Obesity, Bioactive Lipids, and Adipose Tissue Inflammation in Insulin Resistance


Abstract
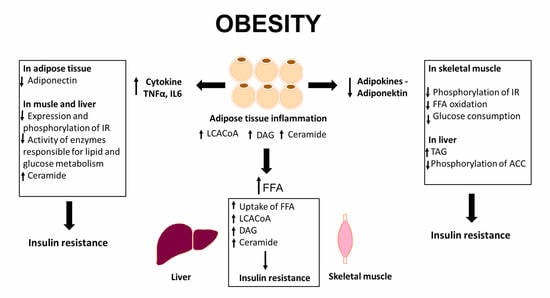
{kind=link}
{kind=link}
1. Introduction
2. Type of Obesity Adipose Tissue Localization and Insulin Resistance
3. Lipid Metabolism in Adipose Tissue
4. Biologically Active Lipids
4.1. Long Chain Acyl-CoA (LCACoA)
4.2. Diacylglycerols (DAG)
4.3. Ceramide (Cer)
5. Obesity, Adipokines and Inflammation
5.1. Adiponectin
5.2. Leptin
5.3. Apelin
5.4. Fibroblast Growth Factor 21 (FGF21)
5.5. Lipocalin 2 (LCN-2)
5.6. Resistin
5.7. Tumor Necrosis Factor α (TNF-α)
5.8. Interleukin 6 (IL6)
6. Summary
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACS | Acyl-CoA synthetase |
| ADA | American Diabetes Association |
| BMI | Body mass index |
| Cer | Ceramide |
| DAG | Diacylglycerols |
| DGAT | diacylglycerol acyltransferase |
| FABPpm | fatty acid-binding protein plasma membrane |
| FATP | fatty acid transport proteins |
| FFA | Free fatty acids |
| GPAT | glycerol-3-phosphate acyltransferase |
| HDL | high-density lipoprotein |
| HSL | hormone-sensitive lipase |
| IR | insulin receptor |
| IRS-1 | insulin receptor substrate-1 |
| LCACoA | long-chain acyl-CoA |
| LDL | low-density lipoprotein |
| LPA | lysophosphatidic acid |
| LPL | lipoprotein lipase |
| PA | phosphatidic acid |
| PAP | phosphatidic acid phosphatase |
| PI3K | phosphoinositide 3-kinases |
| PKA | Protein kinase A |
| PKC | protein kinase C |
| SAT | subcutaneous adipose tissue |
| SPT | serine palmitoyltransferase |
| SVF | stromal vascular fraction |
| TAG | Triacylglycerols |
| TNF-α | tumor necrosis factor-α |
| VAT | visceral adipose tissue |
| VLDL | very low-density lipoproteins |
References
- Garrow, J.S.; Webster, J. Quetelet’s index (W/H2) as a measure of fatness. Int. J. Obes. 1985, 9, 147–153. [Google Scholar]
- Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 8 March 2020).
- Pasco, J.A.; Holloway, K.L.; Dobbins, A.G.; Kotowicz, M.A.; Williams, L.J.; Brennan, S.L. Body mass index and measures of body fat for defining obesity and underweight: A cross-sectional, population-based study. BMC Obes. 2014, 1, 9. [Google Scholar] [CrossRef] [PubMed]
- Herrera, B.M.; Lindgren, C.M. The genetics of obesity. Curr. Diabetes Rep. 2010, 10, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, M.O. Genetics of obesity: What genetic association studies have taught us about the biology of obesity and its complications. Lancet Diabetes Endocrinol. 2018, 6, 223–236. [Google Scholar] [CrossRef]
- Agodi, A.; Maugeri, A.; Kunzova, S.; Sochor, O.; Bauerova, H.; Kiacova, N.; Barchitta, M.; Vinciguerra, M. Association of Dietary Patterns with Metabolic Syndrome: Results from the Kardiovize Brno 2030 Study. Nutrients 2018, 10, 898. [Google Scholar] [CrossRef]
- Maugeri, A.; Barchitta, M.; Favara, G.; La Rosa, M.C.; La Mastra, C.; Magnano San Lio, R.; Agodi, A. Maternal Dietary Patterns Are Associated with Pre-Pregnancy Body Mass Index and Gestational Weight Gain: Results from the “Mamma & Bambino” Cohort. Nutrients 2019, 11, 1308. [Google Scholar] [CrossRef]
- Maugeri, A.; Kunzova, S.; Medina-Inojosa, J.R.; Agodi, A.; Barchitta, M.; Homolka, M.; Kiacova, N.; Bauerova, H.; Sochor, O.; Lopez-Jimenez, F.; et al. Association between eating time interval and frequency with ideal cardiovascular health: Results from a random sample Czech urban population. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 847–855. [Google Scholar] [CrossRef]
- Maugeri, A.; Medina-Inojosa, J.R.; Kunzova, S.; Agodi, A.; Barchitta, M.; Sochor, O.; Lopez-Jimenez, F.; Geda, Y.E.; Vinciguerra, M. Sleep Duration and Excessive Daytime Sleepiness Are Associated with Obesity Independent of Diet and Physical Activity. Nutrients 2018, 10, 1219. [Google Scholar] [CrossRef]
- Obesity and Overweight. Available online: http://www.who.int/mediacentre/factsheets/fs311/en/ (accessed on 8 March 2020).
