DNA Methylation Profiles of Vegans and Non-Vegetarians in the Adventist Health Study-2 Cohort

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Laboratory Analysis and Data Processing
2.3. Statistical Analyses
3. Results
3.1. Methylation Differences in Vegans and Non-Vegetarians
3.1.1. Differential Methylation of Select Regions Associated with Individual Genes
- Proportions of Differentially Methylated Gene Regions
- 2.
- Unique Genes Showing Differential Methylation
3.1.2. Differential Methylation of CpG Sites
- 1.
- Proportions of Differentially Methylated CpG Sites by Region
- 2.
- Genes Associated with Differentially Methylated CpGs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Appendix A
A Method of Power Calculations, Estimating Ability to Detect Differentially Methylated Sites with a Given False Discovery Rate

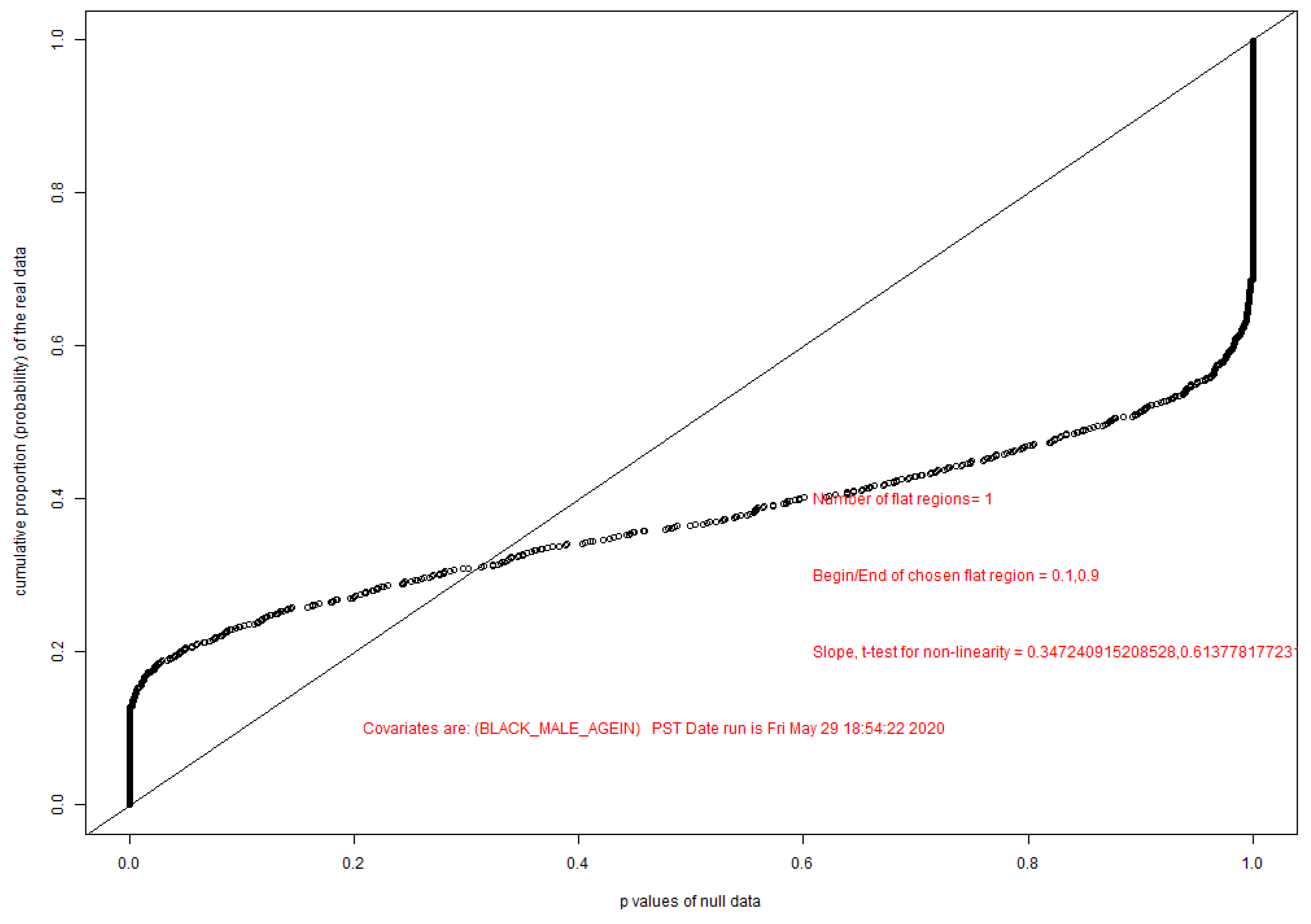
References
- Fraser, G.; Katuli, S.; Anousheh, R.; Knutsen, S.; Herring, P.; Fan, J. Vegetarian diets and cardiovascular risk factors in black members of the Adventist Health Study-2. Public Health Nutr. 2015, 18, 537–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, N.S.; Sabate, J.; Jaceldo-Siegl, K.; Fraser, G.E. Vegetarian dietary patterns are associated with a lower risk of metabolic syndrome: The adventist health study 2. Diabetes Care 2011, 34, 1225–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonstad, S.; Stewart, K.; Oda, K.; Batech, M.; Herring, R.P.; Fraser, G.E. Vegetarian diets and incidence of diabetes in the Adventist Health Study-2. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 292–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tharrey, M.; Mariotti, F.; Mashchak, A.; Barbillon, P.; Delattre, M.; Fraser, G.E. Patterns of plant and animal protein intake are strongly associated with cardiovascular mortality: The Adventist Health Study-2 cohort. Int. J. Epidemiol. 2018, 47, 1603–1612. [Google Scholar] [CrossRef] [PubMed]
- Alshahrani, S.M.; Fraser, G.E.; Sabate, J.; Knutsen, R.; Shavlik, D.; Mashchak, A.; Lloren, J.I.; Orlich, M.J. Red and Processed Meat and Mortality in a Low Meat Intake Population. Nutrients 2019, 11, 622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tantamango-Bartley, Y.; Jaceldo-Siegl, K.; Fan, J.; Fraser, G. Vegetarian diets and the incidence of cancer in a low-risk population. Cancer Epidemiol. Biomark. Prev. 2013, 22, 286–294. [Google Scholar] [CrossRef] [Green Version]
- Orlich, M.J.; Singh, P.N.; Sabate, J.; Fan, J.; Sveen, L.; Bennett, H.; Knutsen, S.F.; Beeson, W.L.; Jaceldo-Siegl, K.; Butler, T.L.; et al. Vegetarian dietary patterns and the risk of colorectal cancers. JAMA Intern. Med. 2015, 175, 767–776. [Google Scholar] [CrossRef]
- Tantamango-Bartley, Y.; Knutsen, S.F.; Knutsen, R.; Jacobsen, B.K.; Fan, J.; Beeson, W.L.; Sabate, J.; Hadley, D.; Jaceldo-Siegl, K.; Penniecook, J.; et al. Are strict vegetarians protected against prostate cancer? Am. J. Clin. Nutr. 2016, 103, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Orlich, M.J.; Jaceldo-Siegl, K.; Sabate, J.; Fan, J.; Singh, P.N.; Fraser, G.E. Patterns of food consumption among vegetarians and non-vegetarians. Br. J. Nutr. 2014, 112, 1644–1653. [Google Scholar] [CrossRef] [Green Version]
- Miles, F.L.; Lloren, J.I.C.; Haddad, E.; Jaceldo-Siegl, K.; Knutsen, S.; Sabate, J.; Fraser, G.E. Plasma, Urine, and Adipose Tissue Biomarkers of Dietary Intake Differ Between Vegetarian and Non-Vegetarian Diet Groups in the Adventist Health Study-2. J. Nutr. 2019, 149, 667–675. [Google Scholar] [CrossRef]
- Field, A.E.; Robertson, N.A.; Wang, T.; Havas, A.; Ideker, T.; Adams, P.D. DNA Methylation Clocks in Aging: Categories, Causes, and Consequences. Mol. Cell 2018, 71, 882–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stylianou, E. Epigenetics of chronic inflammatory diseases. J. Inflamm. Res. 2019, 12, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Anderson, O.S.; Sant, K.E.; Dolinoy, D.C. Nutrition and epigenetics: An interplay of dietary methyl donors, one-carbon metabolism and DNA methylation. J. Nutr. Biochem. 2012, 23, 853–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, M.Z.; Chen, D.; Sun, Y.; Jin, Z.; Christman, J.K.; Yang, C.S. Reversal of hypermethylation and reactivation of p16INK4a, RARbeta, and MGMT genes by genistein and other isoflavones from soy. Clin. Cancer Res. 2005, 11, 7033–7041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Ni, X. ROS-mediated DNA methylation pattern alterations in carcinogenesis. Curr. Drug Targets 2015, 16, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Fang, M.; Lambert, J.D.; Yan, P.; Huang, T.H. Reversal of hypermethylation and reactivation of genes by dietary polyphenolic compounds. Nutr. Rev. 2008, 66 (Suppl. 1), S18–S20. [Google Scholar] [CrossRef] [Green Version]
