Sesamol Alleviates Obesity-Related Hepatic Steatosis via Activating Hepatic PKA Pathway

 , ,
, ,
Abstract

1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Animals and Diet
2.3. Glucose Tolerance Test (GTT) and Insulin Tolerance Test (ITT)
2.4. Serum Parameter Analysis
2.5. Histological Analysis
2.6. Hepatic Parameter Analysis
2.7. Cell Culture and Treatment
2.8. Cellular Oil Red O Staining and Lipid Content
2.9. Quantitative Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
2.10. Western Blot
2.11. Statistical Analysis
3. Results
3.1. Sesamol (SEM) Ameliorated High-Fat Diet (HFD)-Induced Obesity
3.2. SEM Attenuated Metabolic Disorders in Obese Mice
3.3. SEM Alleviated Hepatic Steatosis in Obese Mice
3.4. SEM Promoted Hepatic Lipid Metabolism in Obese Mice
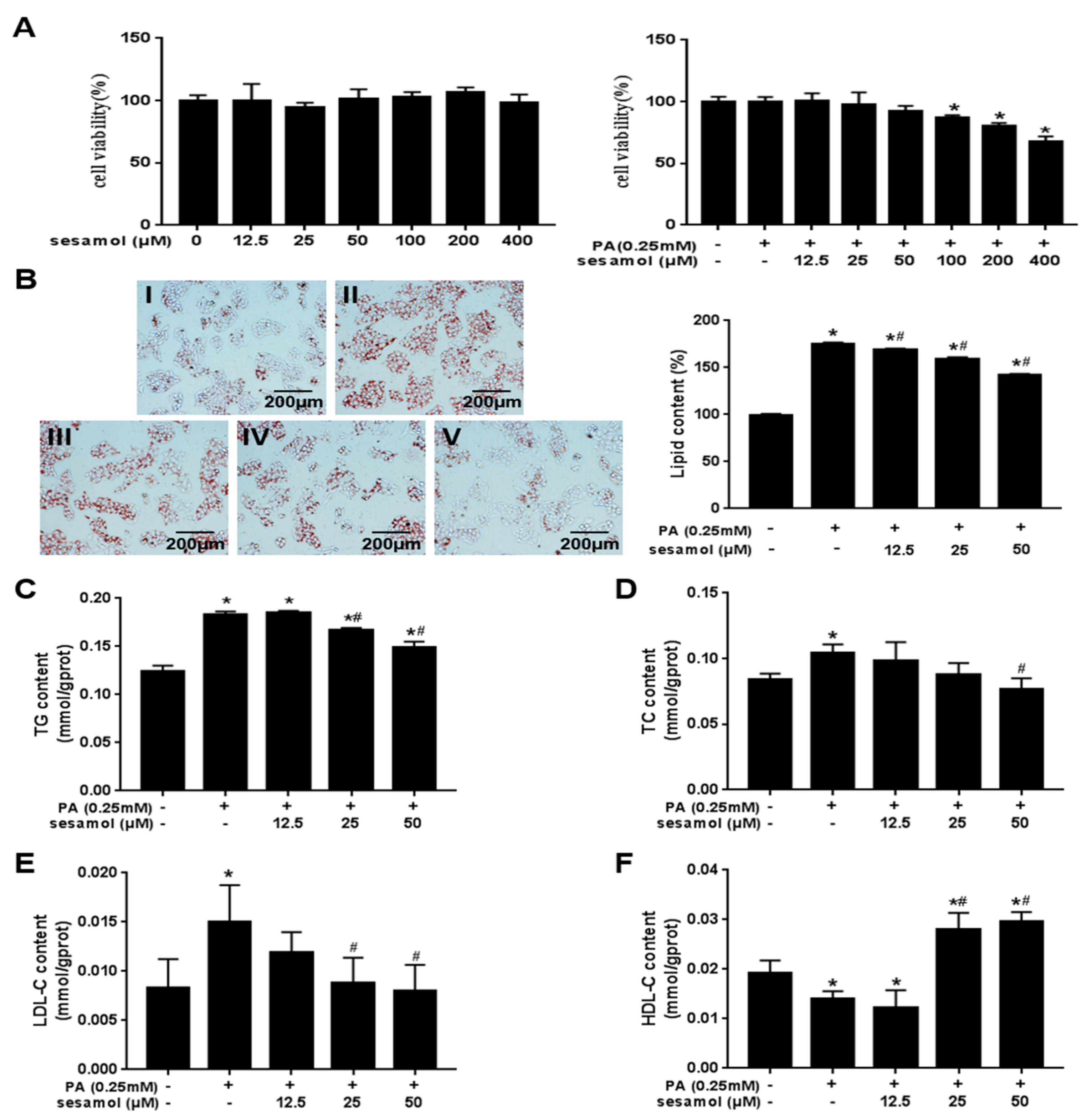
3.5. SEM Reduced Lipid Accumulation in Palmitic Acid (PA)-Treated HepG2 Cells
3.6. SEM Promoted Lipid Metabolism in PA-Treated HepG2 Cells
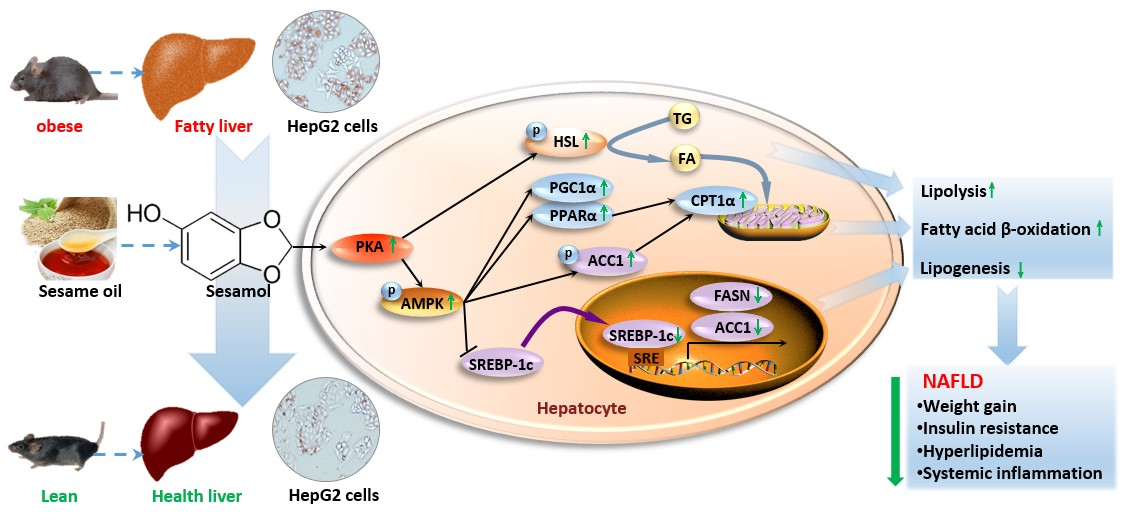
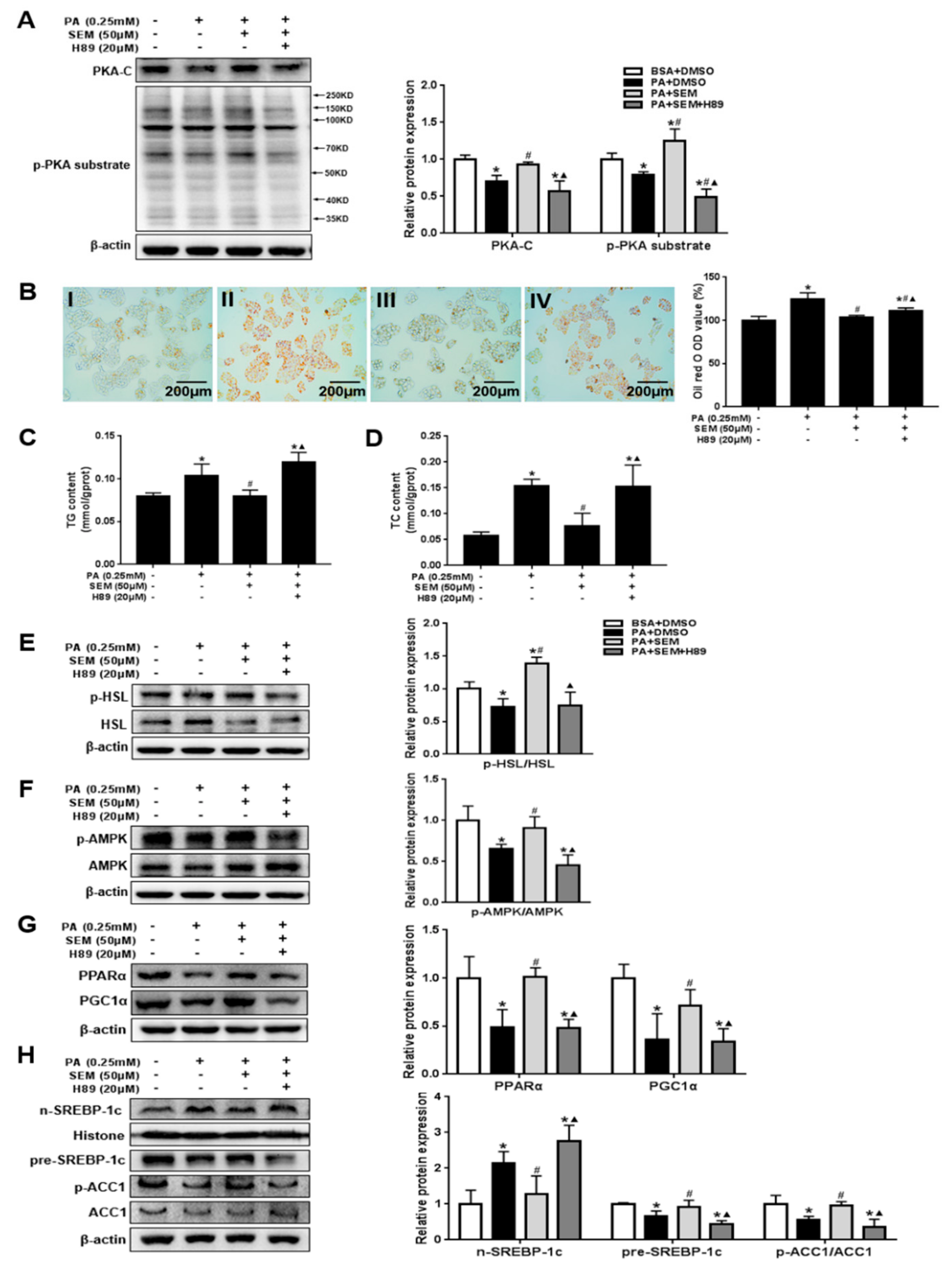
3.7. SEM Regulated Lipid Homeostasis by Activating the Hepatic Protein Kinase A (PKA) Pathway
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Arroyo-Johnson, C.; Mincey, K.D. Obesity Epidemiology Worldwide. Gastroenterol. Clin. North Am. 2016, 45, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Kopelman, P.G. Obesity as a medical problem. Nature 2000, 404, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Thach, T.T.; Kim, Y.J.; Lee, S.J. Olfactory receptor 43 reduces hepatic lipid accumulation and adiposity in mice. Bba Mol. Cell Biol. L 2019, 1864, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Dowman, J.K.; Tomlinson, J.W.; Newsome, P.N. Systematic review: The diagnosis and staging of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis. Aliment Pharmacol. Ther. 2011, 33, 525–540. [Google Scholar] [CrossRef]
- Benhamed, F.; Denechaud, P.D.; Lemoine, M.; Robichon, C.; Moldes, M.; Bertrand-Michel, J.; Ratziu, V.; Serfaty, L.; Housset, C.; Capeau, J.; et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J. Clin. Invest. 2012, 122, 2176–2194. [Google Scholar] [CrossRef]