- Yang, Q.; Vijayakumar, A.; Kahn, B.B. Metabolites as regulators of insulin sensitivity and metabolism. Nat. Rev. Mol. Cell Biol. 2018, 19, 654–672. [Google Scholar] [CrossRef]
- Association, A.D. Diagnosis and classification of diabetes mellitus. Diabetes Care 2009, 32 (Suppl. 1), S62–S67. [Google Scholar] [CrossRef]
- Vague, J. Sexual differentiation. A determinant factor of the forms of obesity. 1947. Obes. Res. 1996, 4, 201–203. [Google Scholar] [CrossRef] [PubMed]
- Ognjanovic, S.; Bao, S.; Yamamoto, S.Y.; Garibay-Tupas, J.; Samal, B.; Bryant-Greenwood, G.D. Genomic organization of the gene coding for human pre-B-cell colony enhancing factor and expression in human fetal membranes. J. Mol. Endocrinol. 2001, 26, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Heymsfield, S.B.; Wadden, T.A. Mechanisms, Pathophysiology, and Management of Obesity. N. Engl. J. Med. 2017, 376, 1492. [Google Scholar] [CrossRef] [PubMed]
- Arner, P. Human fat cell lipolysis: Biochemistry, regulation and clinical role. Best Pract. Res. Clin. Endocrinol. Metab. 2005, 19, 471–482. [Google Scholar] [CrossRef]
- Ibrahim, M.M. Subcutaneous and visceral adipose tissue: Structural and functional differences. Obes. Rev. 2010, 11, 11–18. [Google Scholar] [CrossRef]
- Lee, M.J.; Wu, Y.; Fried, S.K. Adipose tissue heterogeneity: Implication of depot differences in adipose tissue for obesity complications. Mol. Aspects Med. 2013, 34, 1–11. [Google Scholar] [CrossRef]
- Rebuffe-Scrive, M.; Enk, L.; Crona, N.; Lonnroth, P.; Abrahamsson, L.; Smith, U.; Bjorntorp, P. Fat cell metabolism in different regions in women. Effect of menstrual cycle, pregnancy, and lactation. J. Clin. Investig. 1985, 75, 1973–1976. [Google Scholar] [CrossRef]
- Rebuffe-Scrive, M.; Lonnroth, P.; Marin, P.; Wesslau, C.; Bjorntorp, P.; Smith, U. Regional adipose tissue metabolism in men and postmenopausal women. Int. J. Obes. 1987, 11, 347–355. [Google Scholar]
- Tanko, L.B.; Bagger, Y.Z.; Alexandersen, P.; Larsen, P.J.; Christiansen, C. Peripheral adiposity exhibits an independent dominant antiatherogenic effect in elderly women. Circulation 2003, 107, 1626–1631. [Google Scholar] [CrossRef]
- Van Pelt, R.E.; Jankowski, C.M.; Gozansky, W.S.; Schwartz, R.S.; Kohrt, W.M. Lower-body adiposity and metabolic protection in postmenopausal women. J. Clin. Endocrinol. Metab. 2005, 90, 4573–4578. [Google Scholar] [CrossRef]
- Van Pelt, R.E.; Evans, E.M.; Schechtman, K.B.; Ehsani, A.A.; Kohrt, W.M. Contributions of total and regional fat mass to risk for cardiovascular disease in older women. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E1023–E1028. [Google Scholar] [CrossRef] [PubMed]
- Tanko, L.B.; Bruun, J.M.; Alexandersen, P.; Bagger, Y.Z.; Richelsen, B.; Christiansen, C.; Larsen, P.J. Novel associations between bioavailable estradiol and adipokines in elderly women with different phenotypes of obesity: Implications for atherogenesis. Circulation 2004, 110, 2246–2252. [Google Scholar] [CrossRef] [PubMed]
- Blachnio-Zabielska, A.; Baranowski, M.; Zabielski, P.; Gorski, J. Effect of high fat diet enriched with unsaturated and diet rich in saturated fatty acids on sphingolipid metabolism in rat skeletal muscle. J. Cell. Physiol. 2010, 225, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Ravussin, E.; Smith, S.R. Increased fat intake, impaired fat oxidation, and failure of fat cell proliferation result in ectopic fat storage, insulin resistance, and type 2 diabetes mellitus. Ann. N. Y. Acad. Sci. 2002, 967, 363–378. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, T.; Lamendola, C.; Liu, A.; Abbasi, F. Preferential fat deposition in subcutaneous versus visceral depots is associated with insulin sensitivity. J. Clin. Endocrinol. Metab. 2011, 96, E1756–E1760. [Google Scholar] [CrossRef] [PubMed]
- Anthonsen, M.W.; Ronnstrand, L.; Wernstedt, C.; Degerman, E.; Holm, C. Identification of novel phosphorylation sites in hormone-sensitive lipase that are phosphorylated in response to isoproterenol and govern activation properties in vitro. J. Biol. Chem. 1998, 273, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Wolf, G. The mechanism and regulation of fat mobilization from adipose tissue: Desnutrin, a newly discovered lipolytic enzyme. Nutr. Rev. 2005, 63, 166–170. [Google Scholar] [CrossRef]
- Moro, C.; Lafontan, M. Natriuretic peptides and cGMP signaling control of energy homeostasis. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H358–H368. [Google Scholar] [CrossRef]
