Abstract
Vitamin C (ascorbate, ASC) is a critical antioxidant in the body with specific roles in the brain. Despite a recent interest in vitamin C therapies for critical care medicine, little is known about vitamin C regulation during acute inflammation and critical illnesses such as sepsis. Using a cecal slurry (CS) model of sepsis in mice, we determined ASC and inflammatory changes in the brain following the initial treatment. ASC levels in the brain were acutely decreased by approximately 10% at 4 and 24 h post CS treatment. Changes were accompanied by a robust increase in liver ASC levels of up to 50%, indicating upregulation of synthesis beginning at 4 h and persisting up to 7 days post CS treatment. Several key cytokines interleukin 6 (IL-6), interleukin 1β (IL-1β), tumor necrosis factor alpha (TNFα), and chemokine (C-X-C motif) ligand 1 (CXCL1, KC/Gro) were also significantly elevated in the cortex at 4 h post CS treatment, although these levels returned to normal by 48 h. These data strongly suggest that ASC reserves are directly challenged throughout illness and recovery from sepsis. Given the timescale of this response, decreases in cortical ASC are likely driven by hyper-acute neuroinflammatory processes. However, future studies are required to confirm this relationship and to investigate how this deficiency may subsequently impact neuroinflammation.
1. Introduction
Sepsis is estimated to affect more than 30 million people and account for more than 5 million deaths annually [1]. During this critical illness, the robust inflammatory response of the immune system includes release of multiple cytokines and other signaling molecules that can ultimately lead to severe tissue injury and multiple organ failure [2,3,4]. This damage extends to the brain as evidenced by delirium in the majority of patients [5]. Despite the advances in clinical understanding of delirium during sepsis, little is known about the cellular and molecular underpinnings of acute brain dysfunction in critically ill patients. Sepsis patients often exhibit low plasma levels of vitamin C (ascorbate, ASC) [6,7,8,9]. In one study, as many as 88% of sepsis patients had subnormal plasma levels of ASC (<23 µM) and up to 38% had severe ASC deficiency (<11 µM) [10], suggesting high demand for ASC during septic insult. Lower plasma levels of ASC are also associated with increased incidence of multiple organ failure and decreased survival [11]. The brain is particularly susceptible to this dysregulated inflammatory response and to suboptimal ASC levels, because higher oxidative stress levels in the brain are especially damaging to its enriched lipid composition and nutrient-demanding metabolic rate [12,13]. Up to 30% of patients are reported to experience cognitive deficits following recovery from sepsis [14,15,16], and several studies using rodent sepsis models have shown that acute illness damages cognition in surviving animals [17,18,19].
ASC is a critical antioxidant for cellular function and has an emerging role in immune function [20]. Preclinical studies have shown a variety of beneficial effects on the pathophysiological changes in sepsis, including protection against microvascular dysfunction and deficits in vasoconstriction [21,22,23] by preserving tight endothelial barrier function and capillary blood flow [24,25,26]. ASC administration during sepsis also attenuates acute lung injury [27] and improves multiple organ dysfunction syndrome in animal models of sepsis [28,29]. The role of intravenous ASC in clinical practice to improve short term patient recovery is still under clinical investigation [30,31]. ASC accumulates to high levels in the brain via the sodium-dependent vitamin C transporter 2 (SVCT2) in a two-step, energy-dependent process from blood into the choroid plexus cerebral spinal fluid and then into neurons [32]. In the brain, ASC serves two primary roles as a neuroprotector and neuromodulator [32,33,34]. ASC maintains blood–brain barrier integrity by preserving tight endothelial barriers [35,36] and maintaining capillary blood flow [37]. ASC is a critical enzymatic co-factor in neurotransmitter synthesis and DNA methylation [32,38], and is intimately involved in preserving glutamatergic neurotransmission through the glutamate-uptake ASC-release exchange [39,40,41]. Despite the interest in ASC as a treatment for preventing organ failure in sepsis, the roles of ASC in the brain, and the number of patients experiencing cognitive deficits following recovery from the acute trauma, the effects of sepsis on the brain are understudied. Furthermore, the specific role of ascorbate in sepsis-induced brain dysfunction has not been studied. Here, we utilized a cecal slurry (CS) model of sepsis in mice to observe changes in ASC and inflammatory response in the brain during and following sepsis. We hypothesized that ASC is depleted in sepsis, and sought to define a timeline for changes in ASC level and cytokine release, particularly in the brain.
2. Materials and Methods
2.1. Mouse CS or LPS Treatment
All experiments conducted with live mice were reviewed and approved by the Vanderbilt Institutional Animal Care and Use Committee. Cecal slurry (CS) was used to induce acute peritonitis in mice as a model of sepsis as previously described [42,43,44]. C57/Bl6J donor mice at six weeks of age were obtained from Jackson Laboratory (#000664) and euthanized within 7 days of arrival. Cecal contents were collected and resuspended in 5% dextrose at 80 mg/mL, then filtered through a 100 µm filter. Aliquots were stored at -80 °C until ready for use.
All mice for treatment groups were bred in house from C57/Bl6J mice originally obtained from Jackson Laboratory. Mice at 10–12 weeks of age were treated with CS (1.5 mg/g; i.p.) or the vehicle 5% dextrose for control groups. Another widely used model of peripheral inflammatory response utilizes lipopolysaccharide (LPS) administration. For LPS studies, mice at 6–8 weeks of age received LPS (3.75 µg/g; i.p.) or saline. Control and treated mice were distributed across cages and provided with supplemental nutrition on the floor of the cage (DietGel 76A, Clear H2O) to promote survival.