- Hsu, A.; Wong, C.P.; Yu, Z.; Williams, D.E.; Dashwood, R.H.; Ho, E. Promoter de-methylation of cyclin D2 by sulforaphane in prostate cancer cells. Clin. Epigenetics 2011, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- Chai, W.; Morimoto, Y.; Cooney, R.V.; Franke, A.A.; Shvetsov, Y.B.; Le Marchand, L.; Haiman, C.A.; Kolonel, L.N.; Goodman, M.T.; Maskarinec, G. Dietary Red and Processed Meat Intake and Markers of Adiposity and Inflammation: The Multiethnic Cohort Study. J. Am. Coll. Nutr. 2017, 36, 378–385. [Google Scholar] [CrossRef]
- Jiao, L.; Stolzenberg-Solomon, R.; Zimmerman, T.P.; Duan, Z.; Chen, L.; Kahle, L.; Risch, A.; Subar, A.F.; Cross, A.J.; Hollenbeck, A.; et al. Dietary consumption of advanced glycation end products and pancreatic cancer in the prospective NIH-AARP Diet and Health Study. Am. J. Clin. Nutr. 2015, 101, 126–134. [Google Scholar] [CrossRef] [Green Version]
- Papuc, C.; Goran, G.V.; Predescu, C.N.; Nicorescu, V. Mechanisms of Oxidative Processes in Meat and Toxicity Induced by Postprandial Degradation Products: A Review. Compr. Rev. Food Sci. Food Saf. 2017, 16, 96–123. [Google Scholar] [CrossRef]
- Welty, F.K. How do elevated triglycerides and low HDL-cholesterol affect inflammation and atherothrombosis? Curr. Cardiol. Rep. 2013, 15, 400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoekstra, A.S.; de Graaff, M.A.; Briaire-de Bruijn, I.H.; Ras, C.; Seifar, R.M.; van Minderhout, I.; Cornelisse, C.J.; Hogendoorn, P.C.; Breuning, M.H.; Suijker, J.; et al. Inactivation of SDH and FH cause loss of 5hmC and increased H3K9me3 in paraganglioma/pheochromocytoma and smooth muscle tumors. Oncotarget 2015, 6, 38777–38788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keleher, M.R.; Zaidi, R.; Hicks, L.; Shah, S.; Xing, X.; Li, D.; Wang, T.; Cheverud, J.M. A high-fat diet alters genome-wide DNA methylation and gene expression in SM/J mice. BMC Genom. 2018, 19, 888. [Google Scholar] [CrossRef] [Green Version]
- Keleher, M.R.; Zaidi, R.; Shah, S.; Oakley, M.E.; Pavlatos, C.; El Idrissi, S.; Xing, X.; Li, D.; Wang, T.; Cheverud, J.M. Maternal high-fat diet associated with altered gene expression, DNA methylation, and obesity risk in mouse offspring. PLoS ONE 2018, 13, e0192606. [Google Scholar] [CrossRef] [Green Version]
- Masuyama, H.; Mitsui, T.; Nobumoto, E.; Hiramatsu, Y. The Effects of High-Fat Diet Exposure in Utero on the Obesogenic and Diabetogenic Traits Through Epigenetic Changes in Adiponectin and Leptin Gene Expression for Multiple Generations in Female Mice. Endocrinology 2015, 156, 2482–2491. [Google Scholar] [CrossRef] [Green Version]
- Perfilyev, A.; Dahlman, I.; Gillberg, L.; Rosqvist, F.; Iggman, D.; Volkov, P.; Nilsson, E.; Riserus, U.; Ling, C. Impact of polyunsaturated and saturated fat overfeeding on the DNA-methylation pattern in human adipose tissue: A randomized controlled trial. Am. J. Clin. Nutr. 2017, 105, 991–1000. [Google Scholar] [CrossRef] [Green Version]
- Gillberg, L.; Jacobsen, S.C.; Ronn, T.; Brons, C.; Vaag, A. PPARGC1A DNA methylation in subcutaneous adipose tissue in low birth weight subjects--impact of 5 days of high-fat overfeeding. Metabolism 2014, 63, 263–271. [Google Scholar] [CrossRef]
- Brons, C.; Jacobsen, S.; Nilsson, E.; Ronn, T.; Jensen, C.B.; Storgaard, H.; Poulsen, P.; Groop, L.; Ling, C.; Astrup, A.; et al. Deoxyribonucleic acid methylation and gene expression of PPARGC1A in human muscle is influenced by high-fat overfeeding in a birth-weight-dependent manner. J. Clin. Endocrinol. Metab. 2010, 95, 3048–3056. [Google Scholar] [CrossRef]
- ElGendy, K.; Malcomson, F.C.; Lara, J.G.; Bradburn, D.M.; Mathers, J.C. Effects of dietary interventions on DNA methylation in adult humans: Systematic review and meta-analysis. Br. J. Nutr. 2018, 120, 961–976. [Google Scholar] [CrossRef] [Green Version]
- Hastie, T.T.R.; Friedman, J. The elements of statistical learning. In Data Mining, Inference, and Prediction, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 687–692. [Google Scholar]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.; Knutsen, S.F.; Sabate, J.; Haddad, E.; Yan, R.; Fraser, G.E. Feasibility of running clinics to collect biological specimens in a nationwide cohort study--Adventist Health Study-2. Ann. Epidemiol. 2007, 17, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.A.; Lemire, M.; Choufani, S.; Butcher, D.T.; Grafodatskaya, D.; Zanke, B.W.; Gallinger, S.; Hudson, T.J.; Weksberg, R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013, 8, 203–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pidsley, R.; Zotenko, E.; Peters, T.J.; Lawrence, M.G.; Risbridger, G.P.; Molloy, P.; Van Djik, S.; Muhlhausler, B.; Stirzaker, C.; Clark, S.J. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016, 17, 208. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Behnam, E.; Huang, J.; Moffatt, M.F.; Schaid, D.J.; Liang, L.; Lin, X. Fast and robust adjustment of cell mixtures in epigenome-wide association studies with SmartSVA. BMC Genom. 2017, 18, 413. [Google Scholar] [CrossRef]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar] [CrossRef] [Green Version]
