Dietary Advanced Glycation Endproducts Decrease Glucocorticoid Sensitivity In Vitro

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Preparation of Dietary AGEs
2.3. Determination of Endotoxin in Dietary AGEs
2.4. Cell Culture and Exposure
2.5. Quantification of Interleukin-8 (IL8) Release by ELISA
2.6. Quantification of Glucocorticoid Receptor Protein Levels by Western Blot
2.7. Quantification of ROS Levels by DCFH-DA Assay
2.8. Statistics
3. Results
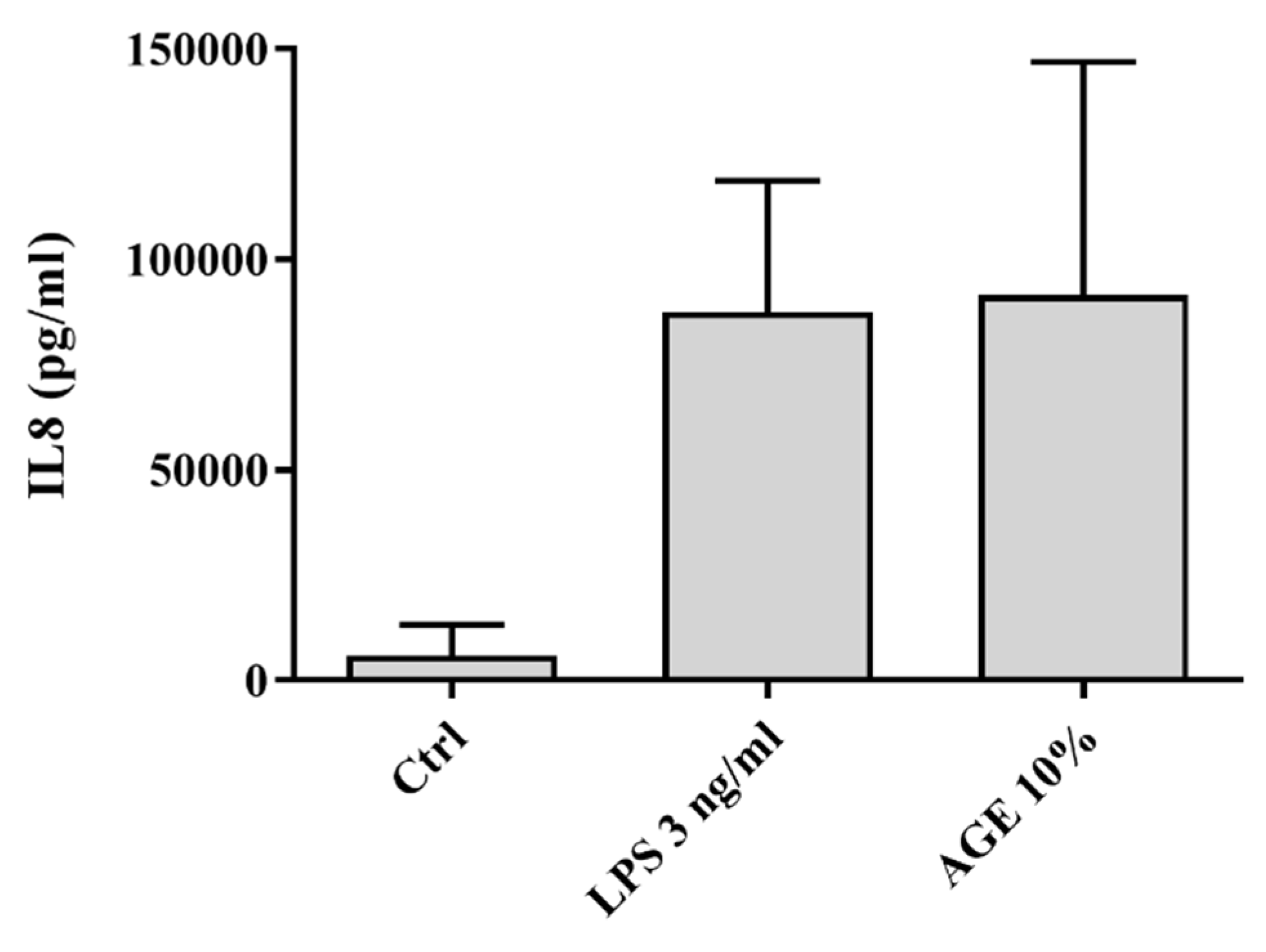
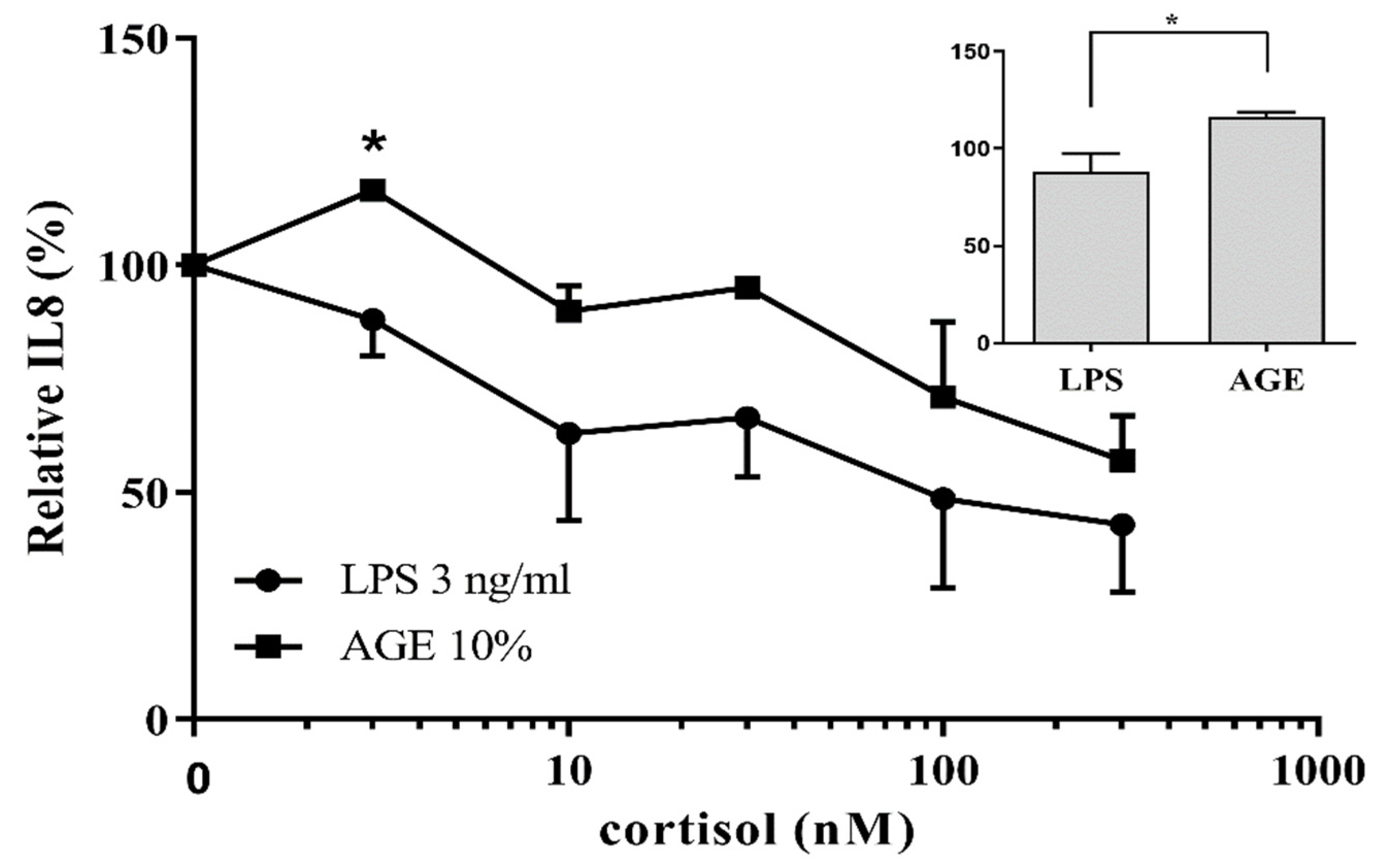
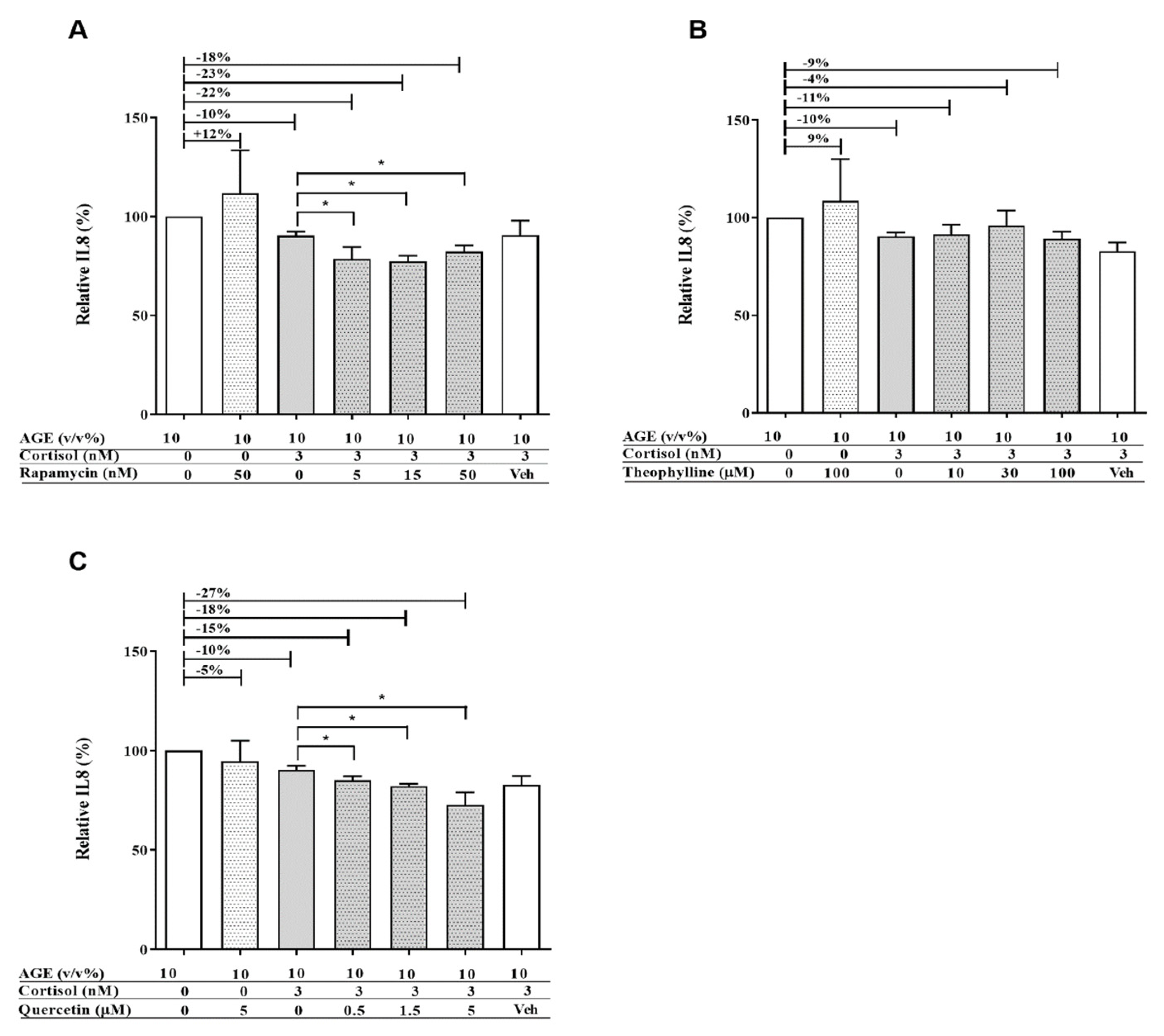
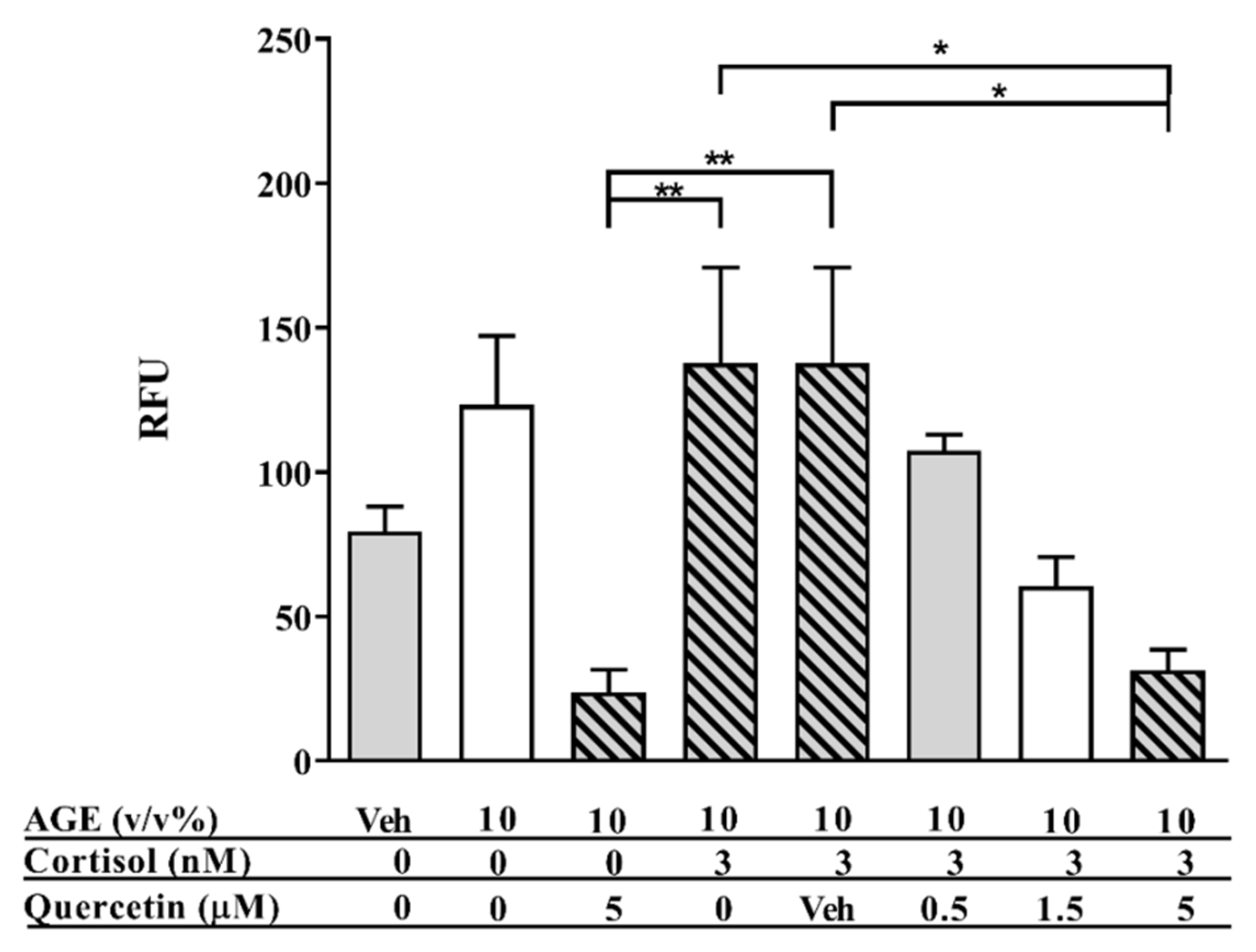
3.1. AGE-Induced Glucocorticoid Resistance
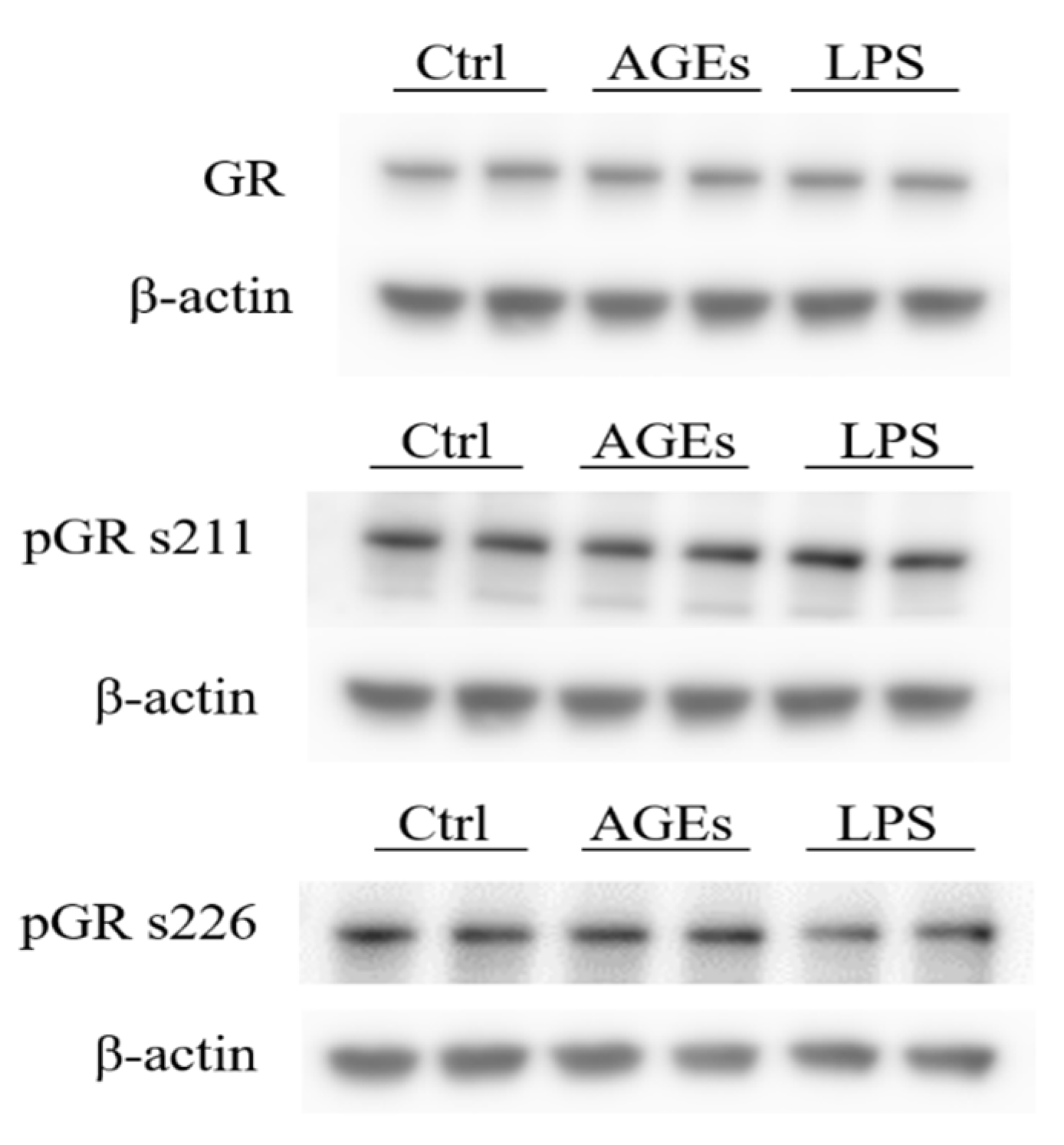
3.2. Phosphorylation of the Glucocorticoid Receptor
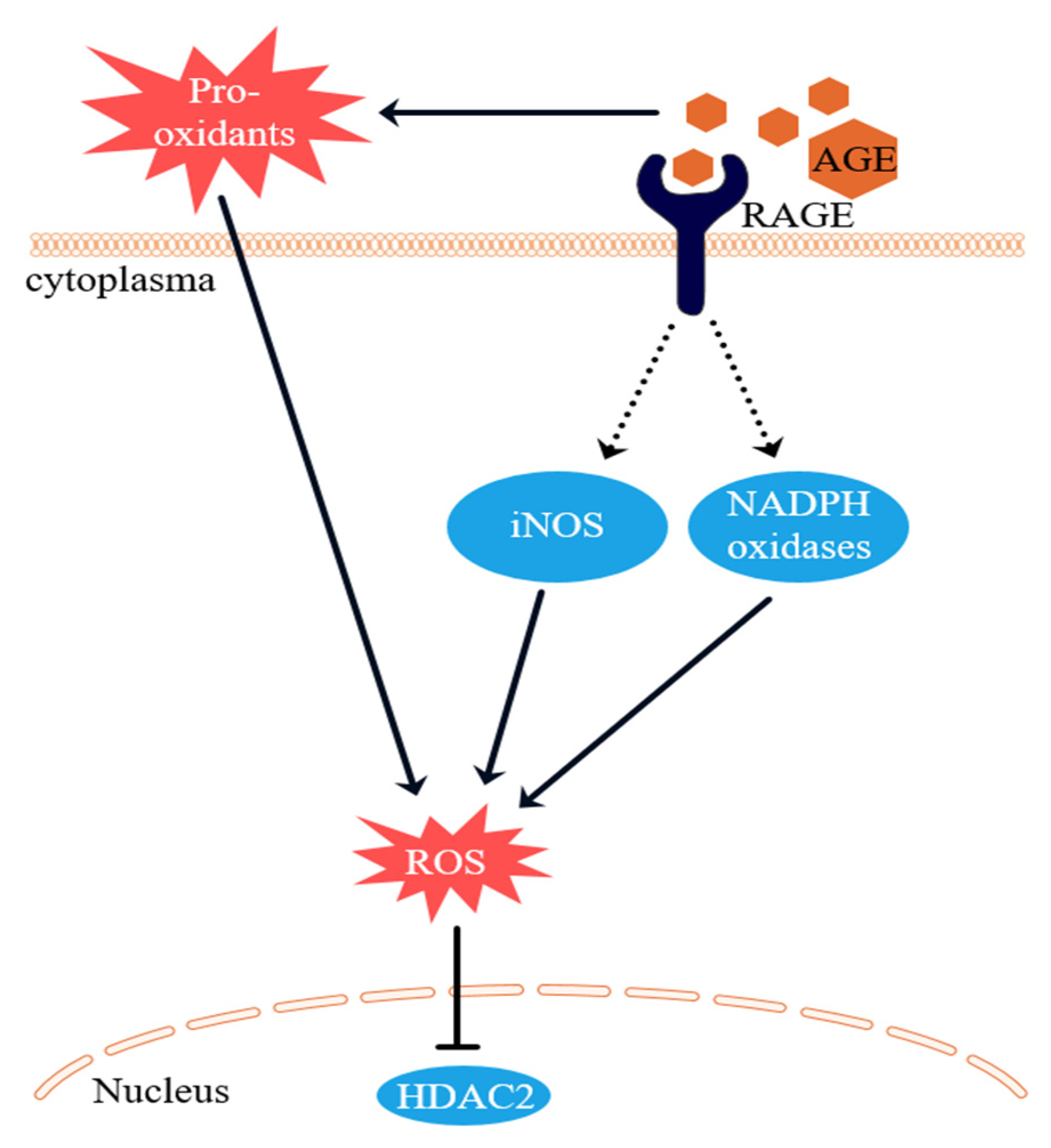
3.3. Intracellular ROS Levels
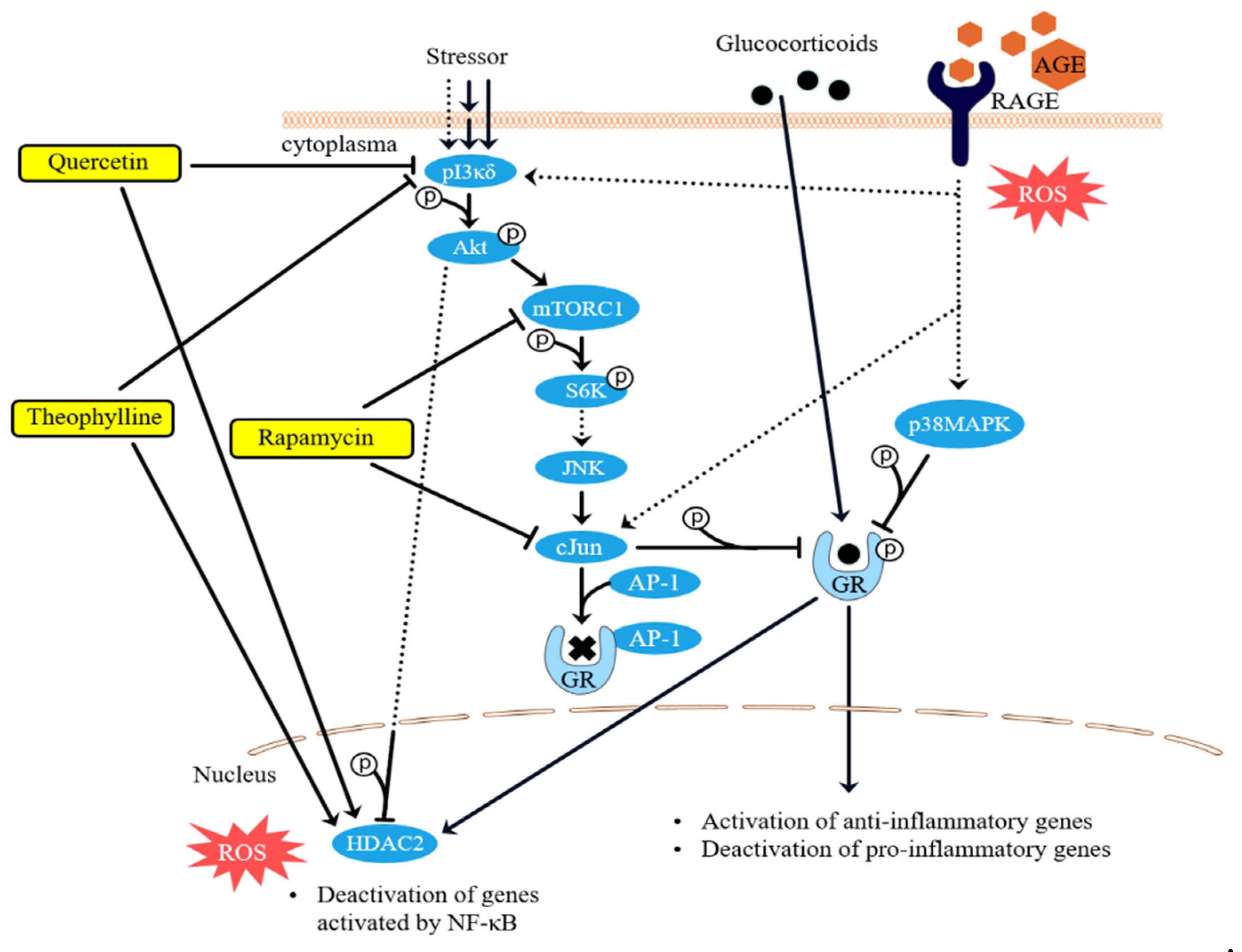
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tung, J.; Loftus, E.V., Jr.; Freese, D.K.; El-Youssef, M.; Zinsmeister, A.R.; Melton, L.J., 3rd; Harmsen, W.S.; Sandborn, W.J.; Faubion, W.A., Jr. A population-based study of the frequency of corticosteroid resistance and dependence in pediatric patients with Crohn’s disease and ulcerative colitis. Inflamm. Bowel Dis. 2006, 12, 1093–1100. [Google Scholar] [CrossRef]
