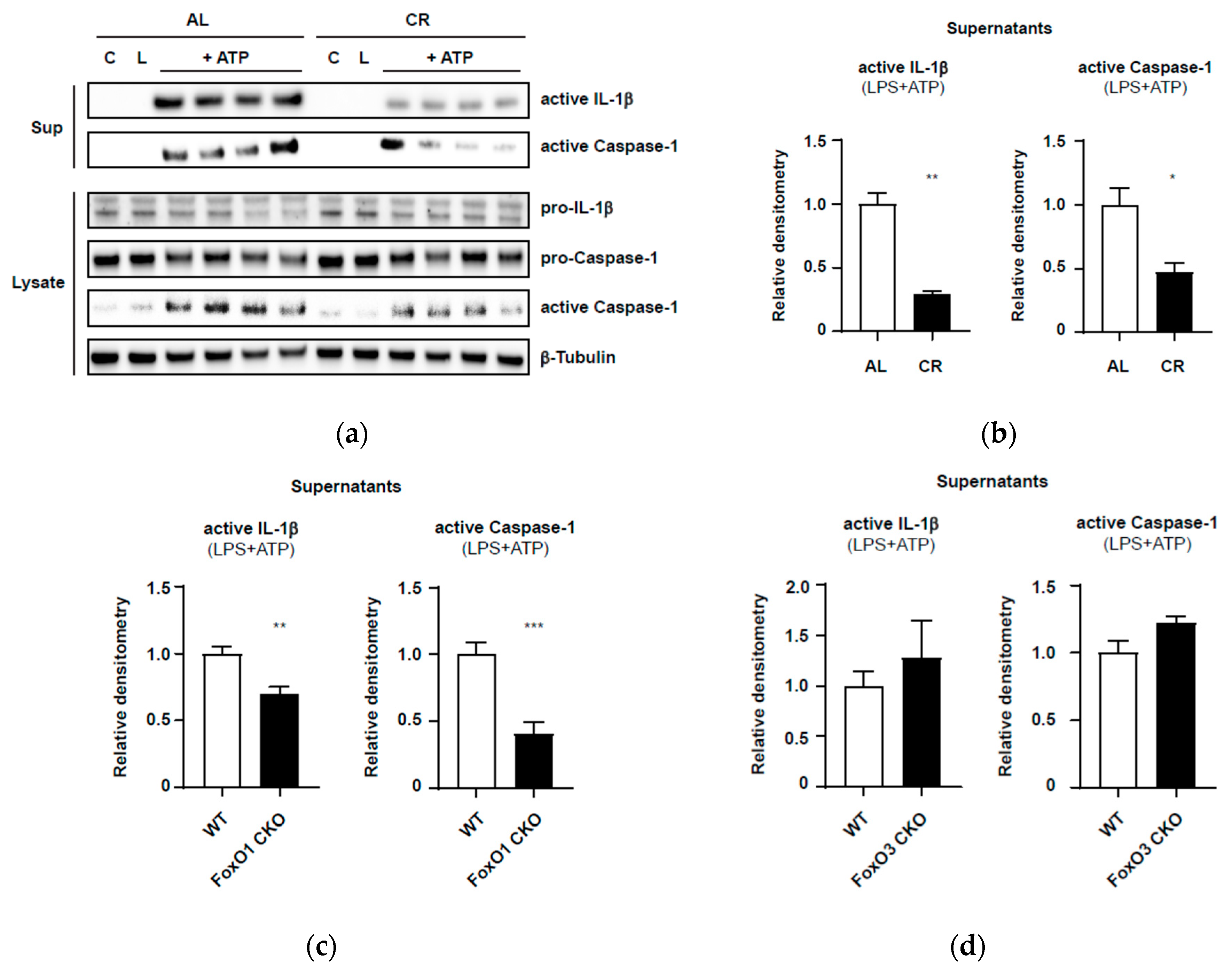
5. Modulation of Inflammaging by Calorie Restriction (CR) via NLRP3 Inflammasome: A Possible Connection with FoxOs
6. MicroRNA (miRNAs) in Inflammasomes and Aging
7. Development of an Assay and Mouse Models for Testing the Hypothesis
8. Perspective
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Calorie-Restricted Mouse Model and Generation of Myeloid-Lineage-Specific Foxo1 and Foxo3a Conditional Knockout Mice
Appendix A.2. Cell-Based NLRP3 Inflammasome Model
Appendix A.2.1. PEC Macrophage Isolation and NLRP3 Inflammasome Activation Model
Appendix A.2.2. Bone Marrow-Derived Macrophage Isolation
Appendix A.2.3. Immunoblot Analysis
References
- Bloom, D.E.; Chatterji, S.; Kowal, P.; Lloyd-Sherlock, P.; McKee, M.; Rechel, B.; Rosenberg, L.; Smith, J.P. Macroeconomic implications of population ageing and selected policy responses. Lancet 2015, 385, 649–657. [Google Scholar] [CrossRef]
- Weindruch, R.; Sohal, R.S. Caloric intake and aging. N. Engl. J. Med. 1997, 337, 986–994. [Google Scholar] [CrossRef]
- Mattison, J.A.; Colman, R.J.; Beasley, T.M.; Allison, D.B.; Kemnitz, J.W.; Roth, G.S.; Ingram, D.K.; Weindruch, R.; De Cabo, R.; Anderson, R.M. Caloric restriction improves health and survival of rhesus monkeys. Nat. Commun. 2017, 8, 14063. [Google Scholar] [CrossRef]
- Dunn, S.E.; Kari, F.W.; French, J.; Leininger, J.R.; Travlos, G.; Wilson, R.; Barrett, J.C. Dietary restriction reduces insulin-like growth factor I levels, which modulates apoptosis, cell proliferation, and tumor progression in p53- deficient mice. Cancer Res. 1997, 57, 4667–4672. [Google Scholar]
- Sonntag, W.E.; Lynch, C.D.; Cefalu, W.T.; Ingram, R.L.; Bennett, S.A.; Thornton, P.L.; Khan, A.S. Pleiotropic effects of growth hormone and insulin-like growth factor (IGF)-1 on biological aging: Inferences from moderate caloric-restricted animals. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 1999, 54, 521–538. [Google Scholar] [CrossRef]
- Sharples, A.P.; Hughes, D.C.; Deane, C.S.; Saini, A.; Selman, C.; Stewart, C.E. Longevity and skeletal muscle mass: The role of IGF signalling, the sirtuins, dietary restriction and protein intake. Aging Cell 2015, 14, 511–523. [Google Scholar] [CrossRef]
- Shimokawa, I. Growth Hormone and IGF-1 Axis in Aging and Longevity. In Hormones in Ageing and Longevity; Springer: Cham, Switzerland, 2017; pp. 91–106. [Google Scholar]
- Komatsu, T.; Park, S.; Hayashi, H.; Mori, R.; Yamaza, H.; Shimokawa, I. Mechanisms of calorie restriction: A review of genes required for the life-extending and tumor-inhibiting effects of calorie restriction. Nutrients 2019, 11, 3068. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; de Luca, M.; Ottaviani, E.; de Benedictis, G. Inflamm-aging: An Evolutionary Perspective on Immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Shishodia, S.; Sandur, S.K.; Pandey, M.K.; Sethi, G. Inflammation and cancer: How hot is the link? Biochem. Pharmacol. 2006, 72, 1605–1621. [Google Scholar] [CrossRef]
- Cesari, M.; Penninx, B.W.J.H.; Newman, A.B.; Kritchevsky, S.B.; Nicklas, B.J.; Sutton-Tyrrell, K.; Rubin, S.M.; Ding, J.; Simonsick, E.M.; Harris, T.B.; et al. Inflammatory Markers and Onset of Cardiovascular Events: Results from the Health ABC Study. Circulation 2003, 108, 2317–2322. [Google Scholar] [CrossRef]