- Seber, G.A.F. Analysis of covariance and missing observations. In Linear Regression Analyses; John Wiley and Sons: New York, NY, USA, 1977. [Google Scholar]
- Freedman, D.; Lane, D. A Nonstochastic Interpretation of Reported Significance Levels. J. Bus. Econ. Stat. 1983, 1, 292–298. [Google Scholar] [CrossRef]
- Scoccianti, C.; Ricceri, F.; Ferrari, P.; Cuenin, C.; Sacerdote, C.; Polidoro, S.; Jenab, M.; Hainaut, P.; Vineis, P.; Herceg, Z. Methylation patterns in sentinel genes in peripheral blood cells of heavy smokers: Influence of cruciferous vegetables in an intervention study. Epigenetics 2011, 6, 1114–1119. [Google Scholar] [CrossRef]
- Greenlee, H.; Ogden Gaffney, A.; Aycinena, A.C.; Koch, P.; Contento, I.; Karmally, W.; Richardson, J.M.; Shi, Z.; Lim, E.; Tsai, W.Y.; et al. Long-term Diet and Biomarker Changes after a Short-term Intervention among Hispanic Breast Cancer Survivors: The inverted exclamation markCocinar Para Su Salud! Randomized Controlled Trial. Cancer Epidemiol. Biomark. Prev. 2016, 25, 1491–1502. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Cruzata, L.; Zhang, W.; McDonald, J.A.; Tsai, W.Y.; Valdovinos, C.; Falci, L.; Wang, Q.; Crew, K.D.; Santella, R.M.; Hershman, D.L.; et al. Dietary modifications, weight loss, and changes in metabolic markers affect global DNA methylation in Hispanic, African American, and Afro-Caribbean breast cancer survivors. J. Nutr. 2015, 145, 783–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Nunez, G.M.; Cabrera-Mulero, R.; Rubio-Martin, E.; Rojo-Martinez, G.; Olveira, G.; Valdes, S.; Soriguer, F.; Castano, L.; Morcillo, S. Methylation levels of the SCD1 gene promoter and LINE-1 repeat region are associated with weight change: An intervention study. Mol. Nutr. Food Res. 2014, 58, 1528–1536. [Google Scholar] [CrossRef] [PubMed]
- Duggan, C.; Xiao, L.; Terry, M.B.; McTiernan, A. No effect of weight loss on LINE-1 methylation levels in peripheral blood leukocytes from postmenopausal overweight women. Obesity 2014, 22, 2091–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondrashov, N.; Pusic, A.; Stumpf, C.R.; Shimizu, K.; Hsieh, A.C.; Ishijima, J.; Shiroishi, T.; Barna, M. Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell 2011, 145, 383–397. [Google Scholar] [CrossRef] [Green Version]
- Travers, A.A.; Kamen, R.I.; Schleif, R.F. Factor necessary for ribosomal RNA synthesis. Nature 1970, 228, 748–751. [Google Scholar] [CrossRef]
- Nazarian, R.; Falcon-Perez, J.M.; Dell’Angelica, E.C. Biogenesis of lysosome-related organelles complex 3 (BLOC-3): A complex containing the Hermansky-Pudlak syndrome (HPS) proteins HPS1 and HPS4. Proc. Natl. Acad. Sci. USA 2003, 100, 8770–8775. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Li, J.; Su, Y.; Fan, Y.; Guo, X.; Li, L.; Su, X.; Rong, R.; Ying, J.; Mo, X.; et al. A novel 3p22.3 gene CMTM7 represses oncogenic EGFR signaling and inhibits cancer cell growth. Oncogene 2014, 33, 3109–3118. [Google Scholar] [CrossRef] [Green Version]
- Uehara, T.; Minoshima, Y.; Sagane, K.; Sugi, N.H.; Mitsuhashi, K.O.; Yamamoto, N.; Kamiyama, H.; Takahashi, K.; Kotake, Y.; Uesugi, M.; et al. Selective degradation of splicing factor CAPERalpha by anticancer sulfonamides. Nat. Chem. Biol. 2017, 13, 675–680. [Google Scholar] [CrossRef]
- Russell, S.E.; Horan, R.M.; Stefanska, A.M.; Carey, A.; Leon, G.; Aguilera, M.; Statovci, D.; Moran, T.; Fallon, P.G.; Shanahan, F.; et al. IL-36alpha expression is elevated in ulcerative colitis and promotes colonic inflammation. Mucosal Immunol. 2016, 9, 1193–1204. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.F.; Wang, J.Y.; Han, W.L. A role for CMTM7 in BCR expression and survival in B-1a but not B-2 cells. Int. Immunol. 2014, 26, 47–57. [Google Scholar] [CrossRef] [Green Version]
- Bennett, E.P.; Mandel, U.; Clausen, H.; Gerken, T.A.; Fritz, T.A.; Tabak, L.A. Control of mucin-type O-glycosylation: A classification of the polypeptide GalNAc-transferase gene family. Glycobiology 2012, 22, 736–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cogan, U.; Kopelman, M.; Mokady, S.; Shinitzky, M. Binding Affinities of Retinol and Related Compounds to Retinol Binding-Proteins. Eur. J. Biochem. 1976, 65, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, R.; Zhong, M.; Kassai, M.; Ter-Stepanian, M.; Sun, H. STRA6-Catalyzed Vitamin A Influx, Efflux, and Exchange. J. Membr. Biol. 2012, 245, 731–745. [Google Scholar] [CrossRef] [Green Version]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acin-Perez, R.; Latorre-Pellicer, A.; Colas, C.; Balsa, E.; Perales-Clemente, E.; Quiros, P.M.; Calvo, E.; Rodriguez-Hernandez, M.A.; et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 2013, 340, 1567–1570. [Google Scholar] [CrossRef]