2.2. Evaluation of Sickness Score
An observer blinded to treatment groups scored the mice on the severity of sepsis using a 12-point scale of sickness severity, where 12 is healthy with normal activity and 0 is moribund [45,46]. In brief, the score is determined by response to finger poke (4 for normal, 3 for decreased, 2 for severely decreased, or 1 for minimal response), signs of encephalopathy (4 for normal, 3 for tremors or staggering, 2 for twisting movements, or 1 for turning), and general appearance (score is decreased by 1 for each display of piloerection, periorbital exudates, respiratory distress, or diarrhea). All mice were monitored closely for 48 h or until mice recovered to a normal score of 12. CS-treated mice that never received a score below 10 or died prior to the assigned timepoint were excluded from analysis.
2.3. Tissue Collection
Mice were anesthetized with isoflurane then euthanized by decapitation at 4, 24, 48 h or 7 days post-injection. Control mice were euthanized at each timepoint and data collapsed into one group. Tissue samples were collected, flash-frozen on dry ice, and stored at –80 °C for further analysis.
2.4. Ascorbic Acid HPLC
Sample extracts were prepared by adding 10 µl extraction buffer (7:2 25% w/v metaphosphoric acid: 100 mM sodium phosphate, 0.05 mM EDTA pH 8.0) per mg of wet tissue to normalize by weight. Samples were homogenized with 0.5 mm ceria-stabilized zirconium oxide beads (Next Advance, Inc.) in a bullet homogenizer, centrifuged at 10,000 rpm, and the clear supernatant transferred into a fresh tube. Concentrations of ASC were measured at 1:100 dilution in triplicate with ion pair HPLC, using tetrapentyl ammonium bromide as the ion pair reagent and electrochemical detection as previously described [47,48].
2.5. Determination of Gene Expression
RNA was extracted using an RNeasy Mini Kit (Qiagen). qPCR was performed using a PrimePCR Probe Assay consisting of iScript cDNA synthesis, PrimePCR Probes (PrimePCR™ Probe Assay: Slc23a1, Mouse), and Sso Advanced Universal Supermix (Bio Rad).
2.6. Measurement of Oxidative Stress Markers
Malondialdehyde, a lipid peroxidation end product, was measured by fluorescent spectrophotometric assay of thiobarbituric acid reactive substances (TBARS) as previously described [49]. Sulfhydryls were measured by reduction of 5,5’-dithiobis (2-nitrobenzoic acid) (DTNB) to 2-nitro-5-thiobenzoate (TNB) anion by thiol groups and spectrophotometric analysis [50,51].
2.7. Measurement of Cytokine Expression
Frozen mouse tissues were homogenized in volumes of RIPA buffer (Thermo Fisher Scientific) normalized by tissue weight. Tissue levels of IFN-y, IL-1b, IL-6, KC/GRO, IL-10, and TNF-alpha were assayed in duplicate using a V-PLEX Custom Mouse Cytokine Kit, (Meso Scale Diagnostics, LLC) according to the manufacturer’s instructions [52].
2.8. Statistical Analyses
Statistical analyses were performed using Graphpad Prism software (version 8.3.0). Data were first checked for equality of group variances using the Brown–Forsyth test and analyzed using parametric statistics. We did not expect any differences in response to CS according to sex, and all data were first analyzed using a multivariate ANOVA analysis, including sex as an additional variable. There were no main effects of sex on any of the key outcomes (sickness score, weight loss, ASC levels) so data were combined for all subsequent analysis. For outcomes following CS treatment, data were analyzed with univariate ANOVA with group (encompassing treatments and time post treatment) as the main independent variable. Significant omnibus ANOVA were followed with Dunnett’s post hoc analyses to test difference from the control group. Independent t-tests were used to test effects of LPS versus treatment with the vehicle. Numerical outliers that likely reflected experimental error were identified and removed using ROUT (Q = 5%). Error bars are shown as SEM or SD as indicated in figure legends.
3. Results
3.1. Cecal Slurry Treatment Induces Acute Peritonitis and Weight Loss
Mice that received cecal slurry (CS) treatment became severely lethargic and exhibited decreased responses to stimuli, signs of encephalopathy, and worsened appearance by 12 h (Figure 1A). Sickness scores decreased as early as 4 h, began to recover by 24 h, and returned to normal appearance and activity by 48 h (Figure 1B; CS Treatment F(1, 63) = 113.8, p < 0.001; Time F(5, 59) = 15.61, p < 0.001). CS treatment also caused significant weight loss that persisted to 48 h, although mice regained weight by 7 days post CS treatment (Figure 1C; CS Treatment F(1, 63) = 121.3, p < 0.001; Time F(5, 59) = 24.06, p < 0.001). Out of 55 CS treated mice, only two mice died before their scheduled timepoint. This mild 1.5 mg/g dose was chosen to optimize survival to 7 days post CS treatment. The 4% mortality rate at this dose is low compared to other studies utilizing this CS model (2.0 mg/g, up to 67% by 48 h) [42,44].
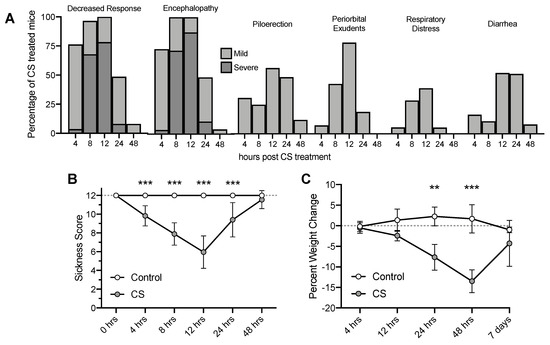
Figure 1.