- Finck, B.N.; Gropler, M.C.; Chen, Z.J.; Leone, T.C.; Croce, M.A.; Harris, T.E.; Lawrence, J.C.; Kelly, D.P. Lipin 1 is an inducible amplifier of the hepatic PGC-1 alpha/PPAR alpha regulatory pathway. Cell Metab. 2006, 4, 199–210. [Google Scholar] [CrossRef]
- Wu, B.N.; Kuo, K.K.; Chen, Y.H.; Chang, C.T.; Huang, H.T.; Chai, C.Y.; Dai, Z.K.; Chen, I.J. Theophylline-Based KMUP-1 Improves Steatohepatitis via MMP-9/IL-10 and Lipolysis via HSL/p-HSL in Obese Mice. Int. J. Mol. Sci. 2016, 17, 1345. [Google Scholar] [CrossRef]
- Lv, S.H.; Qiu, X.C.; Li, J.; Liang, J.Y.; Li, W.D.; Zhang, C.; Zhang, Z.N.; Luan, B. Glucagon-induced extracellular cAMP regulates hepatic lipid metabolism. J. Endocrinol. 2017, 234, 73–87. [Google Scholar] [CrossRef]
- Tian, S.C.; Li, B.L.; Lei, P.; Yang, X.L.; Zhang, X.H.; Bao, Y.P.; Shan, Y.J. Sulforaphane Improves Abnormal Lipid Metabolism via Both ERS-Dependent XBP1/ACC &SCD1 and ERS-Independent SREBP/FAS Pathways. Mol. Nutr. Food Res. 2018, 62, 1700737. [Google Scholar]
- Seo, Y.J.; Kim, K.J.; Choi, J.; Koh, E.J.; Lee, B.Y. Spirulina maxima Extract Reduces Obesity through Suppression of Adipogenesis and Activation of Browning in 3T3-L1 Cells and High-Fat Diet-Induced Obese Mice. Nutrients 2018, 10, 712. [Google Scholar] [CrossRef]
- Fan, L.; Xu, H.Y.; Yang, R.G.; Zang, Y.F.; Chen, J.F.; Qin, H. Combination of Capsaicin and Capsiate Induces Browning in 3T3-L1 White Adipocytes via Activation of the Peroxisome Proliferator-Activated Receptor gamma/beta(3)-Adrenergic Receptor Signaling Pathways. J. Agric. Food Chem. 2019, 67, 6232–6240. [Google Scholar] [CrossRef] [PubMed]
- Chennuru, A.; Saleem, M.T.S. Antioxidant, Lipid Lowering, and Membrane Stabilization Effect of Sesamol against Doxorubicin-Induced Cardiomyopathy in Experimental Rats. Biomed. Res. Int. 2013, 2013, 934239. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.J.; Xie, G.N.; Liu, L.; Fu, Z.J.; Zhang, Z.W.; Teng, L.Z. Sesamol attenuates oxidative stress, apoptosis and inflammation in focal cerebral ischemia/reperfusion injury. Exp. Ther. Med. 2017, 14, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Hemshekhar, M.; Thushara, R.M.; Jnaneshwari, S.; Devaraja, S.; Kemparaju, K.; Girish, K.S. Attenuation of adjuvant-induced arthritis by dietary sesamol via modulation of