- Drummond, D.A.; Wilke, C.O. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell 2008, 134, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Roy, H.; Ibba, M. Molecular biology: Sticky end in protein synthesis. Nature 2006, 443, 41–42. [Google Scholar] [CrossRef]
- Pandolfini, L.; Barbieri, I.; Bannister, A.J.; Hendrick, A.; Andrews, B.; Webster, N.; Murat, P.; Mach, P.; Brandi, R.; Robson, S.C.; et al. METTL1 Promotes let-7 MicroRNA Processing via m7G Methylation. Mol. Cell 2019, 74, 1278–1290.e9. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Wang, G.; Hao, D.; Liu, X.; Wang, D.; Ning, N.; Li, X. Aberrant regulation of the LIN28A/LIN28B and let-7 loop in human malignant tumors and its effects on the hallmarks of cancer. Mol. Cancer 2015, 14, 125. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, A.; Wakamiya, M.; Venkova-Canova, T.; Pandita, R.K.; Aguilera-Aguirre, L.; Sarker, A.H.; Singh, D.K.; Hosoki, K.; Wood, T.G.; Sharma, G.; et al. Neil2-null Mice Accumulate Oxidized DNA Bases in the Transcriptionally Active Sequences of the Genome and Are Susceptible to Innate Inflammation. J. Biol. Chem. 2015, 290, 24636–24648. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef]
- Robert, M.F.; Morin, S.; Beaulieu, N.; Gauthier, F.; Chute, I.C.; Barsalou, A.; MacLeod, A.R. DNMT1 is required to maintain CpG methylation and aberrant gene silencing in human cancer cells. Nat. Genet. 2003, 33, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Grewal, S.I.; Moazed, D. Heterochromatin and epigenetic control of gene expression. Science 2003, 301, 798–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stirzaker, C.; Taberlay, P.C.; Statham, A.L.; Clark, S.J. Mining cancer methylomes: Prospects and challenges. Trends Genet. 2014, 30, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Ustianenko, D.; Chiu, H.S.; Treiber, T.; Weyn-Vanhentenryck, S.M.; Treiber, N.; Meister, G.; Sumazin, P.; Zhang, C. LIN28 Selectively Modulates a Subclass of Let-7 MicroRNAs. Mol. Cell 2018, 71, 271–283.e275. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Su, Y.; Li, T.; Yuan, W.; Mo, X.; Li, H.; He, Q.; Ma, D.; Han, W. CMTM7 knockdown increases tumorigenicity of human non-small cell lung cancer cells and EGFR-AKT signaling by reducing Rab5 activation. Oncotarget 2015, 6, 41092–41107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Mata, R.; Boulter, E.; Burridge, K. The ‘invisible hand’: Regulation of RHO GTPases by RHOGDIs. Nat. Rev. Mol. Cell Biol. 2011, 12, 493–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, A.M.; Rendon, B.E.; Zhao, M.; Qian, M.W.; Bucala, R.; Xin, D.; Mitchell, R.A. Cooperative regulation of non-small cell lung carcinoma angiogenic potential by macrophage migration inhibitory factor and its homolog, D-dopachrome tautomerase. J. Immunol. 2008, 181, 2330–2337. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Li, W.W.; Yang, B.W.; Tao, Z.H.; Sun, H.C.; Wang, L.; Xia, J.L.; Qin, L.X.; Tang, Z.Y.; Fan, J.; et al. Aryl hydrocarbon receptor nuclear translocator is associated with tumor growth and progression of hepatocellular carcinoma. Int. J. Cancer 2012, 130, 1745–1754. [Google Scholar] [CrossRef] [PubMed]
- Pasupuleti, V.; Du, W.; Gupta, Y.; Yeh, I.J.; Montano, M.; Magi-Galuzzi, C.; Welford, S.M. Dysregulated D-dopachrome tautomerase, a hypoxia-inducible factor-dependent gene, cooperates with macrophage migration inhibitory factor in renal tumorigenesis. J. Biol. Chem. 2014, 289, 3713–3723. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.L.; Yoon, D.Y.; Hodge-Bell, K.C.; Bebenek, I.G.; Whitekus, M.J.; Zhang, R.X.; Cochran, A.J.; Huerta-Yepez, S.; Yim, S.H.; Gonzalez, F.J.; et al. The aryl hydrocarbon receptor nuclear translocator (Arnt) is required for tumor initiation by benzo[a]pyrene. Carcinogenesis 2009, 30, 1957–1961. [Google Scholar] [CrossRef]
- Hao, N.; Bhakti, V.L.; Peet, D.J.; Whitelaw, M.L. Reciprocal regulation of the basic helix-loop-helix/Per-Arnt-Sim partner proteins, Arnt and Arnt2, during neuronal differentiation. Nucleic Acids Res. 2013, 41, 5626–5638. [Google Scholar] [CrossRef] [PubMed]
- Bishop, K.S.; Ferguson, L.R. The interaction between epigenetics, nutrition and the development of cancer. Nutrients 2015, 7, 922–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dijk, S.J.; Zhou, J.; Peters, T.J.; Buckley, M.; Sutcliffe, B.; Oytam, Y.; Gibson, R.A.; McPhee, A.; Yelland, L.N.; Makrides, M.; et al. Effect of prenatal DHA supplementation on the infant epigenome: Results from a randomized controlled trial. Clin. Epigenetics 2016, 8, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmoud, A.M.; Ali, M.M. Methyl Donor Micronutrients that Modify DNA Methylation and Cancer Outcome. Nutrients 2019, 11, 608. [Google Scholar] [CrossRef] [Green Version]