Observed sickness scores of mice and percent weight change over time following Cecal Slurry (CS) treatment. (A) Percentage of mice showing sickness behaviors and appearances. (B) Clinical Sickness Scores. (C) Weight loss. ** p < 0.01*** p < 0.001. Error bars plotted as mean ± SD.
3.2. Tissue ASC Concentrations Following CS Treatment
There was a small (~10% at 4 h) but significant decrease in cortical ASC at 4 and 24 h following CS treatment compared to controls (Figure 2A, F(4, 54) = 3.216, p = 0.0193,), indicating consumption of brain ASC reserves. Liver ASC levels were significantly increased following CS treatment (~50% at 24 h) indicating robust upregulation of ASC synthesis (Figure 2B, F(4, 54) = 5.090, p = 0.0015) that persisted to 7 days post CS treatment. No significant differences in ASC levels were observed in peripheral organs of CS-treated mice compared to controls, although levels varied post CS treatment (kidney: Figure 2C, F(4, 54) = 2.153, p = 0.0867; lung: Figure 2D, F(4, 55) = 3.188, p = 0.020). We hypothesized that a decrease in ASC levels in the brain would result in upregulation of SVCT2 expression, though no significant changes were observed in hippocampal SVCT2 expression in response to CS treatment (Figure 2E, F(4, 38) = 2.235, p = 0.0833).
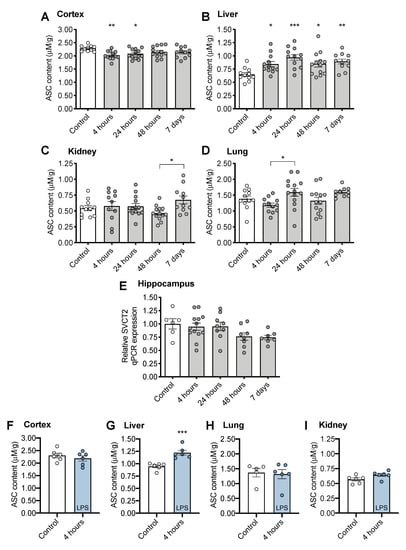
Figure 2.
Tissue ASC (ascorbate) concentrations following CS treatment in (A) cortex, (B) liver, (C) kidney, (D) lung. (E) Sodium-dependent vitamin C transporter 2, SVCT2 gene expression in brain following CS treatment. Tissue ASC concentrations following LPS treatment (F) brain, (G) liver, (H) lung, and (I) kidney. * p < 0.05 ** p < 0.01 *** p < 0.001 from control following significant ANOVA results unless otherwise indicated. Error bars plotted as mean ± SEM.
No decrease in brain ASC level was observed at 4 h following LPS treatment (Figure 2F, t(10) = 0.842, p = 0.4197). However, LPS treatment also induced upregulation of ASC synthesis in liver (Figure 2G, t(10) = 4.913, p < 0.001). No changes were observed in lung (Figure 2H, t(9) = 0.246, p = 0.8111), or kidney (Figure 2I, t(10) = 2.066, p = 0.0658).
3.3. CS Treatment Does Not Induce Changes in Oxidative Stress Measurements
CS treatment did not increase either of the oxidative stress markers malondialdehyde (cortex: Figure 3A, F(4, 33) = 2.092, p = 0.1042; Liver: Figure 3B, F(4, 33) = 1.833, p = 0.1459) or sulfhydryls (cortex: Figure 3C, F(4, 30) = 0.6471, p = 0.6333; Liver: Figure 3D, F(4, 35) = 2.210, p = 0.0880) in the brain.
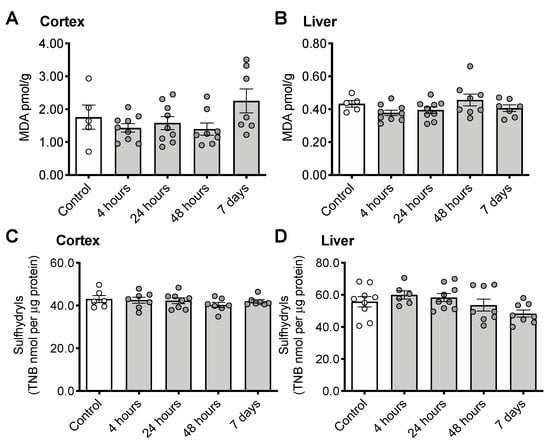
Figure 3.
Indicators of oxidative stress following CS treatment. Malondialdehyde (MDA) in cortex (A) and liver (B) or sulfhydryls in cortex (C) and liver (D). Error bars plotted as mean ± SEM.
3.4. CS Treatment Initiates an Inflammatory Response in the Brain
In cortex, expression of several proinflammatory cytokines was elevated at 4 h post CS treatment including interleukin 6 (IL-6: F(4, 31) = 8.356, p < 0.001), interleukin 1β (IL-1β: F(4, 31) = 9.742, p < 0.001), tumor necrosis factor alpha (TNFα: F(4, 33) = 4.261, p = 0.0069), and chemokine (C-X-C motif) ligand 1 (CXCL1, KC/Gro: F(4, 30) = 7.091, p < 0.001) (Figure 4A–D). Cytokine expression levels returned to normal by 48 h post CS treatment. More modest increases in interferon gamma (INFγ: F(4, 33) = 0.9214, p = 0.4632) and interleukin 10 (IL-10: F(4, 32) = 1.442, p = 0.2430) were not statistically significant (Figure 2E,F).