- Munkholm, P.; Langholz, E.; Davidsen, M.; Binder, V. Frequency of glucocorticoid resistance and dependency in Crohn’s disease. Gut 1994, 35, 360–362. [Google Scholar] [CrossRef]
- Barnes, P.J. Mechanisms and resistance in glucocorticoid control of inflammation. J. Steroid Biochem. Mol. Biol. 2010, 120, 76–85. [Google Scholar] [CrossRef]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory action of glucocorticoids—New mechanisms for old drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef]
- Ito, K.; Barnes, P.J.; Adcock, I.M. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1beta-induced histone H4 acetylation on lysines 8 and 12. Mol. Cell. Biol. 2000, 20, 6891–6903. [Google Scholar] [CrossRef]
- Clark, A.R. MAP kinase phosphatase 1: A novel mediator of biological effects of glucocorticoids? J. Endocrinol. 2003, 178, 5–12. [Google Scholar] [CrossRef]
- Barnes, P.J. Corticosteroid effects on cell signalling. Eur. Respir. J. 2006, 27, 413–426. [Google Scholar] [CrossRef]
- Ito, K.; Yamamura, S.; Essilfie-Quaye, S.; Cosio, B.; Ito, M.; Barnes, P.J.; Adcock, I.M. Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kappaB suppression. J. Exp. Med. 2006, 203, 7–13. [Google Scholar] [CrossRef]