- Moro, C. Natriuretic peptides and fat metabolism. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 645–649. [Google Scholar] [CrossRef]
- Coué, M.; Moro, C. Natriuretic peptide control of energy balance and glucose homeostasis. Biochimie 2016, 124, 84–91. [Google Scholar] [CrossRef]
- Tordjman, J.; Chauvet, G.; Quette, J.; Beale, E.G.; Forest, C.; Antoine, B. Thiazolidinediones block fatty acid release by inducing glyceroneogenesis in fat cells. J. Biol. Chem. 2003, 278, 18785–18790. [Google Scholar] [CrossRef] [PubMed]
- Beale, E.G.; Hammer, R.E.; Antoine, B.; Forest, C. Disregulated glyceroneogenesis: PCK1 as a candidate diabetes and obesity gene. Trends Endocrinol. Metab. 2004, 15, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Sethi, J.K.; Vidal-Puig, A.J. Thematic review series: Adipocyte biology. Adipose tissue function and plasticity orchestrate nutritional adaptation. J. Lipid Res. 2007, 48, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Straczkowski, M.; Kowalska, I.; Nikolajuk, A.; Dzienis-Straczkowska, S.; Kinalska, I.; Baranowski, M.; Zendzian-Piotrowska, M.; Brzezinska, Z.; Gorski, J. Relationship between insulin sensitivity and sphingomyelin signaling pathway in human skeletal muscle. Diabetes 2004, 53, 1215–1221. [Google Scholar] [CrossRef]
- Straczkowski, M.; Kowalska, I.; Baranowski, M.; Nikolajuk, A.; Otziomek, E.; Zabielski, P.; Adamska, A.; Blachnio, A.; Gorski, J.; Gorska, M. Increased skeletal muscle ceramide level in men at risk of developing type 2 diabetes. Diabetologia 2007, 50, 2366–2373. [Google Scholar] [CrossRef]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. Mechanisms for insulin resistance: Common threads and missing links. Cell 2012, 148, 852–871. [Google Scholar] [CrossRef]
- Yu, C.; Chen, Y.; Cline, G.W.; Zhang, D.; Zong, H.; Wang, Y.; Bergeron, R.; Kim, J.K.; Cushman, S.W.; Cooney, G.J.; et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J. Biol. Chem. 2002, 277, 50230–50236. [Google Scholar] [CrossRef]
- Blachnio-Zabielska, A.U.; Chacinska, M.; Vendelbo, M.H.; Zabielski, P. The Crucial Role of C18-Cer in Fat-Induced Skeletal Muscle Insulin Resistance. Cell. Physiol. Biochem. 2016, 40, 1207–1220. [Google Scholar] [CrossRef]
- Zabielski, P.; Chacinska, M.; Charkiewicz, K.; Baranowski, M.; Gorski, J.; Blachnio-Zabielska, A.U. Effect of metformin on bioactive lipid metabolism in insulin-resistant muscle. J. Endocrinol. 2017, 233, 329–340. [Google Scholar] [CrossRef]
- Zabielski, P.; Hady, H.R.; Chacinska, M.; Roszczyc, K.; Gorski, J.; Blachnio-Zabielska, A.U. The effect of high fat diet and metformin treatment on liver lipids accumulation and their impact on insulin action. Sci. Rep. 2018, 8, 7249. [Google Scholar] [CrossRef] [PubMed]
- Zabielski, P.; Daniluk, J.; Hady, H.R.; Markowski, A.R.; Imierska, M.; Górski, J.; Blachnio-Zabielska, A.U. The effect of high-fat diet and inhibition of ceramide production on insulin action in liver. J. Cell. Physiol. 2019, 234, 1851–1861. [Google Scholar] [CrossRef] [PubMed]
- Belfort, R.; Mandarino, L.; Kashyap, S.; Wirfel, K.; Pratipanawatr, T.; Berria, R.; Defronzo, R.A.; Cusi, K. Dose-response effect of elevated plasma free fatty acid on insulin signaling. Diabetes 2005, 54, 1640–1648. [Google Scholar] [CrossRef] [PubMed]
- Bjorntorp, P. Metabolic implications of body fat distribution. Diabetes Care 1991, 14, 1132–1143. [Google Scholar] [CrossRef] [PubMed]
- Hulver, M.W.; Berggren, J.R.; Cortright, R.N.; Dudek, R.W.; Thompson, R.P.; Pories, W.J.; MacDonald, K.G.; Cline, G.W.; Shulman, G.I.; Dohm, G.L.; et al. Skeletal muscle lipid metabolism with obesity. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E741–E747. [Google Scholar] [CrossRef]
- Ellis, B.A.; Poynten, A.; Lowy, A.J.; Furler, S.M.; Chisholm, D.J.; Kraegen, E.W.; Cooney, G.J. Long-chain acyl-CoA esters as indicators of lipid metabolism and insulin sensitivity in rat and human muscle. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E554–E560. [Google Scholar] [CrossRef]
- Adams, J.M., 2nd; Pratipanawatr, T.; Berria, R.; Wang, E.; DeFronzo, R.A.; Sullards, M.C.; Mandarino, L.J. Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes 2004, 53, 25–31. [Google Scholar] [CrossRef]
- Schmitz-Peiffer, C. Protein kinase C and lipid-induced insulin resistance in skeletal muscle. Ann. N. Y. Acad. Sci. 2002, 967, 146–157. [Google Scholar] [CrossRef]
- Cronan, J.E., Jr. In vivo evidence that acyl coenzyme A regulates DNA binding by the Escherichia coli FadR global transcription factor. J. Bacteriol. 1997, 179, 1819–1823. [Google Scholar] [CrossRef][Green Version]