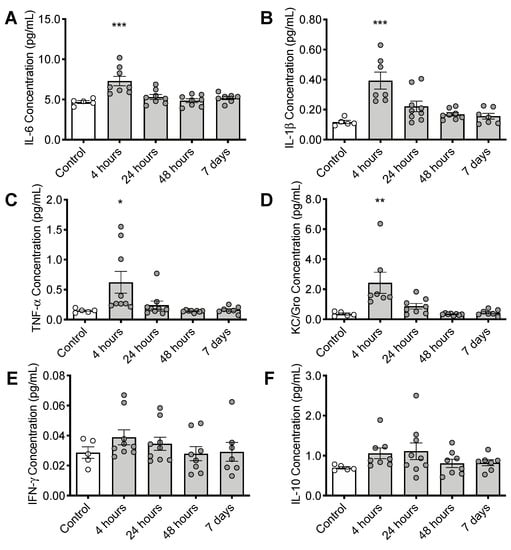
Figure 4.
Timeline of inflammatory changes in the brain following CS treatment. Cortical cytokine levels of (A) interleukin 6 (IL-6), (B) interleukin 1β (IL-1β), (C) tumor necrosis factor alpha (TNFα), and (D) chemokine (C-X-C motif) ligand 1 (CXCL1, KC/Gro), (E) Interferon gamma (INFγ) and (F) interleukin 10 (IL-10) * p < 0.05, ** p < 0.01, *** p < 0.001. Error bars plotted as mean ± SEM.
4. Discussion
Despite significant clinical interest in ASC as a potential therapeutic adjuvant, the role of ASC in sepsis, particularly in the brain, has not been well studied. The present study highlights the potential rapid, although modest, consumption of ASC stores during and following sepsis co-occurring with the associated inflammatory changes that occur in the brain.
Mice and most other rodent species possess the gene encoding gulonolactone oxidase, an enzyme responsible for catalyzing the final step in ASC synthesis in the liver [53,54,55,56]. Synthesis can be upregulated to provide higher tissue levels in the liver under periods of increased physiological need, such as pregnancy [57]. ASC depletion in the brain is rare in non-genetically modified mice, however, increased liver levels indicate upregulation of ASC synthesis to prevent depletion. In the brain, high concentrations are critical for maintaining optimal brain function and preventing oxidative damage [32,33,58]. CS treatment resulted in an approximately 10% decrease in ASC in cortex (Figure 2A) even in this relatively mild sepsis model, and despite upregulation of synthesis in the liver. The two-step transport system of ASC into the brain by SVCT2 from blood into the choroid plexus cerebral spinal fluid and then into neurons allows for preservation of ASC at the expense of other tissues [32]. It is possible that other brain regions, such as the hippocampus, may have different levels of susceptibility to ASC depletion due to varying proximity to the ventricles and choroid plexus, and future studies should address different brain areas, including hippocampus, in analysis.
Over this short time interval, we did not observe decreased ASC levels in most peripheral organs including lung and kidney (Figure 2C,D) and heart, muscle, spleen, and adrenal gland (data not shown). We observed a significant increase in liver ASC by 4 h in both CS- and LPS-treated mice (Figure 2B,G). Our data confirm results of prior work in which an LPS-induced increase in liver ASC was observed from 3 h post LPS treatment [59]. LPS treatment did not fully recapitulate the ASC changes we observed with CS treatment, possibly due to the dose initiating a lower neuroinflammatory effect or different pathways, although this was not directly tested. While no changes were observed in brain in that study [59], the lack of cortical ASC deficiency in the LPS model may be a limitation of intraperitoneal LPS as a model of sepsis. We hypothesize that increased synthesis in the liver provided sufficient circulating levels to replenish (brain) or protect (lung, kidneys) peripheral tissues during the time period and for the dose studied. It took more than 24 h to replenish brain ASC, possibly due to slower brain uptake, since ASC must first accumulate in the cerebral spinal fluid. Future experiments should be performed in genetically modified mice that are, like humans, incapable of synthesizing ASC (e.g., gulonolactone oxidase knockout mice) [47,60], and are therefore unable to upregulate hepatic synthesis to meet increased need. Such models could also be used to establish a timeline of circulating ASC levels in plasma as well as testing whether a clinically relevant ASC pretreatment [61] is capable of protecting against decreased brain ASC, neuroinflammatory changes, or severity of observed sickness. If peripheral ASC administration is unable to change cortical ASC levels, this would suggest blood–brain barrier transport of ASC is limiting early in sepsis or that there is high uptake by other organs. However, if neuroinflammation is improved by pretreatment with ASC, this would suggest that ASC deficiency may be directly associated with sepsis-associated neuroinflammation.
The CS dosing regimen used (1.5 mg/g) causes a mild sickness response, chosen to optimize recovery and survival to 7 days post treatment. Nevertheless, this modest insult was still sufficient to deplete ASC and increase cytokine production in the brain at 4 h after injection (Figure 4), when the mice are just starting to become systemically ill. This finding suggests that the brain is adversely affected by systemic inflammation in the earliest stages of illness. The release of cytokines in the brain at this time indicates activation of brain macrophages and resident microglia, which generate reactive oxygen species. The observed ASC depletion likely indicates ASC plays a primary role in electron donation to neutralize these free radicals, and that the oxidative stress was sufficient to overwhelm ASC recycling capacity. While future studies will need to clarify the relationship between ASC deficiency and neuroinflammation, it is most likely that ASC deficiency is driven by the acute neuroinflammatory response and associated generation of radical species. Oxidative stress is a key component of clinical sepsis [62], and although the global oxidative measurements did not show elevations in brain or liver oxidative stress under these conditions, any reduction in brain ASC and especially upregulation of liver ASC synthesis indicates elevated oxidative challenge. The measures of global tissue MDA may not have been sensitive enough to detect localized increases in oxidative stress in specific cells (endothelium, for example) or tissue compartments. Despite the activated immune response and challenge of brain ASC stores, we did not observe upregulation of the sodium-dependent vitamin C transporter SVCT2 in brain tissue. To confirm the association between ASC consumption and acute inflammatory challenge in a second model, we used LPS to induce endotoxemia and systemic inflammation. LPS treatment was also sufficient to upregulate liver ASC synthesis by 4 h. Whether higher doses of CS or LPS would cause further or prolonged depletion of brain ASC in a more severe illness model should be studied in future experiments.