- Crider, K.S.; Yang, T.P.; Berry, R.J.; Bailey, L.B. Folate and DNA methylation: A review of molecular mechanisms and the evidence for folate’s role. Adv. Nutr. 2012, 3, 21–38. [Google Scholar] [CrossRef] [Green Version]
- Bacalini, M.G.; Boattini, A.; Gentilini, D.; Giampieri, E.; Pirazzini, C.; Giuliani, C.; Fontanesi, E.; Remondini, D.; Capri, M.; Del Rio, A.; et al. A meta-analysis on age-associated changes in blood DNA methylation: Results from an original analysis pipeline for Infinium 450k data. Aging 2015, 7, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Bouchard, L.; Rabasa-Lhoret, R.; Faraj, M.; Lavoie, M.E.; Mill, J.; Perusse, L.; Vohl, M.C. Differential epigenomic and transcriptomic responses in subcutaneous adipose tissue between low and high responders to caloric restriction. Am. J. Clin. Nutr. 2010, 91, 309–320. [Google Scholar] [CrossRef] [Green Version]
- Milagro, F.I.; Campion, J.; Cordero, P.; Goyenechea, E.; Gomez-Uriz, A.M.; Abete, I.; Zulet, M.A.; Martinez, J.A. A dual epigenomic approach for the search of obesity biomarkers: DNA methylation in relation to diet-induced weight loss. FASEB J. 2011, 25, 1378–1389. [Google Scholar] [CrossRef] [Green Version]
- Breton, C.V.; Marsit, C.J.; Faustman, E.; Nadeau, K.; Goodrich, J.M.; Dolinoy, D.C.; Herbstman, J.; Holland, N.; LaSalle, J.M.; Schmidt, R.; et al. Small-Magnitude Effect Sizes in Epigenetic End Points are Important in Children’s Environmental Health Studies: The Children’s Environmental Health and Disease Prevention Research Center’s Epigenetics Working Group. Environ. Health Perspect. 2017, 125, 511–526. [Google Scholar] [CrossRef]
- Li, Y.; Liu, L.; Tollefsbol, T.O. Glucose restriction can extend normal cell lifespan and impair precancerous cell growth through epigenetic control of hTERT and p16 expression. FASEB J. 2010, 24, 1442–1453. [Google Scholar] [CrossRef] [Green Version]
- Tate, P.H.; Bird, A.P. Effects of DNA methylation on DNA-binding proteins and gene expression. Curr. Opin. Genet. Dev. 1993, 3, 226–231. [Google Scholar] [CrossRef]
- Martins, M.C.T.; Jaceldo-Siegl, K.; Orlich, M.; Fan, J.; Mashchak, A.; Fraser, G.E. A New Approach to Assess Lifetime Dietary Patterns Finds Lower Consumption of Animal Foods with Aging in a Longitudinal Analysis of a Health-Oriented Adventist Population. Nutrients 2017, 9, 1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byun, H.M.; Siegmund, K.D.; Pan, F.; Weisenberger, D.J.; Kanel, G.; Laird, P.W.; Yang, A.S. Epigenetic profiling of somatic tissues from human autopsy specimens identifies tissue- and individual-specific DNA methylation patterns. Hum. Mol. Genet. 2009, 18, 4808–4817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crujeiras, A.B.; Diaz-Lagares, A.; Sandoval, J.; Milagro, F.I.; Navas-Carretero, S.; Carreira, M.C.; Gomez, A.; Hervas, D.; Monteiro, M.P.; Casanueva, F.F.; et al. DNA methylation map in circulating leukocytes mirrors subcutaneous adipose tissue methylation pattern: A genome-wide analysis from non-obese and obese patients. Sci. Rep. 2017, 7, 41903. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.C.; Wang, Q.; Yang, H.I.; Tsai, W.Y.; Chen, C.J.; Santella, R.M. Global DNA methylation levels in white blood cells as a biomarker for hepatocellular carcinoma risk: A nested case-control study. Carcinogenesis 2012, 33, 1340–1345. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Vegan | Non-Vegetarian | p | |
|---|---|---|---|
| Participants (n) | 62 | 142 | |
| Sex (%) | 0.57 | ||
| Male | 30 (48.4) | 61 (43) | |
| Female | 32 (51.6) | 81 (57.0) | |
| Age (years) | 67.3 (9.0) | 65.9 (8.4) | 0.28 |
| BMI (kg/m2) | 24.3 (4.6) | 28 (5.1) | <0.001 |
| Ethnicity (%) | 0.50 | ||
| Caucasian | 46 (74.2) | 97 (68.3) | |
| Black | 16 (25.8) | 45 (31.7) |
| Region 2 | Total Genes | Estimated Non-Null | Significantly Hypermethylated | Significantly Hypomethylated | ||||
|---|---|---|---|---|---|---|---|---|
| n | % of All Genes | n3 | % of Region-Specific Total | n | Fold Change 4 | n | Fold Change 4 | |
| All | 18,627 | 100 | 1081 | 5.8 | 4 | 1.02 | 14 | 0.97 |
| Genic/intergenic | ||||||||
| TSS200 | 10,008 | 53.7 | 388 | 3.9 | 4 | 1.03 | 4 | 0.96 |
| TSS1500 | 13,373 | 71.8 | 954 | 7.1 | 2 | 1.03 | 7 | 0.97 |
| 3′ UTR | 1935 | 10.4 | 55 | 2.8 | 1 | 1.04 | 2 | 0.98 |
| 5′ UTR | 7686 | 41.3 | 475 | 6.2 | 2 | 1.02 | 6 | |
| Gene Body | 12,072 | 64.8 | 1100 | 9.1 | 7 | 1.03 | 14 | 0.97 |
| 1st Exon | 6262 | 33.6 | 405 | 6.5 | 1 | 1.07 | 9 | 0.97 |
| Intergenic | 8049 | 43.2 | 449 | 5.6 | 1 | 1.05 | 7 | 0.97 |
| Island-related | ||||||||
| CpG Island | 13,688 | 73.5 | 649 | 4.7 | 4 | 1.03 | 2 | 0.97 |