- Yaney, G.C.; Korchak, H.M.; Corkey, B.E. Long-chain acyl CoA regulation of protein kinase C and fatty acid potentiation of glucose-stimulated insulin secretion in clonal beta-cells. Endocrinology 2000, 141, 1989–1998. [Google Scholar] [CrossRef][Green Version]
- Tomas, E.; Tsao, T.S.; Saha, A.K.; Murrey, H.E.; Zhang Cc, C.; Itani, S.I.; Lodish, H.F.; Ruderman, N.B. Enhanced muscle fat oxidation and glucose transport by ACRP30 globular domain: Acetyl-CoA carboxylase inhibition and AMP-activated protein kinase activation. Proc. Natl. Acad. Sci. USA 2002, 99, 16309–16313. [Google Scholar] [CrossRef] [PubMed]
- Itani, S.I.; Ruderman, N.B.; Schmieder, F.; Boden, G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes 2002, 51, 2005–2011. [Google Scholar] [CrossRef] [PubMed]
- Blachnio-Zabielska, A.U.; Baranowski, M.; Hirnle, T.; Zabielski, P.; Lewczuk, A.; Dmitruk, I.; Gorski, J. Increased bioactive lipids content in human subcutaneous and epicardial fat tissue correlates with insulin resistance. Lipids 2012, 47, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Bednarski, T.; Pyrkowska, A.; Opasinska, A.; Dobrzyn, P. Regulation of cardiac metabolism and function by lipogenic factors. Postepy Hig. Med. Dosw. (Online) 2016, 70, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Oakes, N.D.; Kennedy, C.J.; Jenkins, A.B.; Laybutt, D.R.; Chisholm, D.J.; Kraegen, E.W. A new antidiabetic agent, BRL 49653, reduces lipid availability and improves insulin action and glucoregulation in the rat. Diabetes 1994, 43, 1203–1210. [Google Scholar] [CrossRef]
- Turinsky, J.; O’Sullivan, D.M.; Bayly, B.P. 1,2-Diacylglycerol and ceramide levels in insulin-resistant tissues of the rat in vivo. J. Biol. Chem. 1990, 265, 16880–16885. [Google Scholar] [PubMed]
- Cortright, R.N.; Azevedo, J.L., Jr.; Zhou, Q.; Sinha, M.; Pories, W.J.; Itani, S.I.; Dohm, G.L. Protein kinase C modulates insulin action in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E553–E562. [Google Scholar] [CrossRef]
- Blachnio-Zabielska, A.U.; Hady, H.R.; Markowski, A.R.; Kurianiuk, A.; Karwowska, A.; Górski, J.; Zabielski, P. Inhibition of Ceramide De Novo Synthesis Affects Adipocytokine Secretion and Improves Systemic and Adipose Tissue Insulin Sensitivity. Int. J. Mol. Sci. 2018, 19, 3995. [Google Scholar] [CrossRef]
- Grycel, S.; Markowski, A.R.; Hady, H.R.; Zabielski, P.; Kojta, I.; Imierska, M.; Górski, J.; Blachnio-Zabielska, A.U. Metformin treatment affects adipocytokine secretion and lipid composition in adipose tissues of diet-induced insulin-resistant rats. Nutrition 2019, 63–64, 126–133. [Google Scholar] [CrossRef]
- Kurek, K.; Piotrowska, D.M.; Wiesiolek, P.; Chabowski, A.; Zendzian-Piotrowska, M. Role of sphingolipids in digestive system. Postepy Hig. Med. Dosw. (Online) 2012, 66, 868–875. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K. Serine palmitoyltransferase, a key enzyme of sphingolipid metabolism. Biochim. Biophys. Acta 2003, 1632, 16–30. [Google Scholar] [CrossRef]
- Car, H.; Zendzian-Piotrowska, M.; Fiedorowicz, A.; Prokopiuk, S.; Sadowska, A.; Kurek, K. The role of ceramides in selected brain pathologies: Ischemia/hypoxia, Alzheimer disease. Postepy Hig. Med. Dosw. (Online) 2012, 66, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Kitatani, K.; Idkowiak-Baldys, J.; Hannun, Y.A. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell. Signal. 2008, 20, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- Stephens, J.M.; Pekala, P.H. Transcriptional repression of the GLUT4 and C/EBP genes in 3T3-L1 adipocytes by tumor necrosis factor-alpha. J. Biol. Chem. 1991, 266, 21839–21845. [Google Scholar]
- Stephens, J.M.; Pekala, P.H. Transcriptional repression of the C/EBP-alpha and GLUT4 genes in 3T3-L1 adipocytes by tumor necrosis factor-alpha. Regulations is coordinate and independent of protein synthesis. J. Biol. Chem. 1992, 267, 13580–13584. [Google Scholar]
- Long, S.D.; Pekala, P.H. Lipid mediators of insulin resistance: Ceramide signalling down-regulates GLUT4 gene transcription in 3T3-L1 adipocytes. Biochem. J. 1996, 319 Pt 1, 179–184. [Google Scholar] [CrossRef]
- Holland, W.L.; Summers, S.A. Sphingolipids, insulin resistance, and metabolic disease: New insights from in vivo manipulation of sphingolipid metabolism. Endocr. Rev. 2008, 29, 381–402. [Google Scholar] [CrossRef]
- Blachnio-Zabielska, A.U.; Koutsari, C.; Tchkonia, T.; Jensen, M.D. Sphingolipid content of human adipose tissue: Relationship to adiponectin and insulin resistance. Obesity (Silver Spring) 2012, 20, 2341–2347. [Google Scholar] [CrossRef]