inflammatory mediators, extracellular matrix degrading enzymes and antioxidant status. Eur. J. Nutr. 2013, 52, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- John, J.; Nampoothiri, M.; Kumar, N.; Mudgal, J.; Nampurath, G.K.; Chamallamudi, M.R. Sesamol, a lipid lowering agent, ameliorates aluminium chloride induced behavioral and biochemical alterations in rats. Pharmacogn. Mag. 2015, 11, 327–336. [Google Scholar] [PubMed]
- Liu, Z.G.; Qiao, Q.L.; Sun, Y.L.; Chen, Y.W.; Ren, B.; Liu, X.B. Sesamol ameliorates diet-induced obesity in C57BL/6J mice and suppresses adipogenesis in 3T3-L1 cells via regulating mitochondria-lipid metabolism. Mol. Nutr. Food Res. 2017, 61, 1600717. [Google Scholar] [CrossRef]
- Qin, H.; Xu, H.; Yu, L.; Yang, L.; Lin, C.; Chen, J. Sesamol intervention ameliorates obesity-associated metabolic disorders by regulating hepatic lipid metabolism in high-fat diet-induced obese mice. Food Nutr. Res. 2019, 63. [Google Scholar] [CrossRef]
- Zang, Y.F.; Fang, L.; Chen, J.H.; Huang, R.X.; Qin, H. Improvement of Lipid and Glucose Metabolism by Capsiate in Palmitic Acid-Treated HepG2 Cells via Activation of the AMPK/SIRT1 Signaling Pathway. J. Agric. Food Chem. 2018, 66, 6772–6781. [Google Scholar] [CrossRef]
- Jin, W.; Cui, B.; Li, P.P.; Hua, F.; Lv, X.X.; Zhou, J.C.; Hu, Z.W.; Zhang, X.W. 1,25-Dihydroxyvitamin D3 protects obese rats from metabolic syndrome via promoting regulatory T cell-mediated resolution of inflammation. Acta Pharm. Sin. B 2018, 8, 178–187. [Google Scholar] [CrossRef]
- Benjamin, B.; Wada, Y.; Grundy, S.M.; Szuszkiewicz-Garcia, M.; Vega, G.L. Fatty acid oxidation in normotriglyceridemic men. J. Clin. Lipidol. 2016, 10, 283–288. [Google Scholar] [CrossRef][Green Version]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory Mechanisms in Obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef]
- Liu, T.Y.; Xiong, X.Q.; Ren, X.S.; Zhao, M.X.; Shi, C.X.; Wang, J.J.; Zhou, Y.B.; Zhang, F.; Han, Y.; Gao, X.Y.; et al. FNDC5 Alleviates Hepatosteatosis by Restoring AMPK/mTOR-Mediated Autophagy, Fatty Acid Oxidation, and Lipogenesis in Mice. Diabetes 2016, 65, 3262–3275. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Zhang, P.; Yu, F.; Zhang, Z.; Cai, Q.; Lu, W.D.; Li, B.Y.; Qin, W.D.; Cheng, M.; Wang, H.; et al. Grape seed procyanidin B2 ameliorates hepatic lipid metabolism disorders in db/db mice. Mol. Med. Rep. 2017, 16, 2844–2850. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, H.; Souza, S.C.; Zhang, H.H.; Strissel, K.J.; Christoffolete, M.A.; Kovsan, J.; Rudich, A.; Kraemer, F.B.; Bianco, A.C.; Obin, M.S.; et al. Perilipin promotes hormone-sensitive lipase-mediated adipocyte lipolysis via phosphorylation-dependent and -independent mechanisms. J. Biol. Chem. 2006, 281, 15837–15844. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.H.; Li, H.; Qi, X.Y.; Wang, Y.D.; Xu, C.X.; Liu, G.X.; Wen, G.B.; Liu, J.H. Zinc alpha2 glycoprotein alleviates palmitic acid-induced intracellular lipid accumulation in hepatocytes. Mol. Cell Endocrinol. 2017, 439, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Gaggini, M.; Morelli, M.; Buzzigoli, E.; DeFronzo, R.A.; Bugianesi, E.; Gastaldelli, A. Non-Alcoholic Fatty Liver Disease (NAFLD) and Its Connection with Insulin Resistance, Dyslipidemia, Atherosclerosis and Coronary Heart Disease. Nutrients 2013, 5, 1544–1560. [Google Scholar] [CrossRef]
- Goossens, G.H. The Metabolic Phenotype in Obesity: Fat Mass, Body Fat Distribution, and Adipose Tissue Function. Obes. Facts 2017, 10, 207–215. [Google Scholar] [CrossRef]
- Jones, J.G. Hepatic glucose and lipid metabolism. Diabetologia 2016, 59, 1098–1103. [Google Scholar] [CrossRef]
- Arai, T.; Tanaka, M.; Goda, N. HIF-1-dependent lipin1 induction prevents excessive lipid accumulation in choline-deficient diet-induced fatty liver. Sci. Rep. 2018, 8, 14230. [Google Scholar] [CrossRef]
- Donato, M.T.; Tolosa, L.; Gomez-Lechon, M.J. Culture and Functional Characterization of Human Hepatoma HepG2 Cells. Methods Mol. Biol. 2015, 1250, 77–93. [Google Scholar]
- Kim, M.; Lim, S.J.; Lee, H.J.; Kim, S.Y.; Nho, C.W. Gomisin J Inhibits Oleic Acid-Induced Hepatic Lipogenesis by Activation of the AMPK-Dependent Pathway and Inhibition of the Hepatokine Fetuin-A in HepG2 Cells. J. Agric. Food Chem. 2015, 63, 9729–9739. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Cohen, D.E. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J. Gastroenterol. 