- Bergmann, M.W.; Staples, K.J.; Smith, S.J.; Barnes, P.J.; Newton, R. Glucocorticoid inhibition of granulocyte macrophage-colony-stimulating factor from T cells is independent of control by nuclear factor-kappaB and conserved lymphokine element 0. Am. J. Respir. Cell Mol. Biol. 2004, 30, 555–563. [Google Scholar] [CrossRef]
- Weigel, N.L.; Moore, N.L. Steroid receptor phosphorylation: A key modulator of multiple receptor functions. Mol. Endocrinol. 2007, 21, 2311–2319. [Google Scholar] [CrossRef]
- Matthews, J.G.; Ito, K.; Barnes, P.J.; Adcock, I.M. Defective glucocorticoid receptor nuclear translocation and altered histone acetylation patterns in glucocorticoid-resistant patients. J. Allergy Clin. Immunol. 2004, 113, 1100–1108. [Google Scholar] [CrossRef]
- Szatmary, Z.; Garabedian, M.J.; Vilcek, J. Inhibition of glucocorticoid receptor-mediated transcriptional activation by p38 mitogen-activated protein (MAP) kinase. J. Biol. Chem. 2004, 279, 43708–43715. [Google Scholar] [CrossRef]
- Miller, A.L.; Webb, M.S.; Copik, A.J.; Wang, Y.; Johnson, B.H.; Kumar, R.; Thompson, E.B. p38 Mitogen-activated protein kinase (MAPK) is a key mediator in glucocorticoid-induced apoptosis of lymphoid cells: Correlation between p38 MAPK activation and site-specific phosphorylation of the human glucocorticoid receptor at serine 211. Mol. Endocrinol. 2005, 19, 1569–1583. [Google Scholar] [CrossRef]
- Irusen, E.; Matthews, J.G.; Takahashi, A.; Barnes, P.J.; Chung, K.F.; Adcock, I.M. p38 Mitogen-activated protein kinase-induced glucocorticoid receptor phosphorylation reduces its activity: Role in steroid-insensitive asthma. J. Allergy Clin. Immunol. 2002, 109, 649–657. [Google Scholar] [CrossRef]
- Ito, K.; Hanazawa, T.; Tomita, K.; Barnes, P.J.; Adcock, I.M. Oxidative stress reduces histone deacetylase 2 activity and enhances IL-8 gene expression: Role of tyrosine nitration. Biochem. Biophys. Res. Commun. 2004, 315, 240–245. [Google Scholar] [CrossRef]
- Kirkham, P.; Rahman, I. Oxidative stress in asthma and COPD: Antioxidants as a therapeutic strategy. Pharmacol. Ther. 2006, 111, 476–494. [Google Scholar] [CrossRef]
- Ruijters, E.J.; Haenen, G.R.; Willemsen, M.; Weseler, A.R.; Bast, A. Food-Derived Bioactives Can Protect the Anti-Inflammatory Activity of Cortisol with Antioxidant-Dependent and -Independent Mechanisms. Int. J. Mol. Sci. 2016, 17, 239. [Google Scholar] [CrossRef]