- Kolak, M.; Westerbacka, J.; Velagapudi, V.R.; Wagsater, D.; Yetukuri, L.; Makkonen, J.; Rissanen, A.; Hakkinen, A.M.; Lindell, M.; Bergholm, R.; et al. Adipose tissue inflammation and increased ceramide content characterize subjects with high liver fat content independent of obesity. Diabetes 2007, 56, 1960–1968. [Google Scholar] [CrossRef]
- Chaurasia, B.; Kaddai, V.A.; Lancaster, G.I.; Henstridge, D.C.; Sriram, S.; Galam, D.L.; Gopalan, V.; Prakash, K.N.; Velan, S.S.; Bulchand, S.; et al. Adipocyte Ceramides Regulate Subcutaneous Adipose Browning, Inflammation, and Metabolism. Cell Metab. 2016, 24, 820–834. [Google Scholar] [CrossRef]
- Blachnio-Zabielska, A.U.; Pulka, M.; Baranowski, M.; Nikolajuk, A.; Zabielski, P.; Gorska, M.; Gorski, J. Ceramide metabolism is affected by obesity and diabetes in human adipose tissue. J. Cell. Physiol. 2012, 227, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Mottillo, E.P.; Zhao, J.; Gartung, A.; VanHecke, G.C.; Lee, J.F.; Maddipati, K.R.; Xu, H.; Ahn, Y.H.; Proia, R.L.; et al. Adipocyte lipolysis-stimulated interleukin-6 production requires sphingosine kinase 1 activity. J. Biol. Chem. 2014, 289, 32178–32185. [Google Scholar] [CrossRef] [PubMed]
- Obinata, H.; Hla, T. Sphingosine 1-phosphate and inflammation. Int. Immunol. 2019, 31, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Pyne, S.; Pyne, N.J. Ceramide and sphingosine 1-phosphate in adipose dysfunction. Prog. Lipid Res. 2019, 74, 145–159. [Google Scholar] [CrossRef]
- Samad, F.; Hester, K.D.; Yang, G.; Hannun, Y.A.; Bielawski, J. Altered adipose and plasma sphingolipid metabolism in obesity: A potential mechanism for cardiovascular and metabolic risk. Diabetes 2006, 55, 2579–2587. [Google Scholar] [CrossRef]
- Wang, J.; Badeanlou, L.; Bielawski, J.; Ciaraldi, T.P.; Samad, F. Sphingosine kinase 1 regulates adipose proinflammatory responses and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E756–E768. [Google Scholar] [CrossRef]
- Francisco, V.; Pino, J.; Gonzalez-Gay, M.A.; Mera, A.; Lago, F.; Gómez, R.; Mobasheri, A.; Gualillo, O. Adipokines and inflammation: Is it a question of weight? Br. J. Pharmacol. 2018, 175, 1569–1579. [Google Scholar] [CrossRef]
- Kang, Y.E.; Kim, J.M.; Joung, K.H.; Lee, J.H.; You, B.R.; Choi, M.J.; Ryu, M.J.; Ko, Y.B.; Lee, M.A.; Lee, J.; et al. The Roles of Adipokines, Proinflammatory Cytokines, and Adipose Tissue Macrophages in Obesity-Associated Insulin Resistance in Modest Obesity and Early Metabolic Dysfunction. PLoS ONE 2016, 11, e0154003. [Google Scholar] [CrossRef]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Lotowska, J.M.; Sobaniec-Lotowska, M.E.; Lebensztejn, D.M.; Daniluk, U.; Sobaniec, P.; Sendrowski, K.; Daniluk, J.; Reszec, J.; Debek, W. Ultrastructural Characteristics of Rat Hepatic Oval Cells and Their Intercellular Contacts in the Model of Biliary Fibrosis: New Insights into Experimental Liver Fibrogenesis. Gastroenterol. Res. Pract. 2017, 2017, 2721547. [Google Scholar] [CrossRef]
- Daniluk, U.; Kerros, C.; Tao, R.H.; Wise, J.F.; Ao, X.; Berkova, Z.; Samaniego, F. The peptide derived from the Ig-like domain of human herpesvirus 8 K1 protein induces death in hematological cancer cells. J. Exp. Clin. Cancer Res. 2012, 31, 69. [Google Scholar] [CrossRef] [PubMed]
- Barchetta, I.; Cimini, F.A.; Capoccia, D.; Bertoccini, L.; Ceccarelli, V.; Chiappetta, C.; Leonetti, F.; Di Cristofano, C.; Silecchia, G.; Orho-Melander, M.; et al. Neurotensin Is a Lipid-Induced Gastrointestinal Peptide Associated with Visceral Adipose Tissue Inflammation in Obesity. Nutrients 2018, 10, 526. [Google Scholar] [CrossRef] [PubMed]
- Krstic, J.; Reinisch, I.; Schupp, M.; Schulz, T.J.; Prokesch, A. p53 Functions in Adipose Tissue Metabolism and Homeostasis. Int. J. Mol. Sci. 2018, 19, 2622. [Google Scholar] [CrossRef] [PubMed]
- Goralska, M.; Majewska-Szczepanik, M.; Szczepanik, M. Immunological mechanisms involved in obesity and their role in metabolic syndrome. Postepy Hig. Med. Dosw. (Online) 2015, 69, 1384–1404. [Google Scholar] [PubMed]
- Krist, J.; Wieder, K.; Klöting, N.; Oberbach, A.; Kralisch, S.; Wiesner, T.; Schön, M.R.; Gärtner, D.; Dietrich, A.; Shang, E.; et al. Effects of weight loss and exercise on apelin serum concentrations and adipose tissue expression in human obesity. Obes. Facts 2013, 6, 57–69. [Google Scholar] [CrossRef]