2013, 48, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Kuo, K.K.; Wu, B.N.; Liu, C.P.; Yang, T.Y.; Kao, L.P.; Wu, J.R.; Lai, W.T.; Chen, I.J. Xanthine-based KMUP-1 improves HDL via PPAR/SR-B1, LDL via LDLRs, and HSL via PKA/PKG for hepatic fat loss. J. Lipid Res. 2015, 56, 2070–2084. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, S.Q.; Mihaylova, M.M.; Zheng, B.; Hou, X.Y.; Jiang, B.B.; Park, O.; Luo, Z.J.; Lefai, E.; Shyy, J.Y.J.; et al. AMPK Phosphorylates and Inhibits SREBP Activity to Attenuate Hepatic Steatosis and Atherosclerosis in Diet-Induced Insulin-Resistant Mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Han, J.B.; Li, E.W.; Chen, L.Q.; Zhang, Y.Y.; Wei, F.C.; Liu, J.Y.; Deng, H.T.; Wang, Y.G. The CREB coactivator CRTC2 controls hepatic lipid metabolism by regulating SREBP1. Nature 2015, 524, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Fullerton, M.D.; Galic, S.; Marcinko, K.; Sikkema, S.; Pulinilkunnil, T.; Chen, Z.P.; O’Neill, H.M.; Ford, R.J.; Palanivel, R.; O’Brien, M.; et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 2013, 19, 1649–1654. [Google Scholar] [CrossRef]
- Reid, B.N.; Ables, G.P.; Otlivanchik, O.A.; Schoiswohl, G.; Zechner, R.; Blaner, W.S.; Goldberg, I.J.; Schwabe, R.F.; Chua, S.C.; Huang, L.S. Hepatic overexpression of hormone-sensitive lipase and adipose triglyceride lipase promotes fatty acid oxidation, stimulates direct release of free fatty acids, and ameliorates steatosis. J. Biol. Chem. 2008, 283, 13087–13099. [Google Scholar] [CrossRef]
- Larsson, S.; Jones, H.A.; Goransson, O.; Degerman, E.; Holm, C. Parathyroid hormone induces adipocyte lipolysis via PKA-mediated phosphorylation of hormone-sensitive lipase. Cell Signal 2016, 28, 204–213. [Google Scholar] [CrossRef]
- Gagnon, A.; Antunes, T.T.; Ly, T.; Pongsuwan, P.; Gavin, C.; Lochnan, H.A.; Sorisky, A. Thyroid-stimulating hormone stimulates lipolysis in adipocytes in culture and raises serum free fatty acid levels in vivo. Metabolism 2010, 59, 547–553. [Google Scholar] [CrossRef]
- Li, M.; Xu, C.F.; Shi, J.P.; Ding, J.X.; Wan, X.Y.; Chen, D.H.; Gao, J.G.; Li, C.X.; Zhang, J.; Lin, Y.M.; et al. Fatty acids promote fatty liver disease via the dysregulation of 3-mercaptopyruvate sulfurtransferase/hydrogen sulfide pathway. Gut 2018, 67, 2169–2180. [Google Scholar] [CrossRef]