- van der Lugt, T.; Weseler, A.; Gebbink, W.; Vrolijk, M.; Opperhuizen, A.; Bast, A. Dietary Advanced Glycation Endproducts Induce an Inflammatory Response in Human Macrophages In Vitro. Nutrients 2018, 10, 1868. [Google Scholar] [CrossRef]
- Xie, J.; Mendez, J.D.; Mendez-Valenzuela, V.; Aguilar-Hernandez, M.M. Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell. Signal. 2013, 25, 2185–2197. [Google Scholar] [CrossRef]
- Poulsen, M.W.; Hedegaard, R.V.; Andersen, J.M.; de Courten, B.; Bugel, S.; Nielsen, J.; Skibsted, L.H.; Dragsted, L.O. Advanced glycation endproducts in food and their effects on health. Food Chem. Toxicol. 2013, 60, 10–37. [Google Scholar] [CrossRef]
- Ruiz-Leal, M.; George, S. An In Vitro procedure for evaluation of early stage oxidative stress in an established fish cell line applied to investigation of PHAH and pesticide toxicity. Mar. Environ. Res. 2004, 58, 631–635. [Google Scholar] [CrossRef]
- Bansal, S.; Siddarth, M.; Chawla, D.; Banerjee, B.D.; Madhu, S.V.; Tripathi, A.K. Advanced glycation end products enhance reactive oxygen and nitrogen species generation in neutrophils In Vitro. Mol. Cell. Biochem. 2012, 361, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Adcock, I.M. Glucocorticoid resistance in inflammatory diseases. Lancet 2009, 373, 1905–1917. [Google Scholar] [CrossRef]