- Itoh, N. FGF21 as a Hepatokine, Adipokine, and Myokine in Metabolism and Diseases. Front. Endocrinol. (Lausanne) 2014, 5, 107. [Google Scholar] [CrossRef]
- Unamuno, X.; Gómez-Ambrosi, J.; Rodríguez, A.; Becerril, S.; Frühbeck, G.; Catalán, V. Adipokine dysregulation and adipose tissue inflammation in human obesity. Eur. J. Clin. Investig. 2018, 48, e12997. [Google Scholar] [CrossRef]
- Faraj, M.; Lu, H.L.; Cianflone, K. Diabetes, lipids, and adipocyte secretagogues. Biochem. Cell Biol. 2004, 82, 170–190. [Google Scholar] [CrossRef]
- Chandran, M.; Phillips, S.A.; Ciaraldi, T.; Henry, R.R. Adiponectin: More than just another fat cell hormone? Diabetes Care 2003, 26, 2442–2450. [Google Scholar] [CrossRef]
- Weyer, C.; Funahashi, T.; Tanaka, S.; Hotta, K.; Matsuzawa, Y.; Pratley, R.E.; Tataranni, P.A. Hypoadiponectinemia in obesity and type 2 diabetes: Close association with insulin resistance and hyperinsulinemia. J. Clin. Endocrinol. Metab. 2001, 86, 1930–1935. [Google Scholar] [CrossRef]
- Yang, W.S.; Lee, W.J.; Funahashi, T.; Tanaka, S.; Matsuzawa, Y.; Chao, C.L.; Chen, C.L.; Tai, T.Y.; Chuang, L.M. Weight reduction increases plasma levels of an adipose-derived anti-inflammatory protein, adiponectin. J. Clin. Endocrinol. Metab. 2001, 86, 3815–3819. [Google Scholar] [CrossRef] [PubMed]
- Fain, J.N.; Madan, A.K.; Hiler, M.L.; Cheema, P.; Bahouth, S.W. Comparison of the release of adipokines by adipose tissue, adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology 2004, 145, 2273–2282. [Google Scholar] [CrossRef] [PubMed]
- Lihn, A.S.; Bruun, J.M.; He, G.; Pedersen, S.B.; Jensen, P.F.; Richelsen, B. Lower expression of adiponectin mRNA in visceral adipose tissue in lean and obese subjects. Mol. Cell. Endocrinol. 2004, 219, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Aldhahi, W.; Hamdy, O. Adipokines, inflammation, and the endothelium in diabetes. Curr. Diabetes Rep. 2003, 3, 293–298. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Waki, H.; Terauchi, Y.; Kubota, N.; Hara, K.; Mori, Y.; Ide, T.; Murakami, K.; Tsuboyama-Kasaoka, N.; et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat. Med. 2001, 7, 941–946. [Google Scholar] [CrossRef]
- Diez, J.J.; Iglesias, P. The role of the novel adipocyte-derived hormone adiponectin in human disease. Eur. J. Endocrinol. 2003, 148, 293–300. [Google Scholar] [CrossRef]
- Chen, H.; Montagnani, M.; Funahashi, T.; Shimomura, I.; Quon, M.J. Adiponectin stimulates production of nitric oxide in vascular endothelial cells. J. Biol. Chem. 2003, 278, 45021–45026. [Google Scholar] [CrossRef]
- Xi, L.; Qian, Z.; Xu, G.; Zhou, C.; Sun, S. Crocetin attenuates palmitate-induced insulin insensitivity and disordered tumor necrosis factor-alpha and adiponectin expression in rat adipocytes. Br. J. Pharmacol. 2007, 151, 610–617. [Google Scholar] [CrossRef]
- Montague, C.T.; Prins, J.B.; Sanders, L.; Digby, J.E.; O’Rahilly, S. Depot- and sex-specific differences in human leptin mRNA expression: Implications for the control of regional fat distribution. Diabetes 1997, 46, 342–347. [Google Scholar] [CrossRef]
- Saad, M.F.; Damani, S.; Gingerich, R.L.; Riad-Gabriel, M.G.; Khan, A.; Boyadjian, R.; Jinagouda, S.D.; el-Tawil, K.; Rude, R.K.; Kamdar, V. Sexual dimorphism in plasma leptin concentration. J. Clin. Endocrinol. Metab. 1997, 82, 579–584. [Google Scholar] [CrossRef]
- Margetic, S.; Gazzola, C.; Pegg, G.G.; Hill, R.A. Leptin: A review of its peripheral actions and interactions. Int. J. Obes. Relat Metab. Disord 2002, 26, 1407–1433. [Google Scholar] [CrossRef] [PubMed]
- Dobbins, R.L.; Szczepaniak, L.S.; Zhang, W.; McGarry, J.D. Chemical sympathectomy alters regulation of body weight during prolonged ICV leptin infusion. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E778–E787. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; Paulus, K.; Johren, O.; Lehnert, H. Intranasal leptin reduces appetite and induces weight loss in rats with diet-induced obesity (DIO). Endocrinology 2012, 153, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Fasshauer, M.; Blüher, M. Adipokines in health and disease. Trends Pharmacol. Sci. 2015, 36, 461–470. [Google Scholar] [CrossRef]
- Gourdy, P.; Cazals, L.; Thalamas, C.; Sommet, A.; Calvas, F.; Galitzky, M.; Vinel, C.; Dray, C.; Hanaire, H.; Castan-Laurell, I.; et al. Apelin administration improves insulin sensitivity in overweight men during hyperinsulinaemic-euglycaemic clamp. Diabetes Obes. Metab. 2018, 20, 157–164. [Google Scholar] [CrossRef]