| North Shelf | 2562 | 13.8 | 180 | 7.0 | 2 | 1.03 | 2 | 0.97 |
| North Shore | 8315 | 44.6 | 424 | 5.1 | 1 | 1.03 | 7 | 0.96 |
| South Shelf | 2278 | 12.2 | 146 | 6.4 | 3 | 1.04 | 7 | 0.97 |
| South Shore | 7137 | 38.3 | 441 | 6.2 | 5 | 1.06 | 7 | 0.96 |
| Open Sea | 11,884 | 63.8 | 798 | 6.7 | 2 | 1.04 | 14 | 0.96 |
| Promoter | ||||||||
| All | 9131 | 49.0 | 459 | 5.0 | 2 | 1.05 | 9 | 0.98 |
| CpG Island | 7693 | 41.3 | 60 | 0.8 | 1 | 1.02 | 2 | 0.97 |
| Fold Increase in Sample Size—Hypomethylated Genes | Fold Increase in Sample Size—Hypermethylated Genes | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Region | 1 | 1.5 | 2 | 3 | 4 | 5 | 1 | 1.5 | 2 | 3 | 4 | 5 |
| All | 14 | 28 | 42 | 66 | 79 | 86 | 4 | 29 | 103 | 266 | 377 | 456 |
| Body | 14 | 23 | 37 | 58 | 68 | 91 | 7 | 17 | 65 | 159 | 254 | 309 |
| TSS1500 | 7 | 7 | 24 | 31 | 43 | 49 | 2 | 25 | 108 | 254 | 350 | 411 |
| TSS200 | 4 | 4 | 4 | 12 | 19 | 19 | 4 | 9 | 44 | 141 | 203 | 254 |
| 3′ UTR | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 1 | 10 | 30 | 47 | 57 |
| 5′ UTR | 6 | 6 | 12 | 17 | 26 | 28 | 2 | 15 | 47 | 122 | 172 | 226 |
| 1st Exon | 9 | 10 | 23 | 33 | 37 | 43 | 1 | 10 | 45 | 96 | 142 | 169 |
| Island | 2 | 2 | 12 | 26 | 27 | 33 | 4 | 27 | 98 | 243 | 336 | 397 |
| North Shore | 7 | 7 | 12 | 23 | 24 | 28 | 1 | 8 | 47 | 144 | 222 | 260 |
| South Shore | 7 | 7 | 16 | 24 | 31 | 36 | 5 | 19 | 60 | 124 | 175 | 229 |
| North Shelf | 2 | 2 | 2 | 7 | 7 | 7 | 2 | 4 | 17 | 46 | 69 | 83 |
| South Shelf | 7 | 7 | 7 | 7 | 7 | 7 | 3 | 9 | 21 | 39 | 59 | 71 |
| Promoter Associated | 9 | 9 | 12 | 16 | 17 | 20 | 2 | 12 | 62 | 145 | 214 | 250 |
| Promoter and CpG Island | 2 | 2 | 2 | 5 | 10 | 13 | 1 | 8 | 45 | 112 | 161 | 201 |
| Open Sea | 14 | 20 | 28 | 44 | 58 | 61 | 2 | 11 | 61 | 144 | 218 | 256 |
| Intergenic | 7 | 8 | 19 | 30 | 40 | 51 | 1 | 7 | 40 | 101 | 154 | 185 |
| Gene ID | Gene Symbol | Description | Fold Change |
|---|---|---|---|
| TSS200 | |||
| 341,350 | OVCH1 (1) | ovochymase 1 | 0.93 |
| 8499 | PPFIA2 | PTPRF interacting protein alpha 2 | 0.96 |
| 51,099 | ABHD5 | abhydrolase domain containing 5, lysophosphatidic acid acyltransferase | 0.98 |
| 51,205 | ACP6 | acid phosphatase 6, lysophosphatidic | 0.98 |
| 9874 | TLK1 | tousled-like kinase 1 | 1.02 |
| 10,440 | TIMM17A | translocase of inner mitochondrial membrane 17A | 1.02 |
| 1132 | CHRM4 | cholinergic receptor, muscarinic 4 | 1.03 |
| 9570 | GOSR2 | lgi SNAP receptor complex member 2 | 1.03 |
| TSS1500 | |||
| 151,313 | FAHD2B | fumarylacetoacetate hydrolase domain containing 2B | 0.95 |
| 10,653 | SPINT2 | serine peptidase inhibitor, Kunitz type 2 | 0.96 |
| 100,302,640 | LINC00882 | Long Intergenic Non-Protein Coding RNA 882 | 0.96 |
| 84,838 | ZNF496 (1) | zinc finger protein 496 | 0.96 |
| 4234 | METTL1 | methyltransferase-like 1 | 0.98 |
| 9054 | NFS1 | NFS1 cysteine desulfurase | 0.98 |
| 1263 | PLK3 | polo-like kinase 3 | 0.98 |
| 8546 | AP3B1 | adaptor-related protein complex 3 subunit beta 1 | 1.02 |
| 11,267 | SNF8 | SNF8 subunit of ESCRT-II | 1.04 |
| 3′ UTR | |||
| 79,960 | JADE1 | jade family PHD finger 1 | 0.97 |
| 667 | DST | dystonin | 0.98 |
| 54,820 | NDE1 | nudE neurodevelopment protein 1 | 1.04 |
| 5′ UTR | |||
| 252,969 | NEIL2 | nei-like DNA glycosylase 2 | 0.96 |
| 4147 | MATN2 | matrilin 2 | 0.97 |
| 5928 | RBBP4 | RB binding protein 4, chromatin remodeling factor | 0.97 |
| 3792 | KEL | Kell metallo-endopeptidase (Kell blood group) | 0.97 |
| 9772 | TMEM94 | transmembrane protein 94 | 0.98 |
| 63,924 | CIDEC | cell death-inducing DFFA-like effector c | 0.99 |
| 51,465 | UBE2J1 | ubiquitin conjugating enzyme E2 J1 | 1.02 |
| 1374 | CPT1A | carnitine palmitoyltransferase 1A | 1.03 |
| Body | |||
| 100,037,417 | DDTL | D-dopachrome decarboxylase-like protein | 0.92 |
| 154,007 | SNRNP48 | small nuclear ribonucleoprotein U11/U12 subunit 48 | 0.95 |
| 8120 | AP3B2 | adaptor-related protein complex 3 subunit beta 2 | 0.96 |
| 89,781 | HPS4 | HPS4, biogenesis of lysosomal organelles complex 3 subunit 2 | 0.96 |
| 2668 | GDNF | glial cell line derived neurotrophic factor | 0.96 |
| 56,666 | PANX2 | pannexin 2 | 0.97 |
| 91,010 | FMNL3 | formin-like 3 | 0.98 |
| 117,246 | FTSJ3 | FtsJ RNA 2’-O-methyltransferase 3 | 0.98 |
| 11,052 | CPSF6 | cleavage and polyadenylation-specific factor 6 | 0.98 |
| 149,297 | FAM78B | family with sequence similarity 78 member B | 0.98 |
| 399,671 | HEATR4 | HEAT repeat containing 4 | 0.98 |
| 285,987 | DLX6-AS1 | DLX6 antisense RNA 1 | 0.99 |
| 64,784 | CRTC3 | CREB regulated transcription coactivator 3 | 0.99 |
| 5361 | PLXA1 | plexin A1 | 0.99 |
| Body | |||
| 9915 | ARNT2 | aryl hydrocarbon receptor nuclear translocator 2 | 1.02 |
| 5170 | PDPK1 | 3-phosphoinositide dependent protein kinase 1 | 1.02 |
| 10,610 | ST6GALNAC2 | ST6 N-acetylgalactosaminide alpha-2,6-sialyltransferase 2 | 1.02 |