- Oh, Y.S.; Bae, G.D.; Baek, D.J.; Park, E.Y.; Jun, H.S. Fatty Acid-Induced Lipotoxicity in Pancreatic Beta-Cells During Development of Type 2 Diabetes. Front. Endocrinol. 2018, 9, 384. [Google Scholar] [CrossRef] [PubMed]
- Montagner, A.; Polizzi, A.; Fouche, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Regnier, M.; Lukowicz, C.; Benhamed, F.; et al. Liver PPAR alpha is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 2016, 65, 1202–1214. [Google Scholar] [CrossRef]
- Kim, B.; Woo, M.J.; Park, C.S.; Lee, S.H.; Kim, J.S.; Kim, B.; An, S.; Kim, S.H. Hovenia Dulcis Extract Reduces Lipid Accumulation in Oleic Acid-Induced Steatosis of Hep G2 Cells via Activation of AMPK and PPAR alpha/CPT-1 Pathway and in Acute Hyperlipidemia Mouse Model. Phytother. Res. 2017, 31, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Sathyanarayan, A.; Mashek, M.T.; Ong, K.T.; Wollaston-Hayden, E.E.; Mashek, D.G. ATGL-Catalyzed Lipolysis Regulates SIRT1 to Control PGC-1 alpha/PPAR-alpha Signaling. Diabetes 2015, 64, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Shaw, R.J. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef]
- Li, L.Y.; Xiao, L.N.; Hou, Y.H.; He, Q.; Zhu, J.; Li, Y.X.; Wu, J.X.; Zhao, J.; Yu, S.S.; Zhao, Y. Sestrin2 Silencing Exacerbates Cerebral Ischemia/Reperfusion Injury by Decreasing Mitochondrial Biogenesis through the AMPK/PGC-1 alpha Pathway in Rats. Sci. Rep. 2016, 6, 30272. [Google Scholar] [CrossRef]
- Jung, T.W.; Kim, H.C.; Abd El-Aty, A.M.; Jeong, J.H. Protectin DX ameliorates palmitate- or high-fat diet-induced insulin resistance and inflammation through an AMPK-PPARalpha-dependent pathway in mice. Sci. Rep. 2017, 7, 1397. [Google Scholar] [CrossRef]
- Ford, R.J.; Fullerton, M.D.; Pinkosky, S.L.; Day, E.A.; Scott, J.W.; Oakhill, J.S.; Bujak, A.L.; Smith, B.K.; Crane, J.D.; Blumer, R.M.; et al. Metformin and salicylate synergistically activate liver AMPK, inhibit lipogenesis and improve insulin sensitivity. Biochem. J. 2015, 468, 125–132. [Google Scholar] [CrossRef]