- Ruijters, E.J.; Haenen, G.R.; Weseler, A.R.; Bast, A. The cocoa flavanol (-)-epicatechin protects the cortisol response. Pharmacol. Res. 2014, 79, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Mitani, A.; Azam, A.; Vuppusetty, C.; Ito, K.; Mercado, N.; Barnes, P.J. Quercetin restores corticosteroid sensitivity in cells from patients with chronic obstructive pulmonary disease. Exp. Lung Res. 2017, 43, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Role of HDAC2 in the pathophysiology of COPD. Annu. Rev. Physiol. 2009, 71, 451–464. [Google Scholar] [CrossRef]
- To, Y.; Ito, K.; Kizawa, Y.; Failla, M.; Ito, M.; Kusama, T.; Elliott, W.M.; Hogg, J.C.; Adcock, I.M.; Barnes, P.J. Targeting phosphoinositide-3-kinase-delta with theophylline reverses corticosteroid insensitivity in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2010, 182, 897–904. [Google Scholar] [CrossRef]
- Randall, M.J.; Haenen, G.R.; Bouwman, F.G.; van der Vliet, A.; Bast, A. The tobacco smoke component acrolein induces glucocorticoid resistant gene expression via inhibition of histone deacetylase. Toxicol. Lett. 2016, 240, 43–49. [Google Scholar] [CrossRef]