ASC deficiency is well defined in critical care patient populations [6,7,8,9,10]. Many preclinical studies have shown that early ASC supplementation or IV treatment can protect against vascular and organ dysfunctions associated with sepsis [22,23,26]. One possible explanation for the potential beneficial effects of ASC is that hospitalized patients who develop sepsis are more likely to have underlying ASC deficiency, either as a result of chronic illness, co-morbidities, or poor diet. However, our results and many others suggest that ASC depletion is directly caused by the illness itself, likely due to massive inflammatory challenge, endothelial breakdown, and elevated oxidative stress [35,37]. Although some clinical studies have associated lower plasma levels of ASC with increased incidence of multiple organ failure and decreased survival [11], several phase I clinical studies have shown that IV administration of ASC during sepsis does not improve or worsen short term survival outcomes [8,31,63]. Overall survivability during sepsis is dependent on a variety of factors including antibiotics administration, prior health status, prior injury or illness, and age [64]. While maintenance of ASC levels during sepsis may not directly impact acute survival, it may be critical to protection against inflammatory damage following sepsis, especially in the brain [14]. Future studies will seek to understand how ASC is involved in the acute inflammatory response and the implications for long term cognitive dysfunction following recovery.
Author Contributions
Conceptualization, F.E.H., J.A.B. and D.C.C.; statistical analyses, F.E.H. and D.C.C.; experimental investigation, D.C.C., J.J.J., N.D.P., K.R.K., and A.A.T.; writing—original draft preparation, D.C.C.; writing—review and editing, F.E.H. and J.A.B.; funding acquisition, F.E.H. and J.A.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by R01 HL135849-03 and HL135849-02S1 to Lorraine Ware and Julie Bastarache, and VA Merit I01 CX001610 to James M May.
Acknowledgments
The authors would like to thank Jordyn M Wilcox, Shilpy Dixit, Krista C Paffenroth, and John E Dugan for technical assistance in generating data for this manuscript, and James M May for providing valuable feedback on early versions of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Fleischmann, C.; Scherag, A.; Adhikari, N.K.J.; Hartog, C.S.; Tsaganos, T.; Schlattmann, P.; Angus, D.C.; Reinhart, K. Assessment of global incidence and mortality of hospital-treated sepsis current estimates and limitations. Am. J. Respir. Crit. Care Med. 2016, 193, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, S.O.; Meissner, K.; Mayes, L.M.; Bartels, K. Vitamin C in sepsis. Curr. Opin. Anaesthesiol. 2018, 31, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Ince, C.; Mayeux, P.R.; Nguyen, T.; Gomez, H.; Kellum, J.A.; Ospina-Tascón, G.A.; Hernandez, G.; Murray, P.; De Backer, D. The endothelium in sepsis. Shock 2016, 45, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Biesalski, H.K.; McGregor, G.P. Antioxidant therapy in critical care–Is the microcirculation the primary target? Proc. Crit. Care Med. 2007, 35, S577–S583. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, F.G.; Park, M.; Machado, F.S.; Azevedo, L.C.P. Sepsis-associated encephalopathy: Not just delirium. Clinics 2011, 66, 1825–1831. [Google Scholar] [CrossRef][Green Version]
- Borrelli, E.; Roux-Lombard, P.; Grau, G.E.; Girardin, E.; Ricou, B.; Dayer, J.M.; Suter, P.M. Plasma concentrations of cytokines, their soluble receptors, and antioxidant vitamins can predict the development of multiple organ failure in patients at risk. Crit. Care Med. 1996, 24, 392–397. [Google Scholar] [CrossRef]
- Galley, H.F.; Davies, M.J.; Webster, N.R. Ascorbyl radical formation in patients with sepsis: Effect of ascorbate loading. Free Radic. Biol. Med. 1996, 20, 139–143. [Google Scholar] [CrossRef]
- Fujii, T.; Udy, A.A.; Deane, A.M.; Luethi, N.; Bailey, M.; Eastwood, G.M.; Frei, D.; French, C.; Orford, N.; Shehabi, Y.; et al. Vitamin C, Hydrocortisone and Thiamine in Patients with Septic Shock (VITAMINS) trial: Study protocol and statistical analysis plan. Crit. Care Resusc. 2019, 21, 119–125. [Google Scholar]