- Kalogeropoulos, A.; Georgiopoulou, V.; Psaty, B.M.; Rodondi, N.; Smith, A.L.; Harrison, D.G.; Liu, Y.; Hoffmann, U.; Bauer, D.C.; Newman, A.B.; et al. Inflammatory Markers and Incident Heart Failure Risk in Older Adults. The Health ABC (Health, Aging, and Body Composition) Study. J. Am. Coll. Cardiol. 2010, 55, 2129–2137. [Google Scholar] [CrossRef]
- Rodondi, N.; Marques-Vidal, P.; Butler, J.; Sutton-Tyrrell, K.; Cornuz, J.; Satterfield, S.; Harris, T.; Bauer, D.C.; Ferrucci, L.; Vittinghoff, E.; et al. Markers of atherosclerosis and inflammation for prediction of coronary heart disease in older adults. Am. J. Epidemiol. 2010, 171, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Huuskonen, J.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing Res. Rev. 2008, 7, 83–105. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Bang, E.; Jung, H.J.; Noh, S.G.; Yu, B.P.; Choi, Y.J.; Chung, H.Y. Anti-aging effects of calorie restriction (CR) and CR mimetics based on the senoinflammation concept. Nutrients 2020, 12, 422. [Google Scholar] [CrossRef]
- Campisi, J.; D’Adda Di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Franceschi, C.; Zaikin, A.; Gordleeva, S.; Ivanchenko, M.; Bonifazi, F.; Storci, G.; Bonafè, M. Inflammaging 2018: An update and a model. Semin. Immunol. 2018, 40, 1–5. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; Van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16 Ink4a-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T.; Zhu, Y.; Niedernhofer, L.J.; Robbins, P.D. The Clinical Potential of Senolytic Drugs. J. Am. Geriatr. Soc. 2017, 65, 2297–2301. [Google Scholar] [CrossRef]
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K.; Bohr, V.A.; Misra Sen, J.; Gorospe, M.; et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Klein, D.; Kerscher, S.; West, B.L.; Weis, J.; Katona, I.; Martini, R. Macrophage depletion ameliorates peripheral neuropathy in aging mice. J. Neurosci. 2018, 38, 4610–4620. [Google Scholar] [CrossRef] [PubMed]
- Bouchlaka, M.N.; Sckisel, G.D.; Chen, M.; Mirsoian, A.; Zamora, A.E.; Maverakis, E.; Wilkins, D.E.C.; Alderson, K.L.; Hsiao, H.H.; Weiss, J.M.; et al. Aging predisposes to acute inflammatory induced pathology after tumor immunotherapy. J. Exp. Med. 2013, 210, 2223–2237. [Google Scholar] [CrossRef]
- Hall, B.M.; Balan, V.; Gleiberman, A.S.; Strom, E.; Krasnov, P.; Virtuoso, L.P.; Rydkina, E.; Vujcic, S.; Balan, K.; Gitlin, I.; et al. Aging of mice is associated with p16(Ink4a)- and β-galactosidasepositive macrophage accumulation that can be induced in young mice by senescent cells. Aging 2016, 8, 1294–1315. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune–metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef]
- Dinarello, C.A. Immunological and Inflammatory Functions of the Interleukin-1 Family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Martinon, F.; Mayor, A.; Tschopp, J. The Inflammasomes: Guardians of the Body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef]