| 57,459 | GATAD2B | GATA zinc finger domain containing 2B | 1.03 |
| 3694 | ITGB6 | integrin subunit beta 6 | 1.04 |
| 58 | ACTA1 | actin alpha 1, skeletal muscle | 1.05 |
| 9963 | SLC23A1 | solute carrier family 23 member 2 | 1.06 |
| First Exon | |||
| 2201 | FBN2 | fibrillin 2 | 0.96 |
| 100,303,453 | TSNAX-DISC1 | Disrupted in schizophrenia 1 isoform 51 | 0.96 |
| 10,274 | STAG1 | stromal antigen 1 | 0.97 |
| 252,969 | NEIL2 | nei-like DNA glycosylase 2 | 0.97 |
| 3489 | IGFBP6 | insulin-like growth factor binding protein 6 | 0.97 |
| 5928 | RBBP4 | RB binding protein 4, chromatin remodeling factor | 0.97 |
| 6727 | SRP14 | signal recognition particle 14 | 0.97 |
| 9167 | COX7A2L (1) | cytochrome c oxidase subunit 7A2-like | 0.97 |
| 1802 | DPH2(1) | diphthamide biosynthesis 2 | 0.98 |
| 23,336 | SYNM | Synemin | 1.07 |
| All | |||
| 100,037,417 | DDTL | D-dopachrome tautomerase-like | 0.95 |
| 284,680 | SPATA46 | spermatogenesis associated 46 | 0.96 |
| 100,113,403 | LIN28B-AS1 | LIN28B antisense RNA 1 | 0.96 |
| 64,220 | STRA6 | signaling receptor and transporter of retinol STRA6 | 0.97 |
| 4234 | METTL1 | methyltransferase-like 1 | 0.97 |
| 27,179 | IL36A | interleukin 36 alpha | 0.98 |
| 90,379 | DCAF15 | DDB1 and CUL4 associated factor 15 | 0.98 |
| 89,781 | HPS4 | HPS4, biogenesis of lysosomal organelles complex 3 subunit 2 | 0.98 |
| 136,371 | ASB10 | ankyrin repeat and SOCS box containing 10 | 0.98 |
| 397 | ARHGDIB | Rho GDP dissociation inhibitor beta | 0.98 |
| 6169 | RPL38 | ribosomal protein L38 | 0.98 |
| 55,222 | LRRC20 | leucine rich repeat containing 20 | 0.99 |
| 9491 | PSMF1 | proteasome inhibitor subunit 1 | 0.99 |
| 154,150 | HDGFL1 | HDGF-like 1 | 0.99 |
| 9915 | ARNT2 | aryl hydrocarbon receptor nuclear translocator 2 | 1.02 |
| 219,990 | OOSP2 | oocyte secreted protein 2 | 1.02 |
| 114,757 | CYGB | cytoglobin | 1.02 |
| 7284 | TUFM | Tu translation elongation factor, mitochondrial | 1.02 |
| Intergenic | |||
| 136,371 | ASB10 | Ankyrin repeat and SOCS box protein 10 | 0.95 |
| 100,113,403 | LIN28B-AS1 | LIN28B antisense RNA 1 | 0.96 |
| 23,704 | KCNE4 | Potassium voltage-gated channel subfamily E member 4 | 0.97 |
| 27,319 | BHLHE22 | Class E basic helix-loop-helix protein 22 | 0.97 |
| 9935 | MAFB | Transcription factor MafB | 0.98 |
| 7100 | TLR5 | Toll-like receptor 5 | 0.98 |
| 154,150 | HDGFL1 | Hepatoma-derived growth factor-like protein 1 | 0.99 |
| 284,889 | MIF-AS1 | MIF antisense RNA | 1.05 |
| Gene ID | Gene Symbol | Description | Fold Change |
|---|---|---|---|
| Island | |||
| 80,070 | ADAMTS20 | ADAM metallopeptidase with thrombospondin type 1 motif 20 | 0.95 |
| 101,409,261 | OGFR-AS1 | OGFR Antisense RNA 1 | 0.98 |
| 4609 | MYC | MYC proto-oncogene | 1.01 |
| 161,145 | TMEM229B | transmembrane protein 229B | 1.02 |
| 7284 | TUFM | Tu translation elongation factor, mitochondrial | 1.04 |
| 273 | AMPH | amphiphysin | 1.05 |
| North Shelf | |||
| 4000 | LMNA | lamin A/C | 0.97 |
| 123,041 | SLC24A4 | solute carrier family 24 member 4 | 0.97 |
| 100,534,592 | URGCP-MRPS24 | URGCP-MRPS24 readthrough | 1.02 |
| 116,844 | LRG1 | leucine rich alpha-2-glycoprotein 1 | 1.04 |
| North Shore | |||
| 8022 | LHX3 | LIM homeobox protein 3 | 0.92 |
| 10,653 | SPINT2 | serine peptidase inhibitor, Kunitz type 2 | 0.95 |
| 6169 | RPL38 | ribosomal protein L38 | 0.96 |
| 23,200 | ATP11B | Probable phospholipid-transporting ATPase IF | 0.96 |
| 7707 | ZNF148 | Zinc finger protein 148 | 0.97 |
| 26,156 | RSL1D1 | ribosomal L1 domain containing 1 | 0.97 |
| 9379 | NRXN2 | Neurexin-2 | 0.98 |
| 9480 | ONECUT2 | one cut homeobox 2 | 1.03 |
| South Shelf | |||
| 55,608 | ANKRD10 | ankyrin repeat domain 10 | 0.95 |
| 26,693 | OR2V1 | olfactory receptor family 2 subfamily V member 1 | 0.96 |
| 2180 | ACSL1 | acyl-CoA synthetase long chain family member 1 | 0.96 |
| 399,829 | LINC01168 | long intergenic non-protein coding RNA 1168 | 0.97 |
| 2774 | GNAL | G protein subunit alpha L | 0.97 |
| 64,319 | FBRS | fibrosin | 0.97 |
| 84,286 | TMEM175 | transmembrane protein 175 | 0.97 |
| 140,706 | CCM2L | cerebral cavernous malformation 2-like | 1.04 |
| 1112 | FOXN3 | forkhead box N3 | 1.04 |
| 10,410 | IFITM3 | interferon-induced transmembrane protein 3 | 1.04 |
| South Shore | |||
| 41 | ASIC1 | acid-sensing (proton-gated) ion channel 1 | 0.94 |
| 151,313 | FAHD2B | fumarylacetoacetate hydrolase domain containing 2B | 0.95 |
| 84,838 | ZNF496 (2) | zinc finger protein 496 | 0.95 |
| 116,988 | AGAP3 | ArfGAP with GTPase domain, ankyrin repeat and PH domain 3 | 0.95 |
| 9735 | KNTC1 | kinetochore associated 1 | 0.96 |
| 29,993 | PACSIN1 | protein kinase C and casein kinase substrate in neurons 1 | 0.96 |
| 85,395 | FAM207A | family with sequence similarity 207 member A | 0.97 |