- Kamikubo, R.; Kai, K.J.; Tsuji-Naito, K.; Akagawa, M. beta-Caryophyllene attenuates palmitate-induced lipid accumulation through AMPK signaling by activating CB2 receptor in human HepG2 hepatocytes. Mol. Nutr. Food Res. 2016, 60, 2228–2242. [Google Scholar] [CrossRef]
- Go, G.; Sung, J.S.; Jee, S.C.; Kim, M.; Jang, W.H.; Kang, K.Y.; Kim, D.Y.; Lee, S.; Shin, H.S. In vitro anti-obesity effects of sesamol mediated by adenosine monophosphate-activated protein kinase and mitogen-activated protein kinase signaling in 3T3-L1 cells. Food Sci. Biotechnol. 2017, 26, 195–200. [Google Scholar] [CrossRef]
- Wu, H.M.; Yang, Y.M.; Kim, S.G. Rimonabant, a Cannabinoid Receptor Type 1 Inverse Agonist, Inhibits Hepatocyte Lipogenesis by Activating Liver Kinase B1 and AMP-Activated Protein Kinase Axis Downstream of G alpha(i/o) Inhibition. Mol. Pharmacol. 2011, 80, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Turnham, R.E.; Scott, J.D. Protein kinase A catalytic subunit isoform PRKACA. History, function and physiology. Gene 2016, 577, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.; Narayan, V.P.; Sung, M.K.; Park, T. Piperonal attenuates visceral adiposity in mice fed a high-fat diet: potential involvement of the adenylate cyclase-protein kinase A dependent pathway. Mol. Nutr. Food Res. 2017, 61, 1601124. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Specifies | Gene Name | Primer Sequence (5′ to 3′) | |
|---|---|---|---|
| Forward | Reverse | ||
| Mus musculus | Srebp-1c | AGGCAGAGAGCAGAGATG | AAAGAGAAGAGCCAAGCA |
| Acc1 | AAAACAGGGAGGAAGCAA | TCACCCCGAATAGACAGC | |
| Fasn | ACCTCATTGGTGGTGTGG | CATTGTGTGTGCCTGCTT | |
| Hsl | CCGCTATGTGGCTTCTAA | CACTCCTGGTCGGTTGAT | |
| Cpt1α | CTATTCGTCTTCTGGGAT | GTGTTGGATGGTGTCTGT | |
| Pparα | TCTCCCCACATCCTTTCT | CTGCCGTTGTCTGTCACT | |
| Pgc1α | GCCTTCTTGCTCTTCCTT | ATCCTTTGGGGTCTTTGA | |
| β-actin | CGTGCGTGACATCAAAGA | AAGGAAGGCTGGAAAAGA | |
| Homo sapiens | Srebp-1c | GCAACACAGCAACCAGAA | GAAAGGTGAGCCAGCATC |
| Acc1 | AAGACTGGGTAGAGCGAT | GGGAAACTGACAGAGGAC | |
| Fasn | GCCCAAGGGAAGCACATT | CGAAGCCACCCAGACCAC | |
| Hsl | TGGAGGAGTGCTTCTTCG | GATTCGTTCCCCTGTTGA | |
| Cpt1α | CTACTTCCAGACTTGCCC | ACACCATTTCCATTCCAC | |
| Pparα | TAGGGACAGACTGACACC | CATAACAAAAGATACGGG | |
| Pgc1α | TGCCACCACCATCAAAGA | ACCAAACAGCCGCAGACT | |