- Cao, J.; Li, H.; Chen, L. Targeting drugs to APJ receptor: The prospect of treatment of hypertension and other cardiovascular diseases. Curr. Drug Targets 2015, 16, 148–155. [Google Scholar] [CrossRef]
- Sörhede Winzell, M.; Magnusson, C.; Ahrén, B. The apj receptor is expressed in pancreatic islets and its ligand, apelin, inhibits insulin secretion in mice. Regul. Pept. 2005, 131, 12–17. [Google Scholar] [CrossRef]
- Dray, C.; Knauf, C.; Daviaud, D.; Waget, A.; Boucher, J.; Buléon, M.; Cani, P.D.; Attané, C.; Guigné, C.; Carpéné, C.; et al. Apelin stimulates glucose utilization in normal and obese insulin-resistant mice. Cell Metab. 2008, 8, 437–445. [Google Scholar] [CrossRef]
- Yue, P.; Jin, H.; Aillaud, M.; Deng, A.C.; Azuma, J.; Asagami, T.; Kundu, R.K.; Reaven, G.M.; Quertermous, T.; Tsao, P.S. Apelin is necessary for the maintenance of insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E59–E67. [Google Scholar] [CrossRef]
- Gaich, G.; Chien, J.Y.; Fu, H.; Glass, L.C.; Deeg, M.A.; Holland, W.L.; Kharitonenkov, A.; Bumol, T.; Schilske, H.K.; Moller, D.E. The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab. 2013, 18, 333–340. [Google Scholar] [CrossRef]
- Yan, Q.W.; Yang, Q.; Mody, N.; Graham, T.E.; Hsu, C.H.; Xu, Z.; Houstis, N.E.; Kahn, B.B.; Rosen, E.D. The adipokine lipocalin 2 is regulated by obesity and promotes insulin resistance. Diabetes 2007, 56, 2533–2540. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wu, Y.; Zhang, Y.; Leroith, D.; Bernlohr, D.A.; Chen, X. The role of lipocalin 2 in the regulation of inflammation in adipocytes and macrophages. Mol. Endocrinol. 2008, 22, 1416–1426. [Google Scholar] [CrossRef] [PubMed]
- Catalán, V.; Gómez-Ambrosi, J.; Rodríguez, A.; Ramírez, B.; Silva, C.; Rotellar, F.; Gil, M.J.; Cienfuegos, J.A.; Salvador, J.; Frühbeck, G. Increased adipose tissue expression of lipocalin-2 in obesity is related to inflammation and matrix metalloproteinase-2 and metalloproteinase-9 activities in humans. J. Mol. Med. (Berl.) 2009, 87, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Moschen, A.R.; Adolph, T.E.; Gerner, R.R.; Wieser, V.; Tilg, H. Lipocalin-2: A Master Mediator of Intestinal and Metabolic Inflammation. Trends Endocrinol. Metab. 2017, 28, 388–397. [Google Scholar] [CrossRef]
- Catalán, V.; Gómez-Ambrosi, J.; Rodríguez, A.; Ramírez, B.; Valentí, V.; Moncada, R.; Silva, C.; Salvador, J.; Frühbeck, G. Peripheral mononuclear blood cells contribute to the obesity-associated inflammatory state independently of glycemic status: Involvement of the novel proinflammatory adipokines chemerin, chitinase-3-like protein 1, lipocalin-2 and osteopontin. Genes Nutr. 2015, 10, 460. [Google Scholar] [CrossRef]
- Steppan, C.M.; Bailey, S.T.; Bhat, S.; Brown, E.J.; Banerjee, R.R.; Wright, C.M.; Patel, H.R.; Ahima, R.S.; Lazar, M.A. The hormone resistin links obesity to diabetes. Nature 2001, 409, 307–312. [Google Scholar] [CrossRef]
- Barnes, K.M.; Miner, J.L. Role of resistin in insulin sensitivity in rodents and humans. Curr. Protein Pept. Sci. 2009, 10, 96–107. [Google Scholar] [CrossRef]
- Kaser, S.; Kaser, A.; Sandhofer, A.; Ebenbichler, C.F.; Tilg, H.; Patsch, J.R. Resistin messenger-RNA expression is increased by proinflammatory cytokines in vitro. Biochem. Biophys. Res. Commun. 2003, 309, 286–290. [Google Scholar] [CrossRef]
- Patel, L.; Buckels, A.C.; Kinghorn, I.J.; Murdock, P.R.; Holbrook, J.D.; Plumpton, C.; Macphee, C.H.; Smith, S.A. Resistin is expressed in human macrophages and directly regulated by PPAR gamma activators. Biochem. Biophys. Res. Commun. 2003, 300, 472–476. [Google Scholar] [CrossRef]
- McTernan, P.G.; McTernan, C.L.; Chetty, R.; Jenner, K.; Fisher, F.M.; Lauer, M.N.; Crocker, J.; Barnett, A.H.; Kumar, S. Increased resistin gene and protein expression in human abdominal adipose tissue. J. Clin. Endocrinol. Metab. 2002, 87, 2407. [Google Scholar] [CrossRef]
- McTernan, C.L.; McTernan, P.G.; Harte, A.L.; Levick, P.L.; Barnett, A.H.; Kumar, S. Resistin, central obesity, and type 2 diabetes. Lancet 2002, 359, 46–47. [Google Scholar] [CrossRef]