- Youm, Y.H.; Grant, R.W.; McCabe, L.R.; Albarado, D.C.; Nguyen, K.Y.; Ravussin, A.; Pistell, P.; Newman, S.; Carter, R.; Laque, A.; et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013, 18, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Niraula, A.; Sheridan, J.F.; Godbout, J.P. Microglia Priming with Aging and Stress. Neuropsychopharmacology 2017, 42, 318–333. [Google Scholar] [CrossRef] [PubMed]
- Cavallucci, V.; Fidaleo, M.; Pani, G. Neural Stem Cells and Nutrients: Poised Between Quiescence and Exhaustion. Trends Endocrinol. Metab. 2016, 27, 756–769. [Google Scholar] [CrossRef] [PubMed]
- Obernier, K.; Alvarez-Buylla, A. Neural stem cells: Origin, heterogeneity and regulation in the adult mammalian brain. Development 2019, 146. [Google Scholar] [CrossRef]
- Ming, G.-L.; Song, H. Adult Neurogenesis in the Mammalian Brain: Significant Answers and Significant Questions. Neuron 2011, 70, 687–702. [Google Scholar] [CrossRef]
- Li, J.; Tang, Y.; Cai, D. IKKβ/NF-κB disrupts adult hypothalamic neural stem cells to mediate a neurodegenerative mechanism of dietary obesity and pre-diabetes. Nat. Cell Biol. 2012, 14, 999–1012. [Google Scholar] [CrossRef]
- Zhang, G.; Li, J.; Purkayastha, S.; Tang, Y.; Zhang, H.; Yin, Y.; Li, B.; Liu, G.; Cai, D. Hypothalamic programming of systemic ageing involving IKK-β, NF-κB and GnRH. Nature 2013, 497, 211–216. [Google Scholar] [CrossRef]
- Zhang, Y.; Kim, M.S.; Jia, B.; Yan, J.; Zuniga-Hertz, J.P.; Han, C.; Cai, D. Hypothalamic stem cells control ageing speed partly through exosomal miRNAs. Nature 2017, 548, 52–57. [Google Scholar] [CrossRef]
- Lee, D.A.; Bedont, J.L.; Pak, T.; Wang, H.; Song, J.; Miranda-Angulo, A.; Takiar, V.; Charubhumi, V.; Balordi, F.; Takebayashi, H.; et al. Tanycytes of the hypothalamic median eminence form a diet-responsive neurogenic niche. Nat. Neurosci. 2012, 15, 700–702. [Google Scholar] [CrossRef]
- Gould, E. How widespread is adult neurogenesis in mammals? Nat. Rev. Neurosci. 2007, 8, 481–488. [Google Scholar] [CrossRef]
- Fan, Z.; Lu, M.; Qiao, C.; Zhou, Y.; Ding, J.H.; Hu, G. MicroRNA-7 Enhances Subventricular Zone Neurogenesis by Inhibiting NLRP3/Caspase-1 Axis in Adult Neural Stem Cells. Mol. Neurobiol. 2016, 53, 7057–7069. [Google Scholar] [CrossRef]
- Mattson, M.P. Gene-Diet Interactions in Brain Aging and Neurodegenerative Disorders. Ann. Intern. Med. 2003, 139, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, T.; Chiba, T.; Yamaza, H.; Yamashita, K.; Shimada, A.; Hoshiyama, Y.; Henmi, T.; Ohtani, H.; Higami, Y.; de Cabo, R.; et al. Manipulation of caloric content but not diet composition, attenuates the deficit in learning and memory of senescence-accelerated mouse strain P8. Exp. Gerontol. 2008, 43, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.K.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.B.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W.; et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009, 325, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Duan, W.; Long, J.M.; Ingram, D.K.; Mattson, M.P. Dietary restriction increases the number of newly generated neural cells, and BDNF expression, in the dentate gyrus of rats. J. Mol. Neurosci. 2000, 15, 99–108. [Google Scholar] [CrossRef]
- Lee, J.; Duan, W.; Mattson, M.P. Evidence that brain-derived neurotrophic factor is required for basal neurogenesis and mediates, in part, the enhancement of neurogenesis by dietary restriction in the hippocampus of adult mice. J. Neurochem. 2002, 82, 1367–1375. [Google Scholar] [CrossRef] [PubMed]