- Hudson, E.P.; Collie, J.T.; Fujii, T.; Luethi, N.; Udy, A.A.; Doherty, S.; Eastwood, G.; Yanase, F.; Naorungroj, T.; Bitker, L.; et al. Pharmacokinetic data support 6-hourly dosing of intravenous vitamin C to critically ill patients with septic shock. Crit. Care Resusc. 2019, 21, 236–242. [Google Scholar]
- Carr, A.C.; Rosengrave, P.C.; Bayer, S.; Chambers, S.; Mehrtens, J.; Shaw, G.M. Hypovitaminosis C and vitamin C deficiency in critically ill patients despite recommended enteral and parenteral intakes. Crit. Care 2017, 21, 300. [Google Scholar] [CrossRef] [PubMed]
- Fowler, A.A.; Syed, A.A.; Knowlson, S.; Sculthorpe, R.; Farthing, D.; DeWilde, C.; Farthing, C.A.; Larus, T.L.; Martin, E.; Brophy, D.F.; et al. Phase I safety trial of intravenous ascorbic acid in patients with severe sepsis. J. Transl. Med. 2014, 12, 32. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I. A Cellular Perspective on Brain Energy Metabolism and Functional Imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [PubMed]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Calsavara, A.J.C.; Costa, P.A.; Nobre, V.; Teixeira, A.L. Factors Associated with Short and Long Term Cognitive Changes in Patients with Sepsis. Sci. Rep. 2018, 8, 4509. [Google Scholar] [CrossRef]
- Iwashyna, T.J.; Ely, E.W.; Smith, D.M.; Langa, K.M. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA—J. Am. Med. Assoc. 2010, 304, 1787–1794. [Google Scholar] [CrossRef]
- Annane, D.; Sharshar, T. Cognitive decline after sepsis. Lancet Respir. Med. 2015, 3, 61–69. [Google Scholar] [CrossRef]
- Anderson, S.T.; Commins, S.; Moynagh, P.N.; Coogan, A.N. Lipopolysaccharide-induced sepsis induces long-lasting affective changes in the mouse. Brain. Behav. Immun. 2015, 43, 98–109. [Google Scholar] [CrossRef]
- Zaghloul, N.; Addorisio, M.E.; Silverman, H.A.; Patel, H.L.; Valdés-Ferrer, S.I.; Ayasolla, K.R.; Lehner, K.R.; Olofsson, P.S.; Nasim, M.; Metz, C.N.; et al. Forebrain Cholinergic Dysfunction and Systemic and Brain Inflammation in Murine Sepsis Survivors. Front. Immunol. 2017, 8, 1673. [Google Scholar] [CrossRef]
- Hippensteel, J.A.; Anderson, B.J.; Orfila, J.E.; McMurtry, S.A.; Dietz, R.M.; Su, G.; Ford, J.A.; Oshima, K.; Yang, Y.; Zhang, F.; et al. Circulating heparan sulfate fragments mediate septic cognitive dysfunction. J. Clin. Investig. 2019, 129, 1779–1784. [Google Scholar] [CrossRef]
- Carr, A.C.; Maggini, S. Vitamin C and immune function. Nutrients 2017, 9, 1211. [Google Scholar] [CrossRef]
- Armour, J.; Tyml, K.; Lidington, D.; Wilson, J.X. Ascorbate prevents microvascular dysfunction in the skeletal muscle of the septic rat. J. Appl. Physiol. 2001, 90, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Wilson, J.X.; Tyml, K. Ascorbate protects against impaired arteriolar constriction in sepsis by inhibiting inducible nitric oxide synthase expression. Free Radic. Biol. Med. 2004, 37, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Mckinnon, R.L.; Lidington, D.; Tyml, K. Ascorbate inhibits reduced arteriolar conducted vasoconstriction in septic mouse cremaster muscle. Microcirculation 2007, 14, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Pendem, S.; Teh, S.L.; Sukumaran, D.K.; Wu, F.; Wilson, J.X. Ascorbate protects endothelial barrier function during septic insult: Role of protein phosphatase type 2A. Free Radic. Biol. Med. 2010, 48, 128–135. [Google Scholar] [CrossRef]
- Tyml, K.; Li, F.; Wilson, J.X. Septic impairment of capillary blood flow requires nicotinamide adenine dinucleotide phosphate oxidase but not nitric oxide synthase and is rapidly reversed by ascorbate through an endothelial nitric oxide synthase-dependent mechanism. Crit. Care Med. 2008, 36, 2355–2362. [Google Scholar] [CrossRef]
- Zhou, G.; Kamenos, G.; Pendem, S.; Wilson, J.X.; Wu, F. Ascorbate protects against vascular leakage in cecal ligation and puncture-induced septic peritonitis. Am. J. Physiol.—Regul. Integr. Comp. Physiol. 2012, 302, R409–R416. [Google Scholar] [CrossRef]
- Fisher, B.J.; Seropian, I.M.; Kraskauskas, D.; Thakkar, J.N.; Voelkel, N.F.; Fowler, A.A.; Natarajan, R. Ascorbic acid attenuates lipopolysaccharide-induced acute lung injury. Crit. Care Med. 2011, 39, 1454–1460. [Google Scholar] [CrossRef]
- Fisher, B.J.; Kraskauskas, D.; Martin, E.J.; Farkas, D.; Puri, P.; Massey, H.D.; Idowu, M.O.; Brophy, D.F.; Voelkel, N.F.; Fowler, A.A.; et al. Attenuation of sepsis-induced organ injury in mice by vitamin C. J. Parenter. Enter. Nutr. 2014, 38, 825–839. [Google Scholar] [CrossRef]