- Steppan, C.M.; Brown, E.J.; Wright, C.M.; Bhat, S.; Banerjee, R.R.; Dai, C.Y.; Enders, G.H.; Silberg, D.G.; Wen, X.; Wu, G.D.; et al. A family of tissue-specific resistin-like molecules. Proc. Natl. Acad. Sci. USA 2001, 98, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Saghizadeh, M.; Ong, J.M.; Garvey, W.T.; Henry, R.R.; Kern, P.A. The expression of TNF alpha by human muscle. Relationship to insulin resistance. J. Clin. Investig. 1996, 97, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Spiegelman, B.M. Tumor necrosis factor alpha: A key component of the obesity-diabetes link. Diabetes 1994, 43, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Kern, P.A.; Ranganathan, S.; Li, C.; Wood, L.; Ranganathan, G. Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E745–E751. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Bruun, J.M.; Lihn, A.S.; Verdich, C.; Pedersen, S.B.; Toubro, S.; Astrup, A.; Richelsen, B. Regulation of adiponectin by adipose tissue-derived cytokines: In vivo and in vitro investigations in humans. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E527–E533. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Molecular mechanisms of insulin resistance and the role of the adipocyte. Int. J. Obes. Relat. Metab. Disord 2000, 24 (Suppl. 4), S23–S27. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Peraldi, P.; Budavari, A.; Ellis, R.; White, M.F.; Spiegelman, B.M. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 1996, 271, 665–668. [Google Scholar] [CrossRef]
- Garvey, W.T.; Maianu, L.; Zhu, J.H.; Brechtel-Hook, G.; Wallace, P.; Baron, A.D. Evidence for defects in the trafficking and translocation of GLUT4 glucose transporters in skeletal muscle as a cause of human insulin resistance. J. Clin. Investig. 1998, 101, 2377–2386. [Google Scholar] [CrossRef] [PubMed]
- Papanicolaou, D.A.; Wilder, R.L.; Manolagas, S.C.; Chrousos, G.P. The pathophysiologic roles of interleukin-6 in human disease. Ann. Intern. Med. 1998, 128, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Mohamed-Ali, V.; Goodrick, S.; Rawesh, A.; Katz, D.R.; Miles, J.M.; Yudkin, J.S.; Klein, S.; Coppack, S.W. Subcutaneous adipose tissue releases interleukin-6, but not tumor necrosis factor-alpha, in vivo. J. Clin. Endocrinol. Metab. 1997, 82, 4196–4200. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Eagon, J.C.; Trujillo, M.E.; Scherer, P.E.; Klein, S. Visceral fat adipokine secretion is associated with systemic inflammation in obese humans. Diabetes 2007, 56, 1010–1013. [Google Scholar] [CrossRef]
- Senn, J.J.; Klover, P.J.; Nowak, I.A.; Zimmers, T.A.; Koniaris, L.G.; Furlanetto, R.W.; Mooney, R.A. Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. J. Biol. Chem. 2003, 278, 13740–13746. [Google Scholar] [CrossRef]
- Bastard, J.P.; Jardel, C.; Bruckert, E.; Blondy, P.; Capeau, J.; Laville, M.; Vidal, H.; Hainque, B. Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss. J. Clin. Endocrinol. Metab. 2000, 85, 3338–3342. [Google Scholar] [CrossRef]
- Nishimoto, S.; Fukuda, D.; Higashikuni, Y.; Tanaka, K.; Hirata, Y.; Murata, C.; Kim-Kaneyama, J.R.; Sato, F.; Bando, M.; Yagi, S.; et al. Obesity-induced DNA released from adipocytes stimulates chronic adipose tissue inflammation and insulin resistance. Sci. Adv. 2016, 2, e1501332. [Google Scholar] [CrossRef]
- Revelo, X.S.; Ghazarian, M.; Chng, M.H.; Luck, H.; Kim, J.H.; Zeng, K.; Shi, S.Y.; Tsai, S.; Lei, H.; Kenkel, J.; et al. Nucleic Acid-Targeting Pathways Promote Inflammation in Obesity-Related Insulin Resistance. Cell Rep. 2016, 16, 717–730. [Google Scholar] [CrossRef]
- Pérez, S.; Finamor, I.; Martí-Andrés, P.; Pereda, J.; Campos, A.; Domingues, R.; Haj, F.; Sabater, L.; de-Madaria, E.; Sastre, J. Role of obesity in the release of extracellular nucleosomes in acute pancreatitis: A clinical and experimental study. Int. J. Obes. (Lond.) 2019, 43, 158–168. [Google Scholar] [CrossRef]
- Lo Re, O.; Maugeri, A.; Hruskova, J.; Jakubik, J.; Kucera, J.; Bienertova-Vasku, J.; Oben, J.A.; Kubala, L.; Dvorakova, A.; Ciz, M.; et al. Obesity-induced nucleosome release predicts poor cardio-metabolic health. Clin. Epigenetics 2020, 12, 2. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kojta, I.; Chacińska, M.; Błachnio-Zabielska, A. Obesity, Bioactive Lipids, and Adipose Tissue Inflammation in Insulin Resistance. Nutrients 2020, 12, 1305. https://doi.org/10.3390/nu12051305
Kojta I, Chacińska M, Błachnio-Zabielska A. Obesity, Bioactive Lipids, and Adipose Tissue Inflammation in Insulin Resistance. Nutrients. 2020; 12(5):1305. https://doi.org/10.3390/nu12051305
Chicago/Turabian StyleKojta, Iwona, Marta Chacińska, and Agnieszka Błachnio-Zabielska. 2020. "Obesity, Bioactive Lipids, and Adipose Tissue Inflammation in Insulin Resistance" Nutrients 12, no. 5: 1305. https://doi.org/10.3390/nu12051305
APA StyleKojta, I., Chacińska, M., & Błachnio-Zabielska, A. (2020). Obesity, Bioactive Lipids, and Adipose Tissue Inflammation in Insulin Resistance. Nutrients, 12(5), 1305. https://doi.org/10.3390/nu12051305