| 57,186 | RALGAPA2 | Ral GTPase activating protein catalytic subunit alpha 2 | 1.03 |
| 55,243 | KIRREL1 | kirre-like nephrin family adhesion molecule 1 | 1.03 |
| 11,267 | SNF8 | SNF8 subunit of ESCRT-II | 1.04 |
| 375,061 | FAM89A | family with sequence similarity 89, member A | 1.06 |
| 57,495 | NWD2 | NACHT and WD repeat domain containing 2 | 1.12 |
| Open Sea | |||
| 25,774 | GSTTP1 | glutathione S-transferase theta 4 | 0.72 |
| 402,381 | SOHLH1 | spermatogenesis and oogenesis specific basic helix-loop-helix 1 | 0.96 |
| 3034 | HAL | histidine ammonia-lyase | 0.96 |
| 64,220 | STRA6 | signaling receptor and transporter of retinol STRA6 | 0.97 |
| 134,510 | UBLCP1 | ubiquitin-like domain containing CTD phosphatase 1 | 0.97 |
| 1188 | CLCNKB | Chloride channel protein ClC-Kb | 0.97 |
| 1610 | DAO | D-amino-acid oxidase | 0.98 |
| 397 | ARHGDIB | Rho GDP-dissociation inhibitor 2 | 0.98 |
| 27,179 | IL36A | interleukin 36 alpha | 0.98 |
| 54,989 | ZNF770 | zinc finger protein 770 | 0.98 |
| 83,394 | PITPNM3 | PITPNM family member 3 | 0.98 |
| 79,981 | FRMD1 | FERM domain containing 1 | 0.98 |
| 55,137 | FIGN | fidgetin | 0.99 |
| 114,879 | OSBPL5 | oxysterol binding protein-like 5 | 0.99 |
| 219,990 | OOSP2 | oocyte secreted protein 2 | 1.02 |
| 100,130,673 | LOC100130673 | phosphoribosyl pyrophosphate synthetase 2 pseudogene | 1.06 |
| Promoter | |||
| 55,640 | FLVCR2 | FLVCR heme transporter 2 | 0.95 |
| 79,695 | GALNT12 | polypeptide N-acetylgalactosaminyltransferase 12 | 0.96 |
| 6169 | RPL38 | ribosomal protein L38 | 0.97 |
| 4234 | METTL1 | methyltransferase-like 1 | 0.97 |
| 112,616 | CMTM7 | CKLF-like MARVEL transmembrane domain containing 7 | 0.98 |
| 10,073 | SNUPN | snurportin 1 | 0.98 |
| 80,321 | CEP70 | centrosomal protein 70 | 0.98 |
| 9167 | COX7A2L | cytochrome c oxidase subunit 7A2-like | 0.99 |
| 100,288,142 | NBPF20 | NBPF member 20 | 0.99 |
| 8546 | AP3B1 | adaptor-related protein complex 3, beta 1 subunit | 1.01 |
| 100,506,334 | LINC00649 (1) | long intergenic non-protein coding RNA 649 | 1.08 |
| Islands in Promoter | |||
| 4234 | METTL1 | methyltransferase-like 1 | 0.97 |
| 252,969 | NEIL2 | nei-like DNA glycosylase 2 | 0.97 |
| 200,312 | RNF215 | ring finger protein 215 | 1.02 |
| Region 2 | Total Genes | Estimated Non-Null | Significantly Hypermethylated | Significantly Hypomethylated | ||||
|---|---|---|---|---|---|---|---|---|
| n | % of All CpG Sites | n3 | % of Region-Specific Total | n | Fold Change 4 | n | Fold Change 4 | |
| All | 313,161 | 100 | 27,276 | 8.7 | 6 | 1.06 | 8 | 0.92 |
| Genic/intergenic | ||||||||
| TSS200 | 40,816 | 13.0 | 2355 | 5.8 | 5 | 1.05 | 3 | 0.91 |
| TSS1500 | 57,450 | 18.3 | 3950 | 6.9 | 5 | 1.06 | 8 | 0.93 |
| 3′ UTR | 12,840 | 4.1 | 633 | 4.9 | 3 | 1.04 | 6 | 0.93 |
| 5′ UTR | 43,313 | 13.8 | 3270 | 7.6 | 4 | 1.08 | 3 | 0.95 |
| Gene Body | 114,095 | 36.4 | 9835 | 8.6 | 7 | 1.08 | 6 | 0.95 |
| 1st Exon | 25,662 | 8.2 | 2082 | 8.1 | 1 | 1.04 | 7 | 0.92 |
| Intergenic | 72,524 | 23.2 | 5798 | 8.0 | 9 | 1.06 | 3 | 0.92 |
| Island-related | ||||||||
| CpG Island | 99,350 | 31.7 | 7883 | 7.9 | 4 | 1.07 | 4 | 0.92 |
| North Shelf | 15,106 | 4.8 | 923 | 6.1 | 7 | 1.05 | 6 | 0.93 |
| North Shore | 43,549 | 13.9 | 3542 | 8.1 | 6 | 1.06 | 3 | 0.94 |
| South Shelf | 13,564 | 4.3 | 974 | 7.2 | 9 | 1.06 | 3 | 0.93 |
| South Shore | 33,905 | 10.8 | 2652 | 7.8 | 5 | 1.08 | 7 | 0.93 |
| Open Sea | 107,687 | 34.4 | 8266 | 7.7 | 5 | 1.06 | 5 | 0.93 |
| Promoter | ||||||||
| All | 62,398 | 19.9 | 2308 | 3.7 | 6 | 1.08 | 3 | 0.95 |
| CpG Island | 39,230 | 12.5 | 1713 | 4.4 | 1 | 1.07 | 3 | 0.96 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miles, F.L.; Mashchak, A.; Filippov, V.; Orlich, M.J.; Duerksen-Hughes, P.; Chen, X.; Wang, C.; Siegmund, K.; Fraser, G.E. DNA Methylation Profiles of Vegans and Non-Vegetarians in the Adventist Health Study-2 Cohort. Nutrients 2020, 12, 3697. https://doi.org/10.3390/nu12123697
Miles FL, Mashchak A, Filippov V, Orlich MJ, Duerksen-Hughes P, Chen X, Wang C, Siegmund K, Fraser GE. DNA Methylation Profiles of Vegans and Non-Vegetarians in the Adventist Health Study-2 Cohort. Nutrients. 2020; 12(12):3697. https://doi.org/10.3390/nu12123697
Chicago/Turabian StyleMiles, Fayth L., Andrew Mashchak, Valery Filippov, Michael J. Orlich, Penelope Duerksen-Hughes, Xin Chen, Charles Wang, Kimberly Siegmund, and Gary E. Fraser. 2020. "DNA Methylation Profiles of Vegans and Non-Vegetarians in the Adventist Health Study-2 Cohort" Nutrients 12, no. 12: 3697. https://doi.org/10.3390/nu12123697
APA StyleMiles, F. L., Mashchak, A., Filippov, V., Orlich, M. J., Duerksen-Hughes, P., Chen, X., Wang, C., Siegmund, K., & Fraser, G. E. (2020). DNA Methylation Profiles of Vegans and Non-Vegetarians in the Adventist Health Study-2 Cohort. Nutrients, 12(12), 3697. https://doi.org/10.3390/nu12123697