| β-actin | TTGCGTTACACCCTTTCT | ACCTTCACCGTTCCAGTT | |
| NFD | HFD | HFD+SEM | |
|---|---|---|---|
| Serum TG (mM) | 0.74 ± 0.10 | 1.59 ± 0.42 * | 0.93 ± 0.17 # |
| Serum TC (mM) | 5.90 ± 0.96 | 13.38 ± 3.03 * | 10.03 ± 0.93 # |
| Serum LDL-C (mM) | 0.16 ± 0.03 | 0.24 ± 0.03 * | 0.18 ± 0.03 # |
| Serum HDL-C (mM) | 2.43 ± 0.55 | 2.53 ± 0.15 | 3.79 ± 0.15 *# |
| Serum FFA (μM) | 66.69 ± 2.82 | 76.07 ± 3.40 * | 63.07 ± 2.62 # |
| Serum β-HB (mM) | 0.14 ± 0.02 | 0.11 ± 0.01 * | 0.13 ± 0.01 # |
| Serum TNF-α (pg/mL) | 11.79 ± 0.78 | 13.92 ± 1.72 * | 11.17 ± 0.73 # |
| Serum IL-6 (pg/mL) | 11.07 ± 0.61 | 14.8 ± 1.98 * | 12.07 ± 1.12 # |
| NFD | HFD | HFD+SEM | |
|---|---|---|---|
| Liver TG (mmol/gprot) | 0.16 ± 0.03 | 0.28 ± 0.09 * | 0.13 ± 0.03 # |
| Liver TC (mmol/gprot) | 0.05 ± 0.01 | 0.04 ± 0.01 | 0.04 ± 0.01 |
| Liver FFA (μM) | 3.15 ± 0.43 | 4.98 ± 0.21 * | 4.17 ± 0.36 *# |
| Liver β-HB (mM) | 0.17 ± 0.01 | 0.15 ± 0.01 * | 0.16 ± 0.01 # |
| Serum ALT (U/L) | 14.00 ± 3.98 | 83.11 ± 12.63 * | 21.76 ± 9.41 # |
| Serum AST (U/L) | 17.16 ± 5.18 | 32.07 ± 4.68 * | 23.09 ± 5.09 # |
| Liver TNF-α (ng/mL) | 0.33 ± 0.03 | 0.49 ± 0.07 * | 0.32 ± 0.04 # |
| Liver IL-6 (ng/mL) | 0.06 ± 0.01 | 0.10 ± 0.02 * | 0.03 ± 0.02 # |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, H.-Y.; Yu, L.; Chen, J.-H.; Yang, L.-N.; Lin, C.; Shi, X.-Q.; Qin, H. Sesamol Alleviates Obesity-Related Hepatic Steatosis via Activating Hepatic PKA Pathway. Nutrients 2020, 12, 329. https://doi.org/10.3390/nu12020329
Xu H-Y, Yu L, Chen J-H, Yang L-N, Lin C, Shi X-Q, Qin H. Sesamol Alleviates Obesity-Related Hepatic Steatosis via Activating Hepatic PKA Pathway. Nutrients. 2020; 12(2):329. https://doi.org/10.3390/nu12020329
Chicago/Turabian StyleXu, Hai-Yan, Liang Yu, Ji-Hua Chen, Li-Na Yang, Cui Lin, Xiu-Quan Shi, and Hong Qin. 2020. "Sesamol Alleviates Obesity-Related Hepatic Steatosis via Activating Hepatic PKA Pathway" Nutrients 12, no. 2: 329. https://doi.org/10.3390/nu12020329
APA StyleXu, H.-Y., Yu, L., Chen, J.-H., Yang, L.-N., Lin, C., Shi, X.-Q., & Qin, H. (2020). Sesamol Alleviates Obesity-Related Hepatic Steatosis via Activating Hepatic PKA Pathway. Nutrients, 12(2), 329. https://doi.org/10.3390/nu12020329