- Bondolfi, L.; Ermini, F.; Long, J.M.; Ingram, D.K.; Jucker, M. Impact of age and caloric restriction on neurogenesis in the dentate gyrus of C57BL/6 mice. Neurobiol. Aging 2004, 25, 333–340. [Google Scholar] [CrossRef]
- Hornsby, A.K.E.; Redhead, Y.T.; Rees, D.J.; Ratcliff, M.S.G.; Reichenbach, A.; Wells, T.; Francis, L.; Amstalden, K.; Andrews, Z.B.; Davies, J.S. Short-term calorie restriction enhances adult hippocampal neurogenesis and remote fear memory in a Ghsr-dependent manner. Psychoneuroendocrinology 2016, 63, 198–207. [Google Scholar] [CrossRef]
- Apple, D.M.; Mahesula, S.; Fonseca, R.S.; Zhu, C.; Kokovay, E. Calorie restriction protects neural stem cells from age-related deficits in the subventricular zone. Aging 2019, 11, 115–126. [Google Scholar] [CrossRef]
- Shimokawa, I.; Komatsu, T.; Hayashi, N.; Kim, S.E.; Kawata, T.; Park, S.; Hayashi, H.; Yamaza, H.; Chiba, T.; Mori, R. The life-extending effect of dietary restriction requires Foxo3 in mice. Aging Cell 2015, 14, 707–709. [Google Scholar] [CrossRef]
- Renault, V.M.; Rafalski, V.A.; Morgan, A.A.; Salih, D.A.M.; Brett, J.O.; Webb, A.E.; Villeda, S.A.; Thekkat, P.U.; Guillerey, C.; Denko, N.C.; et al. FoxO3 Regulates Neural Stem Cell Homeostasis. Cell Stem Cell 2009, 5, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Paik, J.H.; Kollipara, R.; Chu, G.; Ji, H.; Xiao, Y.; Ding, Z.; Miao, L.; Tothova, Z.; Horner, J.W.; Carrasco, D.R.; et al. FoxOs Are Lineage-Restricted Redundant Tumor Suppressors and Regulate Endothelial Cell Homeostasis. Cell 2007, 128, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Strassburger, U.; Schips, T.G.; Maier, H.J.; Kloiber, K.; Mannella, F.; Braunstein, K.E.; Holzmann, K.; Ushmorov, A.; Liebau, S.; Boeckers, T.M.; et al. Expression of constitutively active FoxO3 in murine forebrain leads to a loss of neural progenitors. FASEB J. 2012, 26, 4990–5001. [Google Scholar] [CrossRef] [PubMed]
- Meydani, S.N.; Das, S.K.; Pieper, C.F.; Lewis, M.R.; Klein, S.; Dixit, V.D.; Gupta, A.K.; Villareal, D.T.; Bhapkar, M.; Huang, M.; et al. Long-term moderate calorie restriction inhibits inflammation without impairing cell-mediated immunity: A randomized controlled trial in non-obese humans. Aging 2016, 8, 1416–1431. [Google Scholar] [CrossRef]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–189. [Google Scholar] [CrossRef]
- Brandhorst, S.; Choi, I.Y.; Wei, M.; Cheng, C.W.; Sedrakyan, S.; Navarrete, G.; Dubeau, L.; Yap, L.P.; Park, R.; Vinciguerra, M.; et al. A Periodic Diet that Mimics Fasting Promotes Multi-System Regeneration, Enhanced Cognitive Performance, and Healthspan. Cell Metab. 2015, 22, 86–99. [Google Scholar] [CrossRef]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef]
- Traba, J.; Kwarteng-Siaw, M.; Okoli, T.C.; Li, J.; Huffstutler, R.D.; Bray, A.; Waclawiw, M.A.; Han, K.; Pelletier, M.; Sauve, A.A.; et al. Fasting and refeeding differentially regulate NLRP3 inflammasome activation in human subjects. J. Clin. Investig. 2015, 125, 4592–4600. [Google Scholar] [CrossRef]