- Marwick, J.A.; Wallis, G.; Meja, K.; Kuster, B.; Bouwmeester, T.; Chakravarty, P.; Fletcher, D.; Whittaker, P.A.; Barnes, P.J.; Ito, K.; et al. Oxidative stress modulates theophylline effects on steroid responsiveness. Biochem. Biophys. Res. Commun. 2008, 377, 797–802. [Google Scholar] [CrossRef]
- Ito, K.; Lim, S.; Caramori, G.; Cosio, B.; Chung, K.F.; Adcock, I.M.; Barnes, P.J. A molecular mechanism of action of theophylline: Induction of histone deacetylase activity to decrease inflammatory gene expression. Proc. Natl. Acad. Sci. USA 2002, 99, 8921–8926. [Google Scholar] [CrossRef]
- Wautier, M.P.; Chappey, O.; Corda, S.; Stern, D.M.; Schmidt, A.M.; Wautier, J.L. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E685–E694. [Google Scholar] [CrossRef] [PubMed]
- Rowan, S.; Bejarano, E.; Taylor, A. Mechanistic targeting of advanced glycation end-products in age-related diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3631–3643. [Google Scholar] [CrossRef] [PubMed]
- Nowotny, K.; Jung, T.; Hohn, A.; Weber, D.; Grune, T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef] [PubMed]