- Gao, Y.-L.; Lu, B.; Zhai, J.-H.; Liu, Y.-C.; Qi, H.-X.; Yao, Y.; Chai, Y.-F.; Shou, S.-T. The Parenteral Vitamin C Improves Sepsis and Sepsis-Induced Multiple Organ Dysfunction Syndrome via Preventing Cellular Immunosuppression. Mediat. Inflamm. 2017, 2017, 4024672. [Google Scholar] [CrossRef]
- Fujii, T.; Luethi, N.; Young, P.J.; Frei, D.R.; Eastwood, G.M.; French, C.J.; Deane, A.M.; Shehabi, Y.; Hajjar, L.A.; Oliveira, G.; et al. Effect of Vitamin C, Hydrocortisone, and Thiamine vs Hydrocortisone Alone on Time Alive and Free of Vasopressor Support among Patients with Septic Shock: The VITAMINS Randomized Clinical Trial. JAMA—J. Am. Med. Assoc. 2020, 323, 423–431. [Google Scholar] [CrossRef]
- Fowler, A.A.; Truwit, J.D.; Hite, R.D.; Morris, P.E.; Dewilde, C.; Priday, A.; Fisher, B.; Thacker, L.R.; Natarajan, R.; Brophy, D.F.; et al. Effect of Vitamin C Infusion on Organ Failure and Biomarkers of Inflammation and Vascular Injury in Patients with Sepsis and Severe Acute Respiratory Failure: The CITRIS-ALI Randomized Clinical Trial. JAMA—J. Am. Med. Assoc. 2019, 322, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E.; May, J.M. Vitamin C function in the brain: Vital role of the ascorbate transporter SVCT2. Free Radic. Biol. Med. 2009, 46, 719–730. [Google Scholar] [CrossRef]
- Harrison, F.E.; Bowman, G.L.; Polidori, M.C. Ascorbic acid and the brain: Rationale for the use against cognitive decline. Nutrients 2014, 6, 1752. [Google Scholar] [CrossRef] [PubMed]
- Ballaz, S.J.; Rebec, G.V. Neurobiology of vitamin C: Expanding the focus from antioxidant to endogenous neuromodulator. Pharm. Res. 2019, 146, 104321. [Google Scholar] [CrossRef] [PubMed]
- Kuck, J.L.; Bastarache, J.A.; Shaver, C.M.; Fessel, J.P.; Dikalov, S.I.; May, J.M.; Ware, L.B. Ascorbic acid attenuates endothelial permeability triggered by cell-free hemoglobin. Biochem. Biophys. Res. Commun. 2018, 495, 433–437. [Google Scholar] [CrossRef]
- Lin, J.L.; Huang, Y.H.; Shen, Y.C.; Huang, H.C.; Liu, P.H. Ascorbic acid prevents blood-brain barrier disruption and sensory deficit caused by sustained compression of primary somatosensory cortex. J. Cereb. Blood Flow Metab. 2010, 30, 1121–1136. [Google Scholar] [CrossRef]
- Wilson, J.X.; Wu, F. Vitamin C in sepsis. Subcell. Biochem. 2012, 56, 67–83. [Google Scholar]
- Young, J.I.; Züchner, S.; Wang, G. Regulation of the Epigenome by Vitamin C. Annu. Rev. Nutr. 2015, 35, 545–564. [Google Scholar] [CrossRef]
- Wilson, J.X.; Peters, C.E.; Sitar, S.M.; Daoust, P.; Gelb, A.W. Glutamate stimulates ascorbate transport by astrocytes. Brain Res. 2000, 858, 61–66. [Google Scholar] [CrossRef]
- May, J.M. Vitamin C transport and its role in the central nervous system. Subcell. Biochem. 2012, 56, 85–103. [Google Scholar]
- Mi, D.J.; Dixit, S.; Warner, T.A.; Kennard, J.A.; Scharf, D.A.; Kessler, E.S.; Moore, L.M.; Consoli, D.C.; Bown, C.W.; Eugene, A.J.; et al. Altered glutamate clearance in ascorbate deficient mice increases seizure susceptibility and contributes to cognitive impairment in APP/PSEN1 mice. Neurobiol. Aging 2018, 71, 241–254. [Google Scholar] [CrossRef]
- Shaver, C.M.; Paul, M.G.; Putz, N.D.; Landstreet, S.R.; Kuck, J.L.; Scarfe, L.; Skrypnyk, N.; Yang, H.; Harrison, F.E.; de Caestecker, M.P.; et al. Cell-free hemoglobin augments acute kidney injury during experimental sepsis. Am. J. Physiol. Physiol. 2019, 317, F922–F929. [Google Scholar] [CrossRef]
- Meegan, J.E.; Shaver, C.M.; Putz, N.D.; Jesse, J.J.; Landstreet, S.R.; Lee, H.N.R.; Sidorova, T.N.; Brennan McNeil, J.; Wynn, J.L.; Cheung-Flynn, J.; et al. Cell-free hemoglobin increases inflammation, lung apoptosis, and microvascular permeability in murine polymicrobial sepsis. PLoS ONE 2020, 15, e0228727. [Google Scholar] [CrossRef]
- Eric Kerchberger, V.; Bastarache, J.A.; Shaver, C.M.; Nagata, H.; Brennan McNeil, J.; Landstreet, S.R.; Putz, N.D.; Kuang Yu, W.; Jesse, J.; Wickersham, N.E.; et al. Haptoglobin-2 variant increases susceptibility to acute respiratory distress syndrome during sepsis. JCI Insight 2019, 4, 21. [Google Scholar] [CrossRef]
- Su, G.; Atakilit, A.; Li, J.T.; Wu, N.; Luong, J.; Chen, R.; Bhattacharya, M.; Sheppard, D. Effective treatment of mouse sepsis with an inhibitory antibody targeting integrin αvβ5. Crit. Care Med. 2013, 41, 546–553. [Google Scholar] [CrossRef]