- Traba, J.; Geiger, S.S.; Kwarteng-Siaw, M.; Han, K.; Ra, O.H.; Siegel, R.M.; Gius, D.; Sack, M.N. Prolonged fasting suppresses mitochondrial NLRP3 inflammasome assembly and activation via SIRT3-mediated activation of superoxide dismutase 2. J. Biol. Chem. 2017, 292, 12153–12164. [Google Scholar] [CrossRef]
- He, M.; Chiang, H.H.; Luo, H.; Zheng, Z.; Qiao, Q.; Wang, L.; Tan, M.; Ohkubo, R.; Mu, W.C.; Zhao, S.; et al. An Acetylation Switch of the NLRP3 Inflammasome Regulates Aging-Associated Chronic Inflammation and Insulin Resistance. Cell Metab. 2020, 31, 580–591.e5. [Google Scholar] [CrossRef]
- Kenyon, C.; Chang, J.; Gensch, E.; Rudner, A.; Tabtiang, R. A C. elegans mutant that lives twice as long as wild type. Nature 1993, 366, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Ogg, S.; Paradis, S.; Gottlieb, S.; Patterson, G.I.; Lee, L.; Tissenbaum, H.A.; Ruvkun, G. The fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 1997, 389, 994–999. [Google Scholar] [CrossRef]
- Gami, M.S.; Wolkow, C.A. Studies of Caenorhabditis elegans DAF-2/insulin signaling reveal targets for pharmacological manipulation of lifespan. Aging Cell 2006, 5, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Finch, C.E. Evolutionary medicine: From dwarf model systems to healthy centenarians? Science 2003, 299, 1342–1346. [Google Scholar] [CrossRef]
- Burgering, B.M.T.; Kops, G.J.P.L. Cell cycle and death control: Long live Forkheads. Trends Biochem. Sci. 2002, 27, 352–360. [Google Scholar] [CrossRef]
- Greer, E.L.; Brunet, A. Different dietary restriction regimens extend lifespan by both independent and overlapping genetic pathways in C. elegans. Aging Cell 2009, 8, 113–127. [Google Scholar] [CrossRef]
- Yamaza, H.; Komatsu, T.; Wakita, S.; Kijogi, C.; Park, S.; Hayashi, H.; Chiba, T.; Mori, R.; Furuyama, T.; Mori, N.; et al. FoxO1 is involved in the antineoplastic effect of calorie restriction. Aging Cell 2010, 9, 372–382. [Google Scholar] [CrossRef]
- Greer, E.L.; Brunet, A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 2005, 24, 7410–7425. [Google Scholar] [CrossRef]
- Huang, C.R.L.; Burns, K.H.; Boeke, J.D. Active Transposition in Genomes. Annu. Rev. Genet. 2012, 46, 651–675. [Google Scholar] [CrossRef]
- De Cecco, M.; Ito, T.; Petrashen, A.P.; Elias, A.E.; Skvir, N.J.; Criscione, S.W.; Caligiana, A.; Brocculi, G.; Adney, E.M.; Boeke, J.D.; et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 2019, 566, 73–78. [Google Scholar] [CrossRef]
- Chen, L.; Song, Y.; He, L.; Wan, X.; Lai, L.; Dai, F.; Liu, Y.; Wang, Q. MicroRNA-223 promotes type i interferon production in antiviral innate immunity by targeting forkhead box protein O3 (FOXO3). J. Biol. Chem. 2016, 291, 14706–14716. [Google Scholar] [CrossRef]
- Litvak, V.; Ratushny, A.V.; Lampano, A.E.; Schmitz, F.; Huang, A.C.; Raman, A.; Rust, A.G.; Bergthaler, A.; Aitchison, J.D.; Aderem, A. A FOXO3-IRF7 gene regulatory circuit limits inflammatory sequelae of antiviral responses. Nature 2012, 490, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X.; Zhang, X.; Wang, J.; Ma, Y.; Zhang, L.; Cao, X. RNA-binding protein YTHDF3 suppresses interferon-dependent antiviral responses by promoting FOXO3 translation. Proc. Natl. Acad. Sci. USA 2019, 116, 976–981. [Google Scholar] [CrossRef] [PubMed]