- Meja, K.K.; Rajendrasozhan, S.; Adenuga, D.; Biswas, S.K.; Sundar, I.K.; Spooner, G.; Marwick, J.A.; Chakravarty, P.; Fletcher, D.; Whittaker, P.; et al. Curcumin restores corticosteroid function in monocytes exposed to oxidants by maintaining HDAC2. Am. J. Respir. Cell Mol. Biol. 2008, 39, 312–323. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van der Lugt, T.; Weseler, A.R.; Vrolijk, M.F.; Opperhuizen, A.; Bast, A. Dietary Advanced Glycation Endproducts Decrease Glucocorticoid Sensitivity In Vitro. Nutrients 2020, 12, 441. https://doi.org/10.3390/nu12020441
van der Lugt T, Weseler AR, Vrolijk MF, Opperhuizen A, Bast A. Dietary Advanced Glycation Endproducts Decrease Glucocorticoid Sensitivity In Vitro. Nutrients. 2020; 12(2):441. https://doi.org/10.3390/nu12020441
Chicago/Turabian Stylevan der Lugt, Timme, Antje R. Weseler, Misha F. Vrolijk, Antoon Opperhuizen, and Aalt Bast. 2020. "Dietary Advanced Glycation Endproducts Decrease Glucocorticoid Sensitivity In Vitro" Nutrients 12, no. 2: 441. https://doi.org/10.3390/nu12020441
APA Stylevan der Lugt, T., Weseler, A. R., Vrolijk, M. F., Opperhuizen, A., & Bast, A. (2020). Dietary Advanced Glycation Endproducts Decrease Glucocorticoid Sensitivity In Vitro. Nutrients, 12(2), 441. https://doi.org/10.3390/nu12020441