- Manley, M.O.; O’Riordan, M.A.; Levine, A.D.; Latifi, S.Q. Interleukin 10 extends the effectiveness of standard therapy during late sepsis with serum interleukin 6 levels predicting outcome. Shock 2005, 23, 521–526. [Google Scholar]
- Harrison, F.E.; Yu, S.S.; Van Den Bossche, K.L.; Li, L.; May, J.M.; McDonald, M.P. Elevated oxidative stress and sensorimotor deficits but normal cognition in mice that cannot synthesize ascorbic acid. J. Neurochem. 2008, 106, 1198–1208. [Google Scholar] [CrossRef]
- May, J.M.; Qu, Z.C.; Mendiratta, S. Protection and recycling of α-tocopherol in human erythrocytes by intracellular ascorbic acid. Arch. Biochem. Biophys. 1998, 349, 281–289. [Google Scholar] [CrossRef]
- Harrison, F.E.; Hosseini, A.H.; McDonald, M.P.; May, J.M. Vitamin C reduces spatial learning deficits in middle-aged and very old APP/PSEN1 transgenic and wild-type mice. Pharm. Biochem. Behav. 2009, 93, 443–450. [Google Scholar] [CrossRef]
- Sgaravatti, Â.M.; Magnusson, A.S.; Oliveira, A.S.; Mescka, C.P.; Zanin, F.; Sgarbi, M.B.; Pederzolli, C.D.; Wyse, A.T.S.; Wannmacher, C.M.D.; Wajner, M.; et al. Effects of 1,4-butanediol administration on oxidative stress in rat brain: Study of the neurotoxicity of γ-hydroxybutyric acid in vivo. Metab. Brain Dis. 2009, 24, 271–282. [Google Scholar] [CrossRef]
- Aksenov, M.Y.; Markesbery, W.R. Changes in thiol content and expression of glutathione redox system genes in the hippocampus and cerebellum in Alzheimer’s disease. Neurosci. Lett. 2001, 302, 141–145. [Google Scholar] [CrossRef]
- Bastarache, J.A.; Koyama, T.; Wickersham, N.E.; Ware, L.B. Validation of a multiplex electrochemiluminescent immunoassay platform in human and mouse samples. J. Immunol. Methods 2014, 408, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Gabbay, K.H.; Bohren, K.M.; Morello, R.; Bertin, T.; Liu, J.; Vogel, P. Ascorbate synthesis pathway: Dual role of ascorbate in bone homeostasis. J. Biol. Chem. 2010, 285, 19510–19520. [Google Scholar] [CrossRef]
- Levine, M.; Downing, D. New Concepts in the Biology and Biochemistry of Ascorbic Acid. J. Nutr. Med. 1992, 3, 361–362. [Google Scholar] [CrossRef]
- Ching, S.; Mahan, D.C.; Dabrowski, K. Liver L-Gulonolactone Oxidase Activity and Tissue Ascorbic Acid Concentrations in Nursing Pigs and the Effect of Various Weaning Ages. J. Nutr. 2001, 131, 2002–2006. [Google Scholar] [CrossRef][Green Version]
- Ching, S.; Mahan, D.C.; Ottobre, J.S.; Dabrowski, K. Ascorbic Acid Synthesis in Fetal and Neonatal Pigs and in Pregnant and Postpartum Sows. J. Nutr. 2001, 131, 1997–2001. [Google Scholar] [CrossRef]
- Harrison, F.E.; Dawes, S.M.; Meredith, M.E.; Babaev, V.R.; Li, L.; May, J.M. Low vitamin C and increased oxidative stress and cell death in mice that lack the sodium-dependent vitamin C transporter SVCT2. Free Radic. Biol. Med. 2010, 49, 821–829. [Google Scholar] [CrossRef]
- Jackson, T.S.; Xu, A.; Vita, J.A.; Keaney, J.F. Ascorbate prevents the interaction of superoxide and nitric oxide only at very high physiological concentrations. Circ. Res. 1998, 83, 916–922. [Google Scholar] [CrossRef]
- Kuo, S.-M.; Tan, C.-H.; Dragan, M.; Wilson, J.X. Endotoxin Increases Ascorbate Recycling and Concentration in Mouse Liver. J. Nutr. 2005, 135, 2411–2416. [Google Scholar] [CrossRef][Green Version]
- Maeda, N.; Hagihara, H.; Nakata, Y.; Hiller, S.; Wilder, J.; Reddick, R. Aortic wall damage in mice unable to synthesize ascorbic acid. Proc. Natl. Acad. Sci. USA 2000, 97, 841–846. [Google Scholar] [CrossRef]
- Wilson, J.X. Evaluation of Vitamin C for Adjuvant Sepsis Therapy. Antioxid. Redox Signal 2013, 19, 2129–2140. [Google Scholar] [CrossRef] [PubMed]
- Prauchner, C.A. Oxidative stress in sepsis: Pathophysiological implications justifying antioxidant co-therapy. Burns 2017, 43, 471–485. [Google Scholar] [CrossRef]
- Ahn, J.H.; Oh, D.K.; Huh, J.W.; Lim, C.M.; Koh, Y.; Hong, S.B. Vitamin C alone does not improve treatment outcomes in mechanically ventilated patients with severe sepsis or septic shock: A retrospective cohort study. J. Thorac. Dis. 2019, 11, 1562–1570. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, A.; Evans, L.E.; Alhazzani, W.; Levy, M.M.; Antonelli, M.; Ferrer, R.; Kumar, A.; Sevransky, J.E.; Sprung, C.L.; Nunnally, M.E.; et al. Surviving Sepsis Campaign: International Guidelines for Management of Sepsis and Septic Shock: 2016. Crit. Care Med. 2017, 45, 486–552. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).