- Hattori, N. Expression, regulation and biological actions of growth hormone (GH) and ghrelin in the immune system. Growth Horm. IGF Res. 2009, 19, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Spadaro, O.; Goldberg, E.L.; Camell, C.D.; Youm, Y.H.; Kopchick, J.J.; Nguyen, K.Y.; Bartke, A.; Sun, L.Y.; Dixit, V.D. Growth Hormone Receptor Deficiency Protects against Age-Related NLRP3 Inflammasome Activation and Immune Senescence. Cell Rep. 2016, 14, 1571–1580. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: MicroRNA biogenesis pathways and their regulation. Nat. Cell Biol. 2009, 11, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef]
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011, 12, 99–110. [Google Scholar] [CrossRef]
- Stenvang, J.; Petri, A.; Lindow, M.; Obad, S.; Kauppinen, S. Inhibition of microRNA function by antimiR oligonucleotides. Silence 2012, 3, 1. [Google Scholar] [CrossRef]
- Thorsen, S.B.; Obad, S.; Jensen, N.F.; Stenvang, J.; Kauppinen, S. The therapeutic potential of MicroRNAs in cancer. Cancer J. 2012, 18, 275–284. [Google Scholar] [CrossRef]
- Van Rooij, E.; Purcell, A.L.; Levin, A.A. Developing MicroRNA therapeutics. Circ. Res. 2012, 110, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Rooij, E.; Kauppinen, S. Development of micro RNA therapeutics is coming of age. EMBO Mol. Med. 2014, 6, 851–864. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Carmell, M.A.; Rivas, F.V.; Marsden, C.G.; Thomson, J.M.; Song, J.J.; Hammond, S.M.; Joshua-Tor, L.; Hannon, G.J. Argonaute2 is the catalytic engine of mammalian RNAi. Science 2004, 305, 1437–1441. [Google Scholar] [CrossRef] [PubMed]
- Mori, R.; Tanaka, K.; Shimokawa, I. Identification and functional analysis of inflammation-related miRNAs in skin wound repair. Dev. Growth Differ. 2018, 60, 306–315. [Google Scholar] [CrossRef]
- Kerckhove, M.; Tanaka, K.K.K.K.; Umehara, T.; Okamoto, M.; Kanematsu, S.; Hayashi, H.; Yano, H.; Nishiura, S.; Tooyama, S.; Matsubayashi, Y.; et al. Targeting miR-223 in neutrophils enhances the clearance of Staphylococcus aureus in infected wounds. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Johnnidis, J.B.; Harris, M.H.; Wheeler, R.T.; Stehling-Sun, S.; Lam, M.H.; Kirak, O.; Brummelkamp, T.R.; Fleming, M.D.; Camargo, F.D. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature 2008, 451, 1125–1129. [Google Scholar] [CrossRef]
- Xu, W.; Wang, Y.; Ma, Y.; Yang, J. MiR-223 plays a protecting role in neutrophilic asthmatic mice through the inhibition of NLRP3 inflammasome. Respir. Res. 2020, 21, 116. [Google Scholar] [CrossRef]
- Olivieri, F.; Rippo, M.R.; Monsurrò, V.; Salvioli, S.; Capri, M.; Procopio, A.D.; Franceschi, C. MicroRNAs linking inflamm-aging, cellular senescence and cancer. Ageing Res. Rev. 2013, 12, 1056–1068. [Google Scholar] [CrossRef]
- Saunders, L.R.; Sharma, A.D.; Tawney, J.; Nakagawa, M.; Okita, K.; Yamanaka, S.; Willenbring, H.; Verdin, E. miRNAs regulate SIRT1 expression during mouse embryonic stem cell differentiation and in adult mouse tissues. Aging 2010, 2, 415–431. [Google Scholar] [CrossRef]
- Bazzoni, F.; Rossato, M.; Fabbri, M.; Gaudiosi, D.; Mirolo, M.; Mori, L.; Tamassia, N.; Mantovani, A.; Cassatella, M.A.; Locati, M. Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc. Natl. Acad. Sci. USA 2009, 106, 5282–5287. [Google Scholar] [CrossRef]
- Hackl, M.; Brunner, S.; Fortschegger, K.; Schreiner, C.; Micutkova, L.; Mück, C.; Laschober, G.T.; Lepperdinger, G.; Sampson, N.; Berger, P.; et al. miR-17, miR-19b, miR-20a, and miR-106a are down-regulated in human aging. Aging Cell 2010, 9, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Philippe, L.; Alsaleh, G.; Pichot, A.; Ostermann, E.; Zuber, G.; Frisch, B.; Sibilia, J.; Pfeffer, S.; Bahram, S.; Wachsmann, D.; et al. MiR-20a regulates ASK1 expression and TLR4-dependent cytokine release in rheumatoid fibroblast-like synoviocytes. Ann. Rheum. Dis. 2013, 72, 1071–1079. [Google Scholar] [CrossRef]
- Ma, X.; Becker Buscaglia, L.E.; Barker, J.R.; Li, Y. MicroRNAs in NF-κB signaling. J. Mol. Cell Biol. 2011, 3, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, F.; Spazzafumo, L.; Santini, G.; Lazzarini, R.; Albertini, M.C.; Rippo, M.R.; Galeazzi, R.; Abbatecola, A.M.; Marcheselli, F.; Monti, D.; et al. Age-related differences in the expression of circulating microRNAs: MiR-21 as a new circulating marker of inflammaging. Mech. Ageing Dev. 2012, 133, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Dellago, H.; Preschitz-Kammerhofer, B.; Terlecki-Zaniewicz, L.; Schreiner, C.; Fortschegger, K.; Chang, M.W.F.; Hackl, M.; Monteforte, R.; Kühnel, H.; Schosserer, M.; et al. High levels of oncomiR-21 contribute to the senescence-induced growth arrest in normal human cells and its knock-down increases the replicative lifespan. Aging Cell 2013, 12, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Martinez, I.; Cazalla, D.; Almstead, L.L.; Steitz, J.A.; DiMaio, D. miR-29 and miR-30 regulate B-Myb expression during cellular senescence. Proc. Natl. Acad. Sci. USA 2011, 108, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Paone, A.; Calore, F.; Galli, R.; Gaudio, E.; Santhanam, R.; Lovat, F.; Fadda, P.; Mao, C.; Nuovo, G.J.; et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc. Natl. Acad. Sci. USA 2012, 109. [Google Scholar] [CrossRef]
- Feng, X.; Wang, H.; Ye, S.; Guan, J.; Tan, W.; Cheng, S.; Wei, G.; Wu, W.; Wu, F.; Zhou, Y. Up-Regulation of microRNA-126 May Contribute to Pathogenesis of Ulcerative Colitis via Regulating NF-kappaB Inhibitor IκBα. PLoS ONE 2012, 7, e52782. [Google Scholar] [CrossRef]
- Oglesby, I.K.; Bray, I.M.; Chotirmall, S.H.; Stallings, R.L.; O’Neill, S.J.; McElvaney, N.G.; Greene, C.M. miR-126 Is Downregulated in Cystic Fibrosis Airway Epithelial Cells and Regulates TOM1 Expression. J. Immunol. 2010, 184, 1702–1709. [Google Scholar] [CrossRef]
- Mocharla, P.; Briand, S.; Giannotti, G.; Dörries, C.; Jakob, P.; Paneni, F.; Lüscher, T.; Landmesser, U. AngiomiR-126 expression and secretion from circulating CD34+ and CD14+ PBMCs: Role for proangiogenic effects and alterations in type 2 diabetics. Blood 2013, 121, 226–236. [Google Scholar] [CrossRef]
- Zampetaki, A.; Willeit, P.; Tilling, L.; Drozdov, I.; Prokopi, M.; Renard, J.M.; Mayr, A.; Weger, S.; Schett, G.; Shah, A.; et al. Prospective study on circulating microRNAs and risk of myocardial infarction. J. Am. Coll. Cardiol. 2012, 60, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Wang, P.; Lin, L.; Liu, X.; Ma, F.; An, H.; Wang, Z.; Cao, X. MicroRNA-146a Feedback Inhibits RIG-I-Dependent Type I IFN Production in Macrophages by Targeting TRAF6, IRAK1, and IRAK2. J. Immunol. 2009, 183, 2150–2158. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, F.; Lazzarini, R.; Recchioni, R.; Marcheselli, F.; Rippo, M.R.; Di Nuzzo, S.; Albertini, M.C.; Graciotti, L.; Babini, L.; Mariotti, S.; et al. MiR-146a as marker of senescence-Associated pro-inflammatory status in cells involved in vascular remodelling. Age 2013, 35, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- Rippo, M.R.; Olivieri, F.; Monsurrò, V.; Prattichizzo, F.; Albertini, M.C.; Procopio, A.D. MitomiRs in human inflamm-aging: A hypothesis involving miR-181a, miR-34a and miR-146a. Exp. Gerontol. 2014, 56, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Tezcan, G.; Martynova, E.V.; Gilazieva, Z.E.; McIntyre, A.; Rizvanov, A.A.; Khaiboullina, S.F. MicroRNA post-transcriptional regulation of the NLRP3 inflammasome in immunopathologies. Front. Pharmacol. 2019, 10, 451. [Google Scholar] [CrossRef] [PubMed]
- Ekiz, H.A.; Ramstead, A.G.; Lee, S.-H.; Nelson, M.C.; Bauer, K.M.; Wallace, J.A.; Hu, R.; Round, J.L.; Rutter, J.; Drummond, M.J.; et al. T Cell–Expressed microRNA-155 Reduces Lifespan in a Mouse Model of Age-Related Chronic Inflammation. J. Immunol. 2020, 204, 2064–2075. [Google Scholar] [CrossRef]
- Yang, D.; Wang, X.; Gao, H.; Chen, B.; Si, C.; Wang, S. Downregulation of miR-155-5p facilitates enterovirus 71 replication through suppression of type I IFN response by targeting FOXO3/IRF7 pathway. Cell Cycle 2020, 19, 179–192. [Google Scholar] [CrossRef]
- Gimenes-Junior, J.; Owuar, N.; Vari, H.R.; Li, W.; Xander, N.; Kotnala, S.; Sajjan, U.S. FOXO3a regulates rhinovirus-induced innate immune responses in airway epithelial cells. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Sadatomi, D.; Nakashioya, K.; Mamiya, S.; Honda, S.; Kameyama, Y.; Yamamura, Y.; Tanimura, S.; Takeda, K. Mitochondrial function is required for extracellular ATP-induced NLRP3 inflammasome activation. J. Biochem. 2017, 161, 503–512. [Google Scholar] [CrossRef]
- Mori, R.; Tanaka, K.K.; De Kerckhove, M.; Okamoto, M.; Kashiyama, K.; Tanaka, K.K.; Kim, S.; Kawata, T.; Komatsu, T.; Park, S.; et al. Reduced FOXO1 expression accelerates skin wound healing and attenuates scarring. Am. J. Pathol. 2014, 184, 2465–2479. [Google Scholar] [CrossRef]
- Clausen, B.E.; Burkhardt, C.; Reith, W.; Renkawitz, R.; Förster, I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999, 8, 265–277. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.-E.; Mori, R.; Shimokawa, I. Does Calorie Restriction Modulate Inflammaging via FoxO Transcription Factors? Nutrients 2020, 12, 1959. https://doi.org/10.3390/nu12071959
Kim S-E, Mori R, Shimokawa I. Does Calorie Restriction Modulate Inflammaging via FoxO Transcription Factors? Nutrients. 2020; 12(7):1959. https://doi.org/10.3390/nu12071959
Chicago/Turabian StyleKim, Sang-Eun, Ryoichi Mori, and Isao Shimokawa. 2020. "Does Calorie Restriction Modulate Inflammaging via FoxO Transcription Factors?" Nutrients 12, no. 7: 1959. https://doi.org/10.3390/nu12071959
APA StyleKim, S.-E., Mori, R., & Shimokawa, I. (2020). Does Calorie Restriction Modulate Inflammaging via FoxO Transcription Factors? Nutrients, 12(7), 1959. https://doi.org/10.3390/nu12071959